
Abstract
A 1-month-old Purebred Spanish Horse (PSH) foal presented with progressive hepatic failure culminating in death. Hepatic lesions were consistent with congenital hepatic fibrosis (CHF). Genetic studies in the PKHD1 gene in the affected foal revealed that it was heterozygous for the 2 previously described single-nucleotide polymorphisms (SNPs) linked to CHF in Swiss Franches-Montagnes (SFM) horses. In addition, 2 novel mutations were detected, the foal being homozygous for one of them and heterozygous for the other. Genetic studies in a healthy PSH population (n = 35) showed a 3-fold higher genotypic frequency for PKHD1 SNP g.49,630,834G>A and a 5-fold higher genotypic frequency for PKHD1 SNP g.49,597,760A>T compared with those reported for SFM horses. SNPs in the PKHD1 gene in CHF-affected SFM horses might not fully explain the CHF observed in the PSH. Other mutations in the PKHD1 gene could play a more important role in the PSH.
Congenital hepatic fibrosis (CHF) is a developmental disorder of the biliary system caused by a congenital defect in the remodeling of the ductal plate at the level of the interlobular ducts, resulting in persistence of excessive abnormal embryonic bile duct structures, irregular portal veins, and progressive fibrosis of the portal tracts, with or without macroscopic cystic dilation of intrahepatic bile ducts. 6,10 CHF has been described mostly in humans as well as sporadically reported in different species, including dogs, 4,11 cats, 2,18 calves, 3,9,14 and a nonhuman primate. 16 In horses, CHF has been described only in foals of the Swiss Franches-Montagnes (SFM) breed, and it consists of a lethal, monogenic autosomal recessive inherited disease linked to 2 mutations in the polycystic kidney and hepatic disease 1 (PKHD1) gene. 7,8,13
The objective of this work was to describe a CHF case in a Purebred Spanish Horse (PSH) foal and to test the 2 mutations in the PKHD1 gene associated with CHF, in both the affected PSH foal and in a healthy population of related PSHs (n = 5). In addition, genic and genotypic frequencies for the mentioned mutations were calculated in unrelated PSHs (n = 30).
A 1-month-old PSH foal presented with a clinical history of profuse diarrhea with acholic feces during its first week of life, followed by constipation until death. On physical examination, the animal showed dehydration, jaundice, and neurological signs such as depression, incoordination, and apparent blindness. The abdominal ultrasound showed hepatomegaly and a heterogeneous hyperechogenicity, compatible with severe fibrosis. Intravenous antibiotic therapy and rehydration were established, but the animal died a few hours later.
Postmortem examination was performed by the referring veterinarian. Grossly, the liver was enlarged, with increased consistency and a 2-fold increase in weight. The hepatic surface showed diffuse gray discoloration (Fig. 1), and the section exhibited prominent bridging fibrosis (Fig. 2) with a small cyst filled with bile-like fluid (Fig. 2, inset). The small intestine was dilated and full of acholic feces.
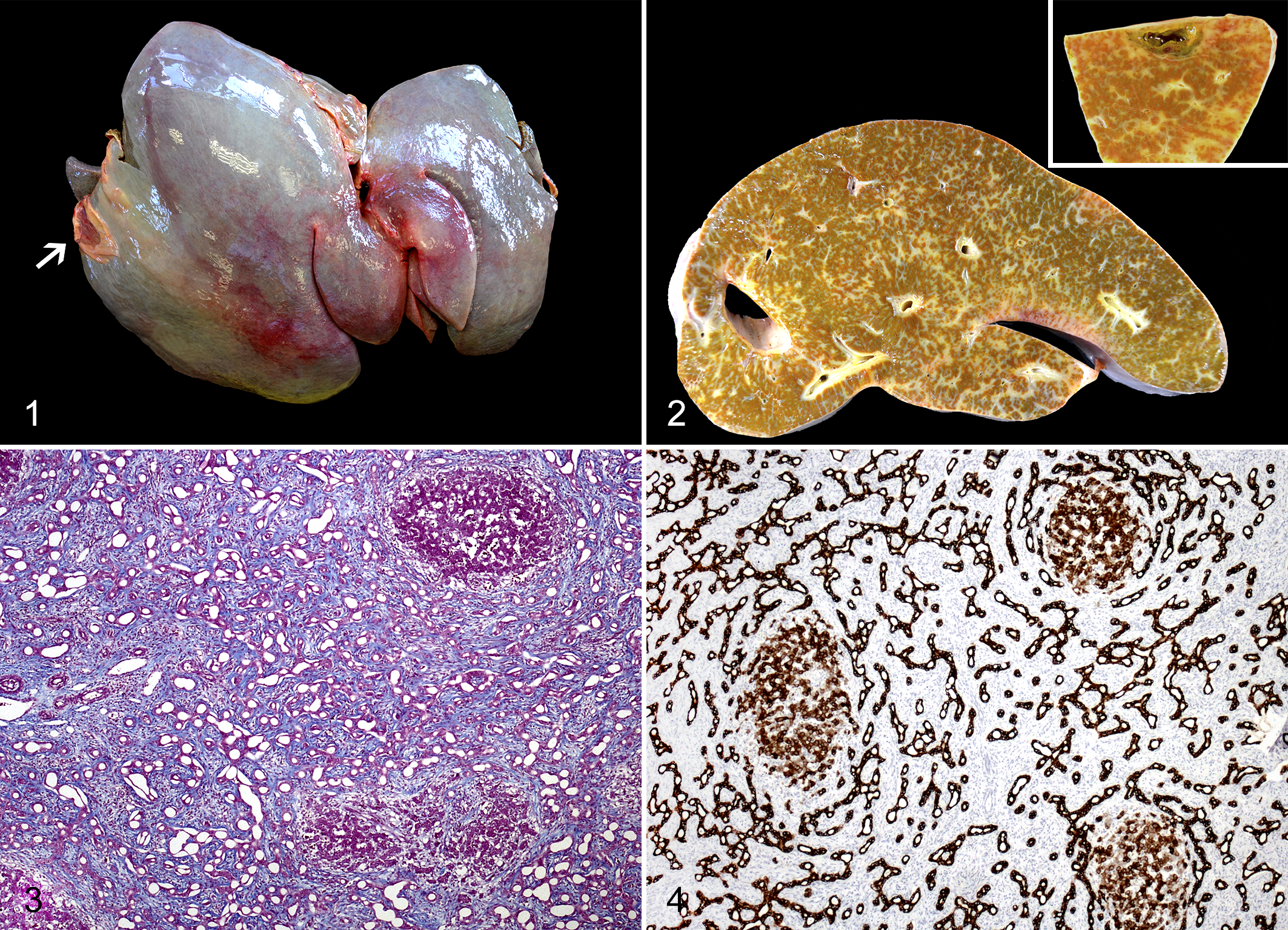
Samples from liver, spleen, kidney, and small intestine were collected at necropsy and routinely processed for histopathological examination. Sections from liver and kidney samples were stained with Masson’s trichrome, and sections from liver were immunostained against pancytokeratin (AE1/AE3; 1:500; Dako, Carpinteria, CA, USA) and Ki-67 (1:100; Dako). Microscopically, the liver showed a severe, diffuse portal-to-portal bridging fibrosis with abundant deposition of collagen fibers and maintenance of the limiting plate (Fig. 3), intermixed with variable number of fibrocytes and fibroblasts together with scarce lymphoplasmacytic inflammation. Scattered within the fibrous stroma, there were numerous small and irregularly shaped bile duct profiles forming a complex, strongly pancytokeratin-positive branching network (Fig. 4). Bile duct profiles were lined by a single layer of cuboidal to low columnar epithelial cells (Fig. 5) with rare mitoses. Most biliary epithelial cells were negative for Ki-67 staining, although randomly distributed, scattered positive cells were rarely observed (Suppl. Fig. S1). Bile duct profiles were multifocally accompanied by arteriolar proliferation and mostly absent or hypoplastic portal veins (Fig. 6). Remaining hepatocytes showed different degrees of degenerative and necrotic changes and a diffuse, moderate, but less intense cytoplasmic positivity for pancytokeratin than biliary ducts (Fig. 4). Hepatocytes lacked nuclear staining for Ki-67. The subcapsular cyst corresponded to a cavity occasionally lined by flattened epithelium and surrounded by necrohemorrhagic hepatic tissue (Fig. 7). A single hepatic regenerative nodule was approximately 1.5 cm in diameter and partially surrounded by a band of fibrous tissue (Fig. 8). Within this nodule, mild to moderate perivascular fibrosis with arteriolar proliferation in the portal tracts was observed. The kidney presented scarce, multifocal, variable-sized microscopic cysts lined by flattened epithelium within the cortex and mild interstitial fibrosis (Suppl. Fig. S2). There was diffuse goblet cell metaplasia within the small intestinal mucosa. No changes in the spleen were observed. Final morphological diagnosis was diffuse and severe portal-to-portal bridging hepatic fibrosis with numerous embryonic bile duct profiles, consistent with CHF.
DNA was extracted from liver tissue in the affected foal by proteinase K treatment and from blood of related (n = 5) and nonrelated PSHs (n = 30) by the Illustra Blood GenomicPrep Mini Spin Kit (GE Healthcare Life Sciences, Buckinghamshire, United Kingdom). The affected foal pedigree and parental relationships were confirmed by analyzing 10 standardized microsatellites for animal genetic identification as recommended by the International Society for Animal Genetics (ISAG). Genotypes were obtained by capillary electrophoresis (ABI Prism 3130; Applied Biosystems, Foster City, CA, USA) using as a control a pretested ISAG sample. Two fragments containing the functional single-nucleotide polymorphism (SNP) mutations associated with CHF were amplified using specific primers (Suppl. Table S1). Both mutations, g.49,630,834G>A and g.49,597,760A>T (NCBI GenBank Accession number NW_001867389), located in exons 37 and 43, respectively, were genotyped by sequencing (ABI Prism 3130; Applied Biosystems).
The affected foal was heterozygous for both mutations. The pedigree study detected a paternal origin of the pathologic variant (Suppl. Fig. S3) while the maternal relatives did not carry the mutated alleles. In addition, 2 novel SNPs were detected in both fragment 37 and fragment 43. SNPs g.49.630.818C>T (intron 36) and g.49.630.951T>C (exon 37) were detected in fragment 37, the foal being double homozygous (TT and CC, respectively). SNPs g.49.597.693G>T (exon 43) and g.49.597.820A>G (intron 43) were detected in fragment 43, the foal being heterozygous for both polymorphic positions. All mentioned SNPs were also tested in the subset of 35 above-mentioned PSHs. Allele and genotype frequencies are summarized in Table 1.
Allele and Genotype Frequencies for SNPs Detected in Fragment 37 and Fragment 36 of the PKHD1Gene in Purebred Spanish Horses (n = 35).a
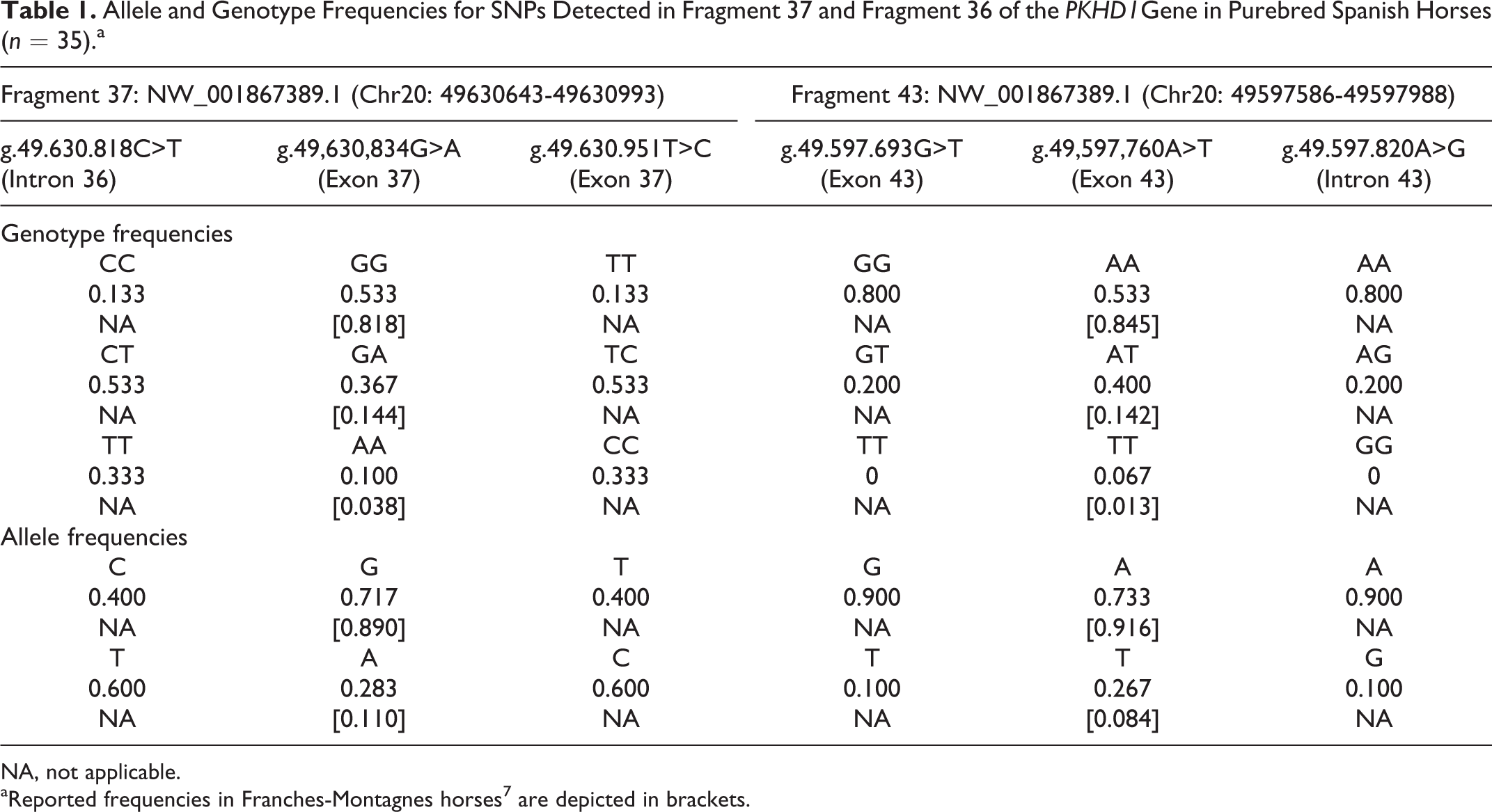
NA, not applicable.
aReported frequencies in Franches-Montagnes horses 7 are depicted in brackets.
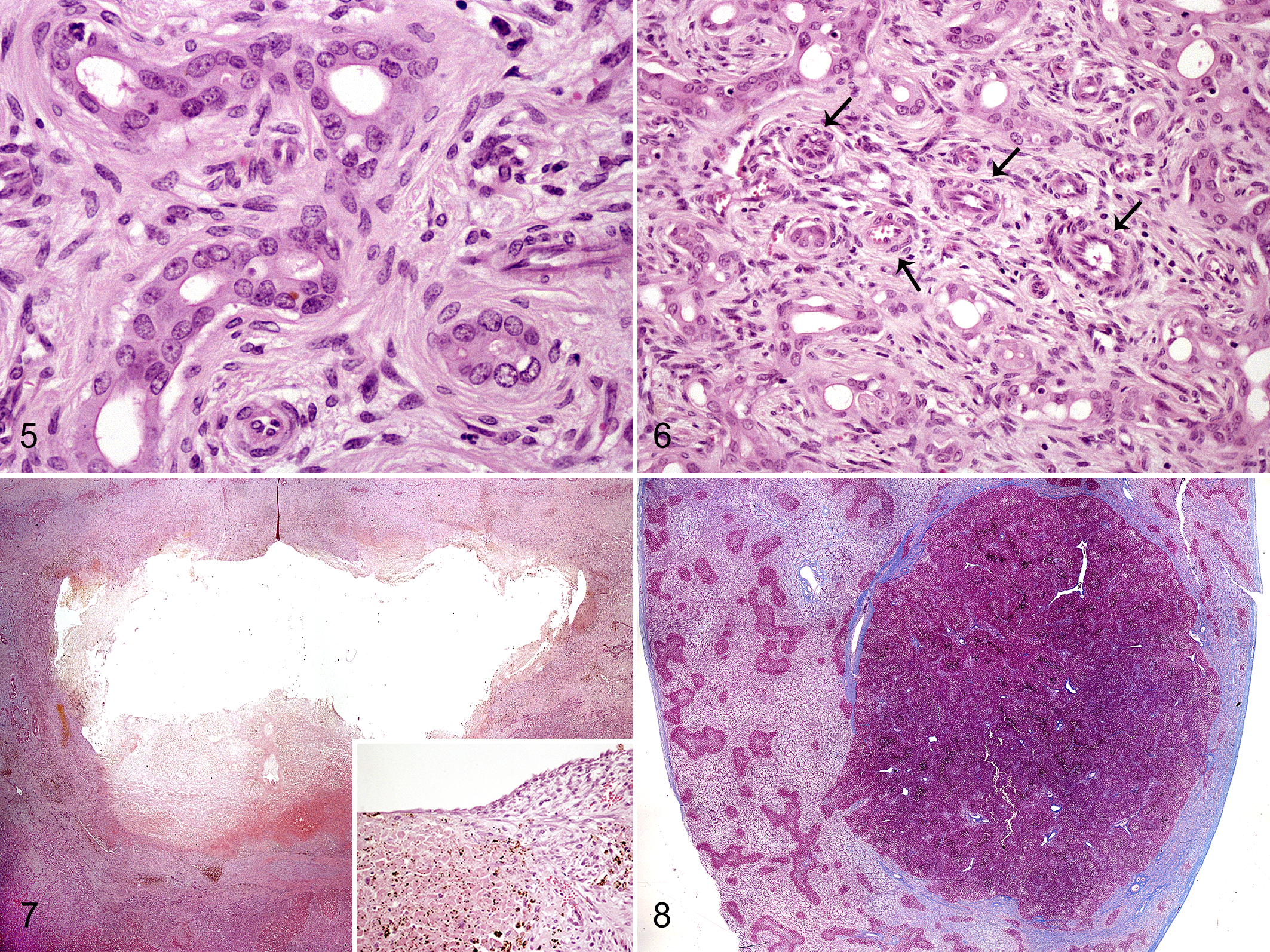
The present CHF case shows identical histological hepatic changes as those previously reported in foals 8,13 and other species: 2 –4,9,16,17 extensive portal-to-portal bridging fibrosis with numerous irregular and interlobular bile ducts, portal vein abnormalities, and compensatory arteriolar proliferation. Nodular regeneration is an unusual event in CHF, and it has not been reported in cases of CHF in domestic animals. In fact, the lack of nodular regeneration has been used as criteria to allow differentiation of this congenital condition from acquired chronic liver disease. 4,5 Regenerative nodules usually result from compensatory hyperplasia of surviving hepatocytes due to hepatic injury, atrophy, and/or fibrosis. 5 The regenerative nodule observed in this case might have resulted from the intrinsic proliferative capacity of remaining hepatocytes in response to parenchymal injury and/or to augmented arterialization and portal hypertension, as suggested in humans with CHF. 19 Cystic dilation of bile ducts has been previously reported in foals with CHF, and the rupture observed in the present case might have responded to alterations of biliary flow. 8,13
The clinical picture depicted by the present case is consistent with a congenital progressive liver disease, hypothetically leading to hepatic encephalopathy, as no samples from the central nervous system were available. Differential diagnoses might include biliary atresia, chronic extrahepatic biliary obstruction, and congenital pyrrolizidine alkalosis. 1,12,15 As previously noted, one of the most common findings in cases of CHF in several species is hypoplasia and paucity of intrahepatic portal venules with concurrent arterial proliferation. Neither chronic extrahepatic biliary obstruction nor congenital pyrrolizidine alkaloid toxicity show these changes, but these vascular abnormalities might accompany biliary atresia in both foals and humans. 15 The presence and patency of the common bile duct were not specifically checked at necropsy, and the severity of acholic feces could suggest some type of biliary obstruction. As biliary atresia may be associated with other ductal plate malformations, we cannot exclude its presence accompanying the pathological scenario in this case. 5,10
CHF in SFM horses has been strongly associated with 2 missense variants in the PKHD1 gene in chromosome 20 (ECA20), suggesting SFM horses could be a model for the liver-restricted autosomal recessive polycystic kidney and hepatic disease in humans. These missense mutations consisted of 2 SNPs: g.49,630,834G>A and g.49,597,760A>T (NCBI GenBank Accession number NW_001867389), respectively, located in exons 37 and 43 of the PKHD1 gene, predicted to cause the nonconservative changes p.H2038Y and p.I2282 N on the protein level. 7 To our knowledge, those 2 PKHD1 pathogenic variants have never been studied in the PSH. Our genetics studies indicate that the affected foal was heterozygous for both mutation sites. All CHF cases in SFM horses were homozygous for both mutation sites, except for 1 affected SFM foal that was double heterozygous. As previously speculated, other independent mutations on the PKHD1 gene could be related to the expression of the disease. 7 We found a 3-fold higher genotypic frequency for PKHD1 SNP: g.49,630,834G>A mutation and a 5-fold higher genotypic frequency for PKHD1 SNP: g.49,597,760A>T in our population compared with those genotypic frequencies reported in SFM horses. 7 Considering this and the fact that 85.7% of the homozygous SFM horses were CHF affected, an increased prevalence of the disease should be expected in the PSH population. However, this is the first time the disease has been described in a PSH foal. Therefore, other SNPs in coding or noncoding regions or epigenetic regulation mechanisms could also be involved in the expression of the disease in horses.
In conclusion, we have described a case of CHF in a PSH foal and performed genetic analyses in this case and in related and nonrelated PSHs, indicating that SNPs in the PKHD1 gene previously linked to the disease in SFM horses might not fully explain the appearance of the process in the PSH. Instead, other mutations in the PKHD1 gene could have a more important role in the PSH. This disease should be considered a differential diagnosis in cases of PSH newborn foals with fatal hepatic clinical signs.
Supplemental Material
Supplemental Material, DS1_VET_10.1177_0300985817754122 - Congenital Hepatic Fibrosis in a Purebred Spanish Horse Foal: Pathology and Genetic Studies on PKHD1 Gene Mutations
Supplemental Material, DS1_VET_10.1177_0300985817754122 for Congenital Hepatic Fibrosis in a Purebred Spanish Horse Foal: Pathology and Genetic Studies on PKHD1 Gene Mutations by Jéssica Molín, Javier Asín, Arantzazu Vitoria, Arianne Sanz, Marina Gimeno, Antonio Romero, Javier Sánchez, Pedro Pinczowski, Francisco J. Vázquez, Clementina Rodellar, and Lluís Luján in Veterinary Pathology
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Supplementary material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.