
Abstract
This study examines apoptosis and viral neuropathogenesis in a murine model infected with vesicular stomatitis virus (VSV). VSV induces apoptotic cell death in cultured cell lines, raising the possibility that apoptosis of infected neurons and other target cells may contribute to disease and mortality. To determine whether or not VSV induces apoptosis in neural tissues, mice were inoculated intranasally with VSV. At 24, 48, 72, 96, and 120 hours postinfection, brain tissues were assayed for the presence of viral RNA by in situ hybridization and viral antigen by immunohistochemistry. Apoptosis was identified by in situ terminal deoxynucleotidyl transferase-mediated deoxyuridine triphosphate nick end labeling and electron microscopy. Viral replication and lesions were observed predominantly in central nervous system neurons. Apoptotic cell death was restricted to the same regions of the brain in which infected cells and tissue injury were identified. Results suggest that VSV-induced apoptosis is a mechanism causing cell death, tissue injury, and mortality in VSV-infected mice.
Keywords
Introduction
Vesicular stomatitis virus (VSV), a member of the Rhabdoviridae family, is an enveloped, negative-sense RNA virus, which naturally infects cattle, horses, and swine, resulting in the formation of vesicular lesions. 5,8,24 VSV also affects humans, causing flulike symptoms. Occasionally, neurologic signs may accompany infection. VSV may cause a fatal meningoencephalitis in experimentally infected mice. 8,14 It has been reported that intranasal inoculation of mice with VSV results in encephalitis, ventriculitis, and focal necrosis of gray matter. 4,10,13,14,21,23 Microscopic lesions consist of widespread nonsuppurative meningoencephalitis characterized by gliosis and lymphocytic perivascular cuffing. After intranasal instillation, VSV produces olfactory bulb infection, followed by infection of neuroanatomic systems that are neuromodulatory in function. 3,13,21
Studies using immunohistochemistry (IHC) and in situ hybridization (ISH) after intranasal inoculation of VSV have indicated that the virus passes transsynaptically using both anterograde and retrograde transports from the olfactory bulb to different regions of the brain. 3,14 More recently, it was reported that VSV has oncolytic activity when inoculated intravenously in immunocompromised mice. 1 In vitro experiments and studies with immunocompromised mice have demonstrated that virus induces cytolysis through caspase-dependent and -independent programmed cell death. 1
Apoptosis is characterized by cell shrinkage, loss of normal cell-to-cell contact, chromatin condensation, cellular fragmentation, and DNA degradation, which produces oligonucleosome-sized nuclear fragments that form a specific ladder pattern on agarose gels. 11,15 Apoptosis plays a protective role by eliminating cells that might prove harmful if they were to survive, e.g., removing cells harboring mutations after irradiation or chemical stimulation, which could lead to the development of neoplasia. Apoptosis protects against the spread of virus to uninfected cells, 7,12,20 thus limiting infection.
Little is known about events involved in the neuropathogenesis of VSV. Limited data exist on the role of apoptosis toward VSV-induced neurologic disease in the mouse. 1,12,17 To better understand the neuropathogenesis, we studied the occurrence of apoptosis in neurons after VSV infection of immunocompetent mice and mapped the progression of viral infection in the mouse brain.
Materials and Methods
Viruses
The pathogenic vesicular stomatitis virus New Jersey (VSV-NJ) USA/95 (USDA No. 32608) was isolated in the southern USA during the 1995 disease outbreak involving horses. The virus was obtained from the National Veterinary Services Laboratory, Animal and Plant Health Inspection Services, US Department of Agriculture, Ames, Iowa.
Experimental infection of mice
Twenty specific pathogen-free male Swiss Webster mice, 3–4 weeks old, were used throughout the study. Mice were inoculated intranasally with 20 µl of a viral suspension containing 107. 3 TCID50/ml (tissue culture infectivity dose) administered equally to both nostrils. Four uninfected mice, which were matched with the principal in age and breed, were used as controls. At 24, 48, 72, 96, and 120 hours postinfection (hpi), four mice were given a lethal intraperitoneal dose of 5 mg of pentobarbital sodium (Nembutal; Abbott Laboratories, North Chicago, IL) in serum-free Roswell Park Memorial Institute medium (GIBCO BRL, Life Technologies, Inc., Gaithersburg, MD). Each mouse was perfused transcardially with 20–30 ml of 4% paraformaldehyde. Whole brain was removed and postfixed in 4% paraformaldehyde for 5 hours. Sections of brain were paraffin embedded, sectioned, stained with hematoxylin and eosin, and examined by light microscopy. Appropriate sections were prepared and further examined by terminal deoxynucleotidyltransferase–mediated deoxyuridine triphosphate (dUTP)nick end labeling (TUNEL), transmission electron microscopy (TEM), IHC, and ISH.
Preparation of tissues for histopathology, IHC, ISH, and TUNEL assay
For light microscopy, IHC, ISH, and TUNEL staining, tissue sections (4 µm) were adhered to Superfrost/plus slides (Fisher Scientific, Pittsburgh, PA). Deparaffinization was done by heating the sections for 20 minutes at 65 C. Sections were rehydrated through graded alcohols and washed with phosphate-buffered saline (PBS) (pH 7.4).
IHC for VSV antigen expression
IHC was performed essentially as described in previous studies. 25 Briefly, tissue sections were treated with 3% hydrogen peroxide in PBS for 20 minutes, followed by washes in PBS and digestion with 0.05% protease XIV (Sigma Chemical Co., St. Louis, MO) for 6 minutes at 37 C. After several washes in PBS, sections were incubated in a blocking solution of 5% normal goat serum in PBS for 30 minutes at room temperature (RT) and then again incubated overnight at 4 C with VSV-specific bovine convalescent serum (diluted 1 : 500 in PBS) directed against VSV structural protein. After washing with PBS, slides were incubated with goat anti-bovine immunoglobulin G (linking antibody) for 40 minutes and alkaline-phosphatase–conjugated antibody for 20 minutes at RT. After the last wash in PBS, slides were incubated with alkaline phosphatase substrate until color was detected. The color reaction was stopped by washing the slides in distilled water. Slides were counterstained with Mayer's hematoxylin.
TUNEL assay
All experiments for evaluating the presence of fragmented DNA by in situ reaction were performed using the TUNEL technique 25 on sections from VSV-infected mice as reported previously. Paraffin-embedded sections were treated with 20 µg/ml proteinase-K in 0.1 M PBS for 20 minutes at 37 C. After washing in PBS, sections were covered with 50 µl of the TUNEL reaction mixture (Boehering Mannheim, Indianapolis, IN), which contained terminal deoxynucleotidyl transferase (TdT) and fluorescein–dUTP, and incubated under a coverslip in humidified chamber for 1 hour at 37 C. The reaction was stopped by washing slides in PBS. Slides were incubated with the anti–fluorescein-alkaline phosphatase conjugate (Roche Diagnostic Corporation, Indianapolis, IN) diluted 1 : 3 in 100 mM Tris-HCl, 150 mM NaCl (pH 7.5), and 1% blocking reagent for 40 minutes at RT. After three 15-minute washes in PBS, sections were stained by incubation with a chromogenic substrate 5-bromo-4-chloro-3-indolyl phosphate (X-phosphate/BCIP) and nitro blue tetrazolium salt (NBT) for 5–15 minutes at RT and counterstained with 0.5% methyl green.
ISH for VSV RNA
ISH was performed as described in previous studies. 25 Briefly, deproteinization was performed in 0.2 N HCl for 20 minutes at RT. This was followed by washing with diethyl pyrocarbonate (DEPC)-treated water for 1 minute and digestion with proteinase-K (GIBCO BRL, Life Technologies, Inc., Gaithersburg, MD) at 20 µg/ml for brain tissues for 20 minutes in PBS. After digestion, tissues were fixed in 4% paraformaldehyde in PBS for 5 minutes. After rinsing with PBS, slides were acetylated in 200 ml of 0.1 mM triethanolamine-HCl buffer (pH 8.0) to which 0.5 ml of acetic anhydride (0.25%) was added. After 5 minutes, another 0.5 ml of acetic anhydride was added for an additional 5 minutes. Slides were then rinsed in 2× standard saline citrate (SSC) (1× SSC is 150 mM NaCl plus 15 mM sodium citrate [pH 7.0]). The prehybridization mixture contained 50% deionized formamide, 4× SSC, 10% dextran sulfate, 1× Denhardt's solution (0.02% Ficoll 400, 0.02% polyvinylpyrrolidone, 0.02% bovine serum albumin), 2 mM ethylenediaminetetraacetic acid, and 500 µg of salmon testis DNA per milliliter. Slides were covered with 200 µl of prehybridization mixture and incubated in a humidified chamber for 1 hour at 65 C. Labeled probes used for ISH were selected from the G and L genes, 2 which were 596 and 474 bp in length, respectively. Hybridization probes (G and L genes) were produced by reverse transcription–polymerase chain reaction (PCR) as described previously. 2 The probe was labeled via random priming reaction with digoxigenin-dUTP ([DIG]; Boehringer Mannheim Corp., Indianapolis, IN). The complementary DNA (cDNA) probes represented a region of the VSV genome in G and L genes. Cocktail probes, VSV-G + VSV-L genes, were used to specifically target VSV in tissue sections. Labeled DNA cocktail probes (0.1 ng/µl) were diluted in 300 µl of the prehybridization mixture, heated for 10 minutes in a 95 C heating block, and quenched on ice before being applied to the sections. Slides were rinsed briefly in 2× SSC, and 60 µl of the VSV probe cocktail mixture for tissues was applied to each slide. Hybridization was performed overnight at 42 C. After hybridization, tissue sections were washed twice in 4× SSC for 5 minutes at RT, once in 2× SSC for 10 minutes at 56 C, once in 0.2× SSC containing 60% formamide for 10 minutes at 56 C, twice in 2× SSC for 5 minutes at RT, twice in 0.2× SSC for 5 minutes at RT, and once in buffer I (100 mM maleic acid, 150 mM NaCl [pH 7.5]) for 5 minutes at RT. Immunohistologic detection was performed by using anti–digoxigenin immunoglobulin conjugated with alkaline phosphatase (Boehringer Mannheim) (diluted to 1 : 500), which was incubated for 2 hours at RT. Sections were then incubated with color substrate solution for 2–4 hours in the dark. The reaction was stopped with a distilled water rinse. Sections were counterstained with 0.5% methyl green.
Ultrastructural procedures
Uninfected and VSV-infected brain tissues obtained at necropsy were fixed with a solution containing 2.5% glutaraldehyde in 0.1 M sodium cacodylate (pH 7.4) for 24 hours at 4 C. Tissues were postfixed with 2% osmium tetroxide, stained en bloc with 2% uranyl acetate, dehydrated with ethanol, and embedded in Spurr's resin. Sections were cut, collected on slot grids, stained with lead citrate, and examined with a Phillips 410 electron microscope at 80 kV.
Results
Clinical signs of mice and histologic findings
All mice that were given VSV developed clinical signs within 72–120 hpi, which included ataxia, hyperexcitability, tremors, circling, and paralysis. The most striking microscopic lesions in the central nervous system (CNS) consisted of nonsuppurative meningoencephalitis and often were most severe in the brain stem (Fig. 1). Nonsuppurative meningoencephalitis was observed along the olfactory bulb and telencephalon (forebrain) and through the brain stem (pons). In all experimental groups, nonsuppurative meningoencephalitis and apoptotic cell debris were detected in the telencephalon and brain stem (Fig. 2). In the brain stem, affected neurons were vacuolated, and many were pyknotic. The severity and extent of neuronal damage correlated directly with time.
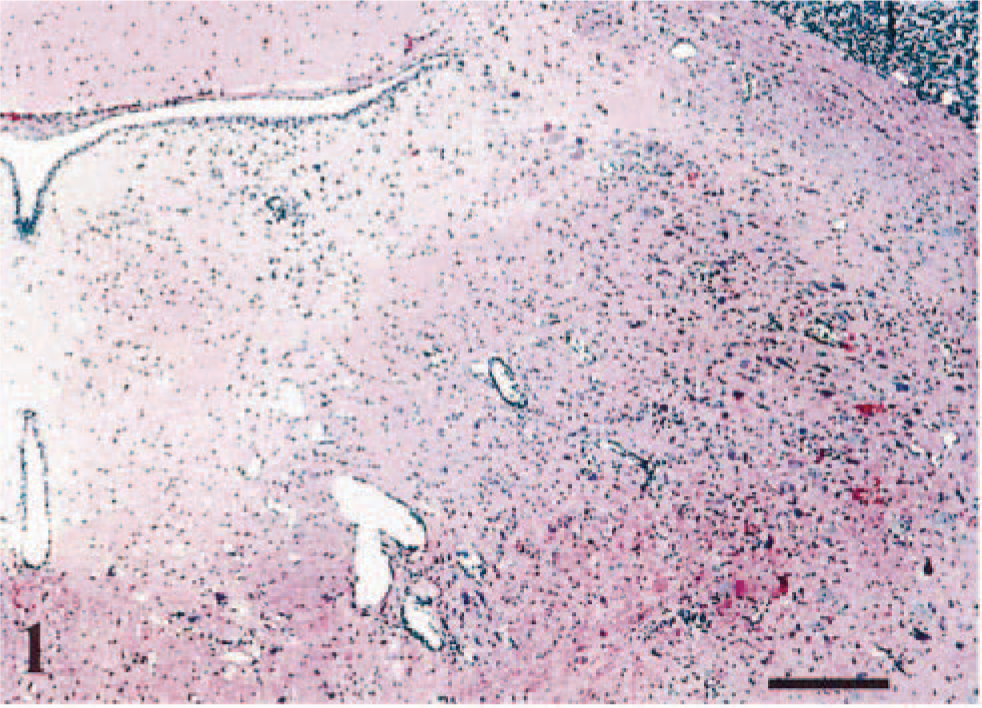
Swiss Webster mice, intranasally inoculated with VSV-NJ.
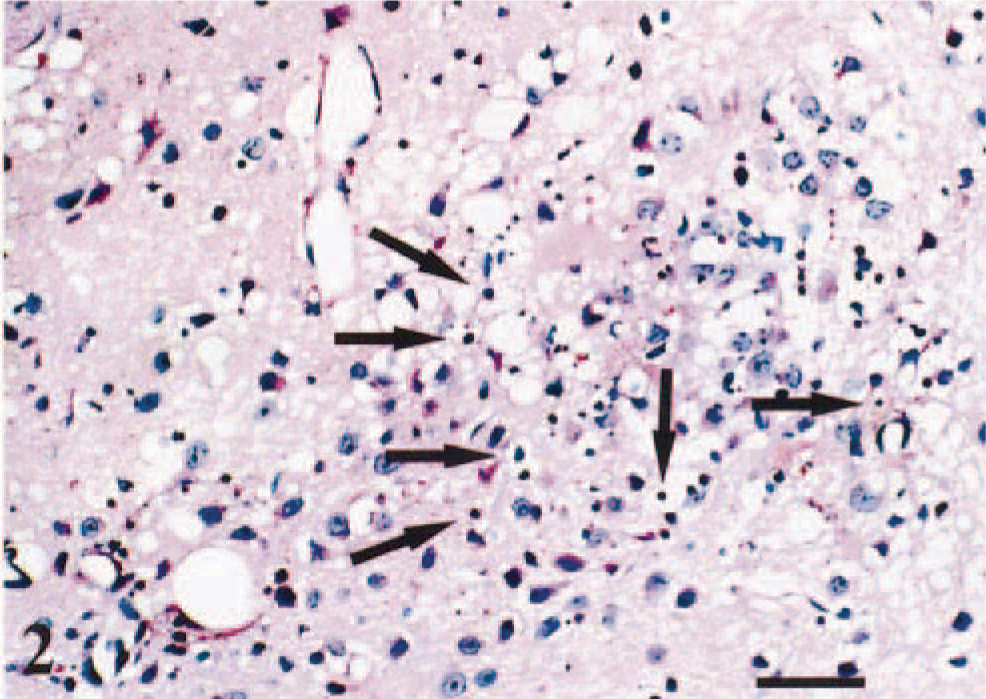
Brain stem. Apoptotic bodies (arrows) are noted in the MVe. Diffuse gliosis and degeneration are evident, 96 hpi. HE. Bar = 46 µm.
Neuronal tracing by IHC and ISH
Detection of signal by IHC (Fig. 3) and ISH (Fig. 4) was similar. However, IHC had lower specificity and a relatively higher degree of background staining. Viral RNA staining detected by ISH was specifically located in the cytoplasm of neurons. VSV-RNA was first detected in the olfactory bulb at 48 hpi (data not shown).
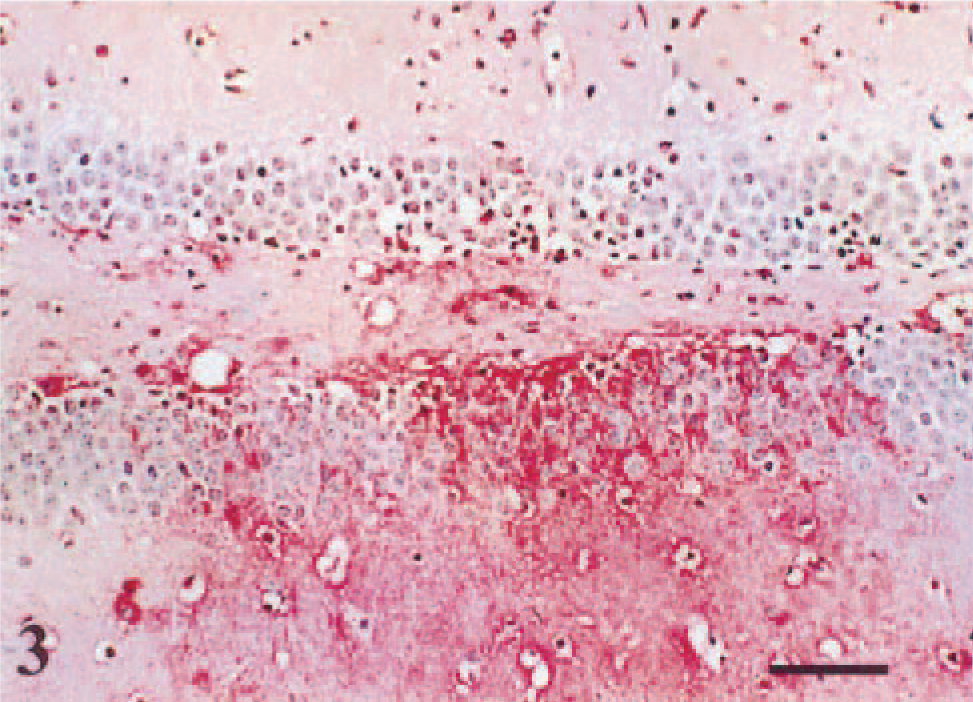
Serial sections of the VSV-NJ–infected brain tissue.
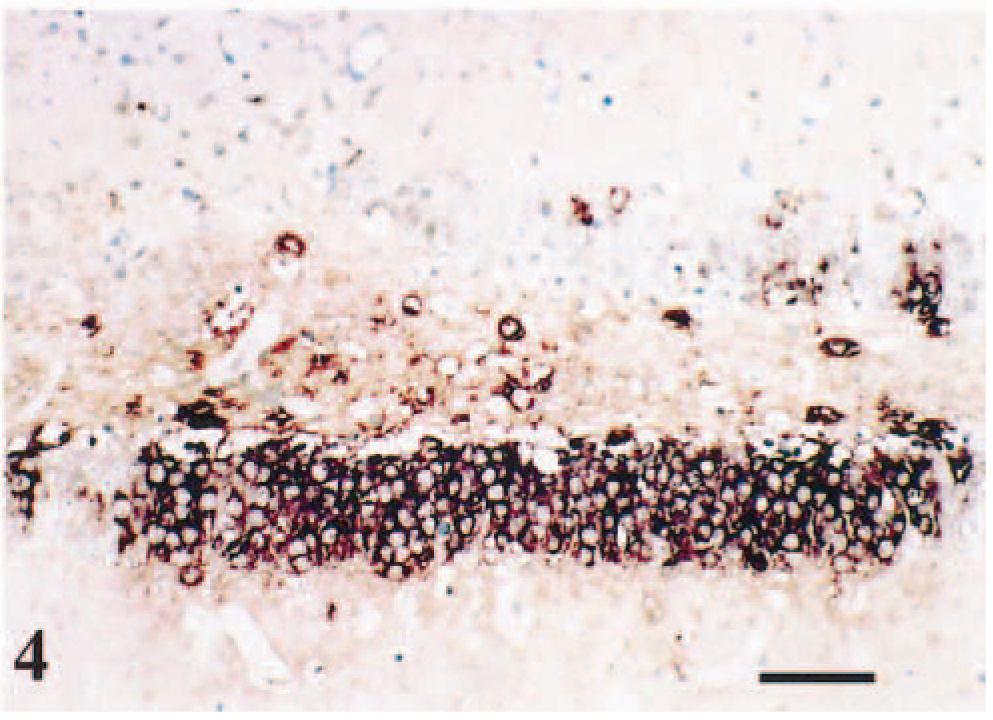
Pir in midbrain. Serial sections of the cells in Pir in panel in
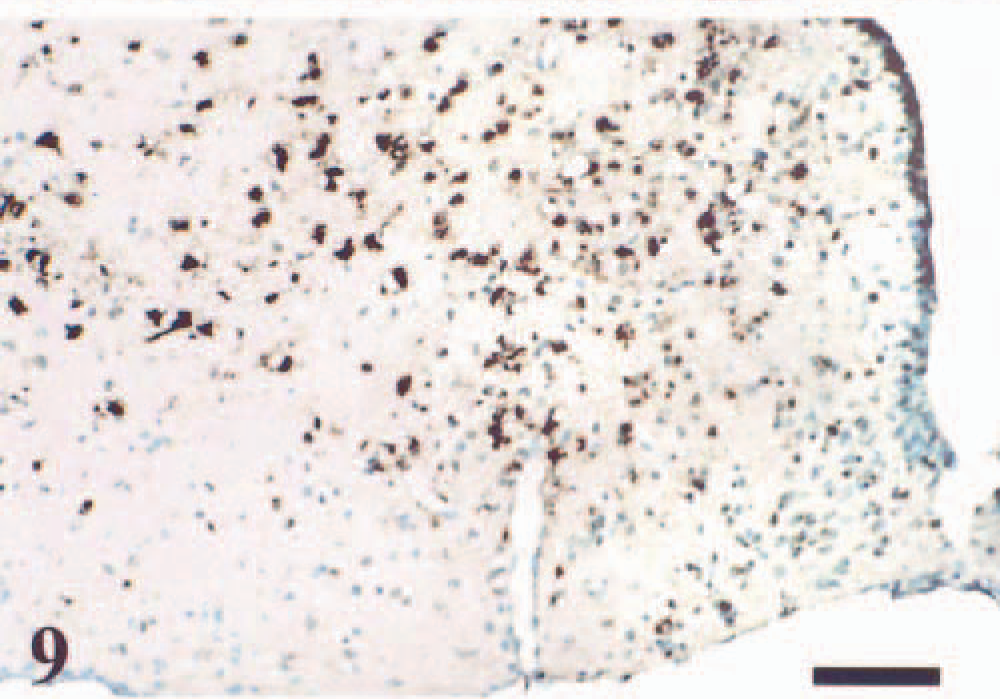
On the basis of representative coronal section of signals by IHC and ISH over time, we reconstructed VSV transneuronal passage in mouse CNS (Figs. 5–(7). At 72 hpi, a significant number of VSV-positive neurons were detected by ISH in the olfactory tubercle in the telencephalon (Fig. 6); piriform cortex (Pir), dorsal endopirform nucleus, and anterior hypothalamic nucleus in the diencephalon (Fig. 9); and medial vestibular nucleus (MVe) and prepositus hypoglossal nucleus (PrH) in the brain stem (Fig. 10). Between 96 and 120 hpi, the number and staining intensity of infected neurons increased progressively with time. In particular, the forebrain, cortical neuroepithelium, posterolateral cortical amygdaloid nucleus, and Pir had intense staining in comparison with other areas of the brain (Fig. 11). Individual neurons had prominent intracytoplasmic as well as slight intranuclear staining by ISH (Fig. 12). At this stage (between 96 and 120 hpi), neither VSV-RNA–positive nor VSV antigens were observed in spinal trigeminal tracts (SPVT) of the brain stem or spinal tract (SPV).
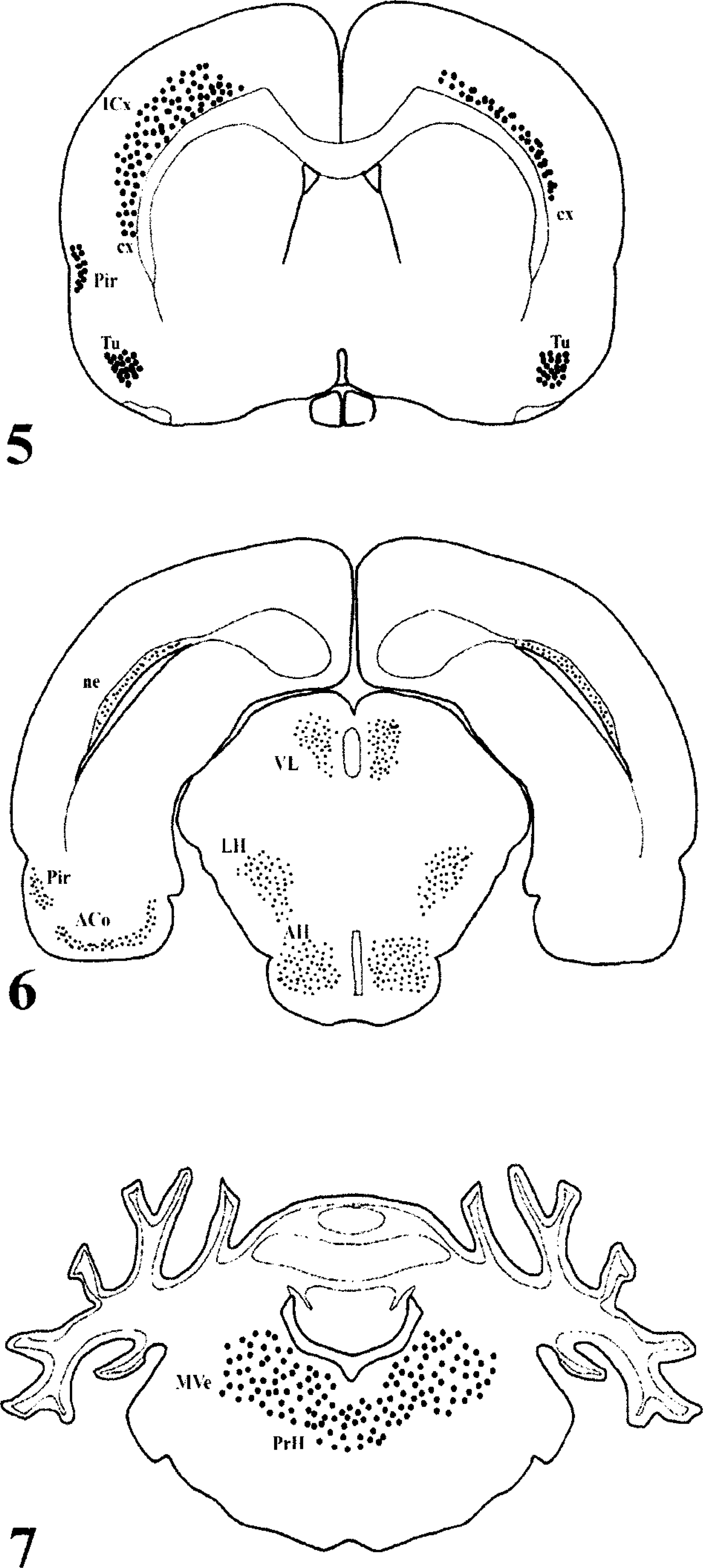
Schematic representation of coronal sections of a mouse brain in which we have marked areas of VSV-NJ–infected cells and apoptotic cells, and tissue damage. ACo, anterior cortical amygdaloid nu; AH, anterior hypothalamic nucleus; cx, cortical neuroepithelium; ICx, intermediate cortical layer; LH, lateral hypothalamic area; MVe, medial vestibular nu; Ne, neuroepithelium; Pir, piriform cortex; PrH, prepositus hypoglossal nu; Tu, olfactory tubercle; VL, ventrolateral thalamus nucleus.
Forebrain (telencephalon). VSV RNA is detected in cytoplasm of neurons in olfactory tubercle (Tu) in the telencephalon. ISH, methyl green counterstain, 72 hpi. Bar = 60 µm.
Diencephalon. VSV RNA is detected in cytoplasm of anterior hypothalamic nucleus (AH) in the diencephalon. ISH, methyl green counterstain, 72 hpi. Bar = 100 µm.
Brain stem (pons). Numerous VSV RNAs are detected in the MVe. ISH, methyl green counterstain, 72 hpi. Bar = 47 µm.
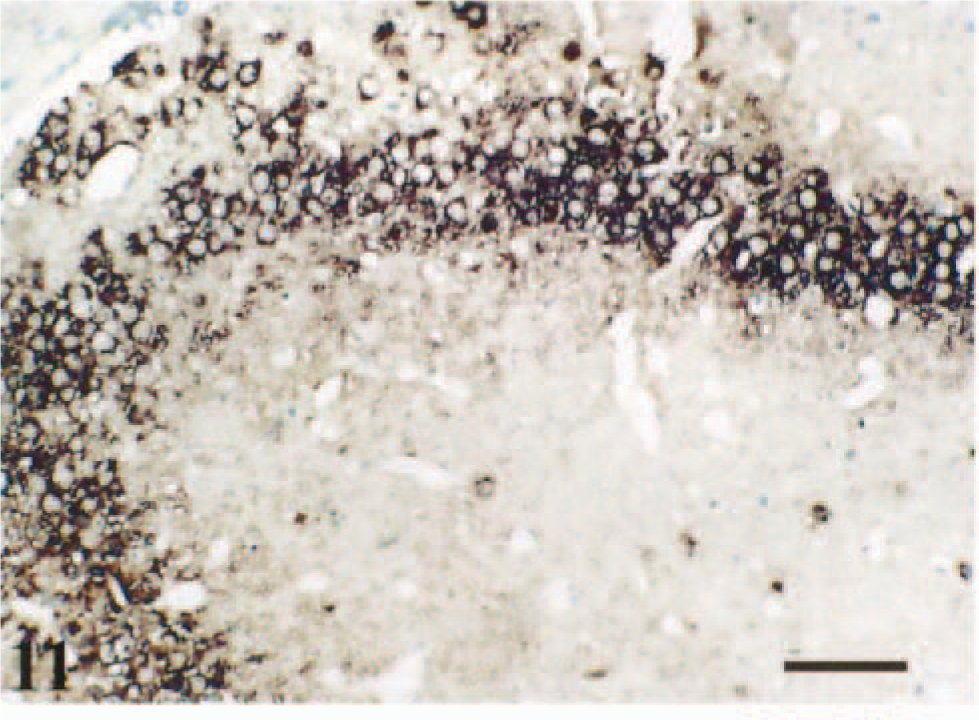
Forebrain (telencephalon). VSV RNA is detected in posterolateral cortical amygdaloid nucleus (PLCo) and Pir. ISH, methyl green counterstain, 96 hpi. Bar = 60 µm.
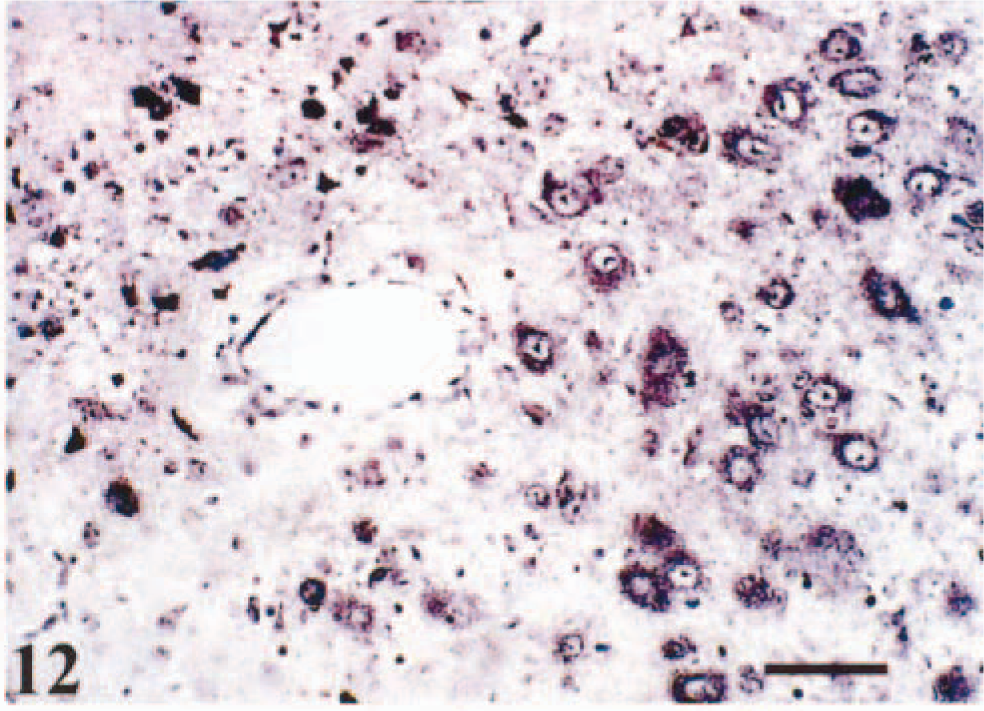
Forebrain (telencephalon). VSV RNA is detected in cytoplasm and nucleus of neuron in the forebrain. ISH, methyl green counterstain, 120 hpi. Bar = 46 µm.
TUNEL labeling and electron microscopy (EM) analysis in the VSV-infected brain
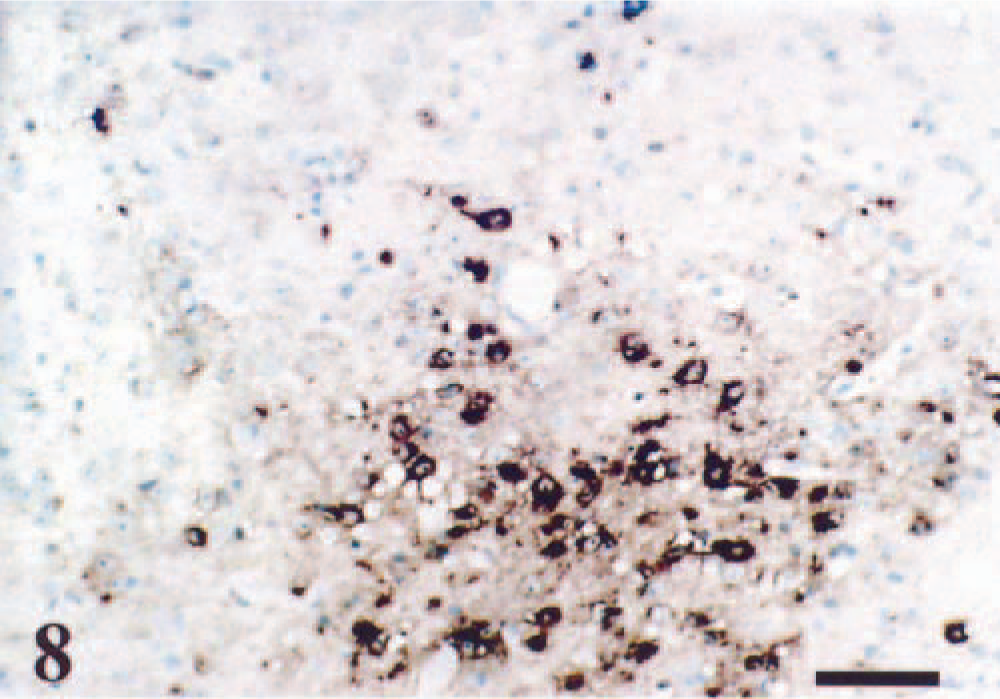
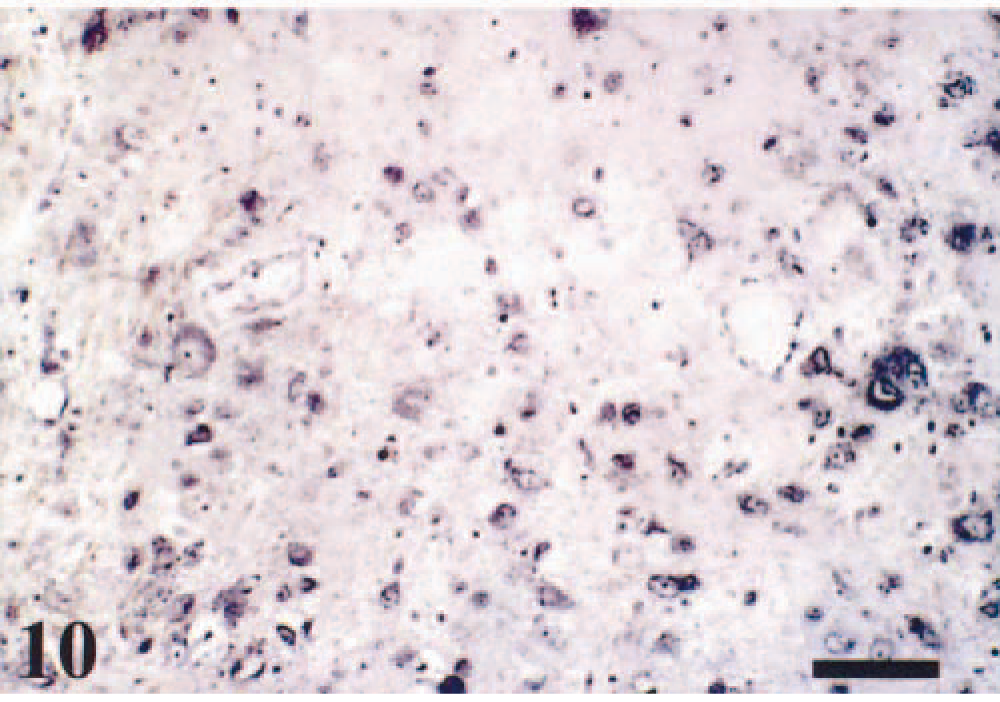
VSV-induced apoptosis in mouse brain was evaluated by detection of DNA fragmentation using TUNEL assay and electron microscopy. None of the sections from mock-infected mice had TUNEL-positive signal (data not shown). In VSV infected mice, the TUNEL assay indicated that infected neurons and many adjacent glial cells had nuclei that exhibited morphologic changes typical of apoptosis. The majority of TUNEL-positive cells were observed in mice with moderate to severe meningoencephalitis. TUNEL-positive signals were detected in the same vicinity where large numbers of VSV-RNA–positive neurons and glial cells were concentrated as evidenced by ISH. In sections of brain from animals that developed paralysis after VSV infection, VSV antigen- or VSV-RNA–positive neurons were diffusely scattered throughout the brain, including the olfactory bulb, telencephalon, and midbrain. TUNEL-positive neurons were detected in olfactory bulb sections obtained from VSV-infected mice at 48 hpi and, mostly, were confined to the olfactory glomerular region (Fig. 13). After intranasal infection, viral RNA and apoptotic cells were observed in brain stem. At 96 hpi, numerous TUNEL-positive cells were observed in the MVe and PrH in the brain stem (Fig. 14). The brain stem and telencephalon contained cells that were positive for TUNEL but negative for VSV viral RNA.
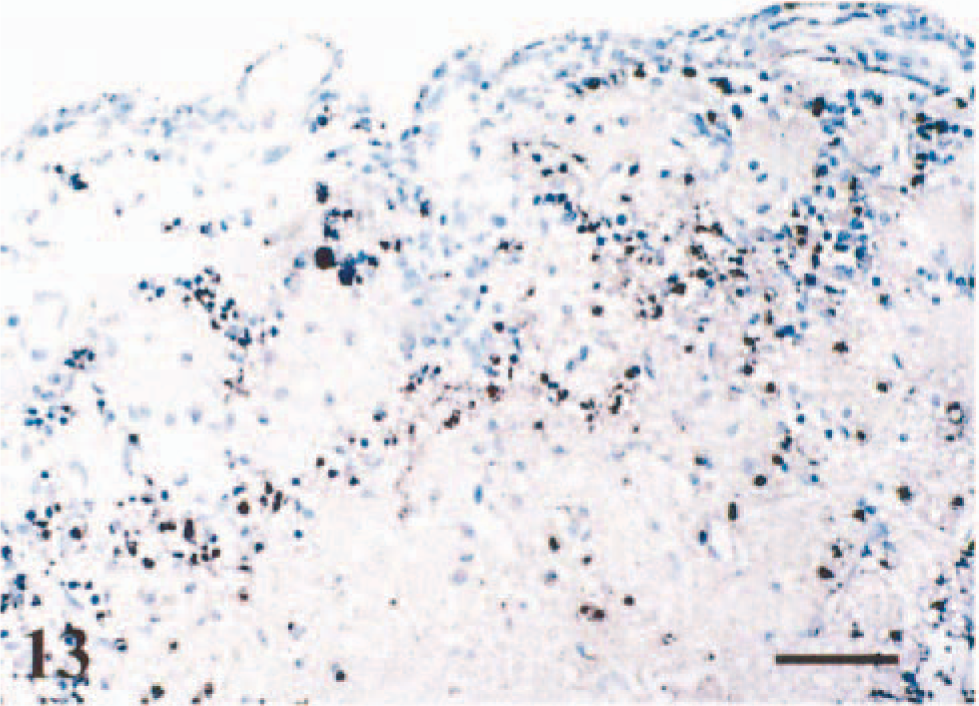
VSV-NJ–infected mice. Apoptosis was detected by TUNEL assay in mouse brain. Olfactory bulb. TUNEL-positive cells were detected in olfactory glomerular region. Methyl green counterstain, 48 hpi. Bar = 60 µm.
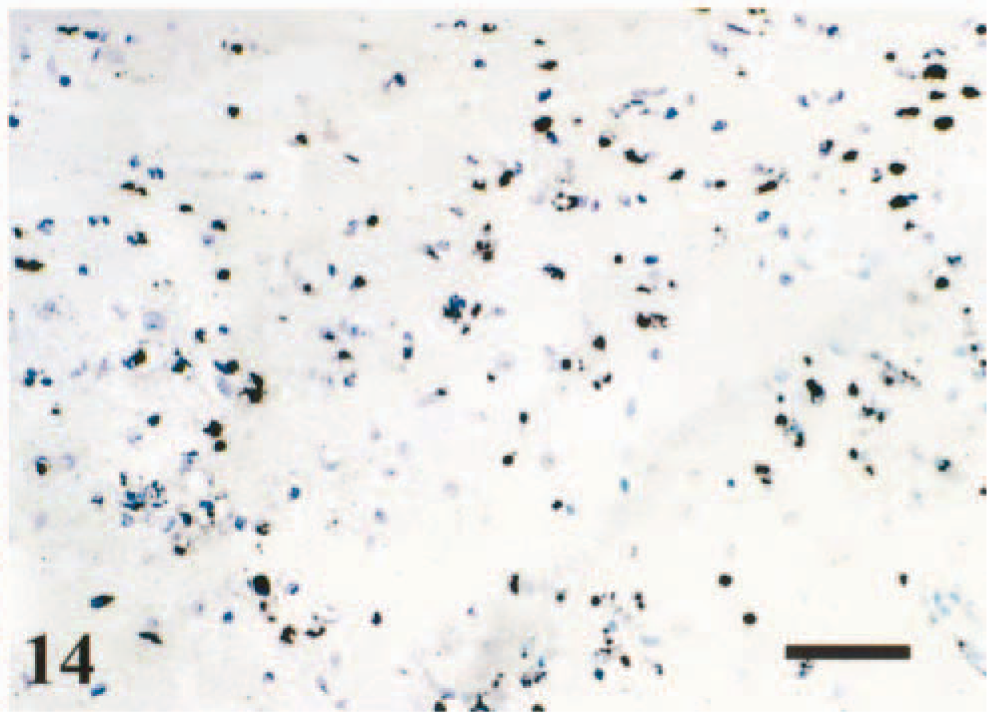
Brain stem. TUNEL-positive cells contain a dark precipitate in the nucleus and cytoplasm in the neuron. Methyl green counterstain, 96 hpi. Bar = 60 µm.
Ultrastructural changes typical of apoptosis were observed in infected mice. At 72 hpi, apoptosis was evident in glial cells in the brain stem. Affected glial cells had condensation of chromatin accompanied by cytoplasmic shrinkage (Fig. 15). At 120 hpi, there was indentation and discrete fragmentation of nuclei. Neuronal shrinkage, membrane blebbing, and abnormal convolution of the nucleus were observed. Condensation of chromatin was accompanied by swelling and distortion of endoplasmic reticulum (Fig. 16).
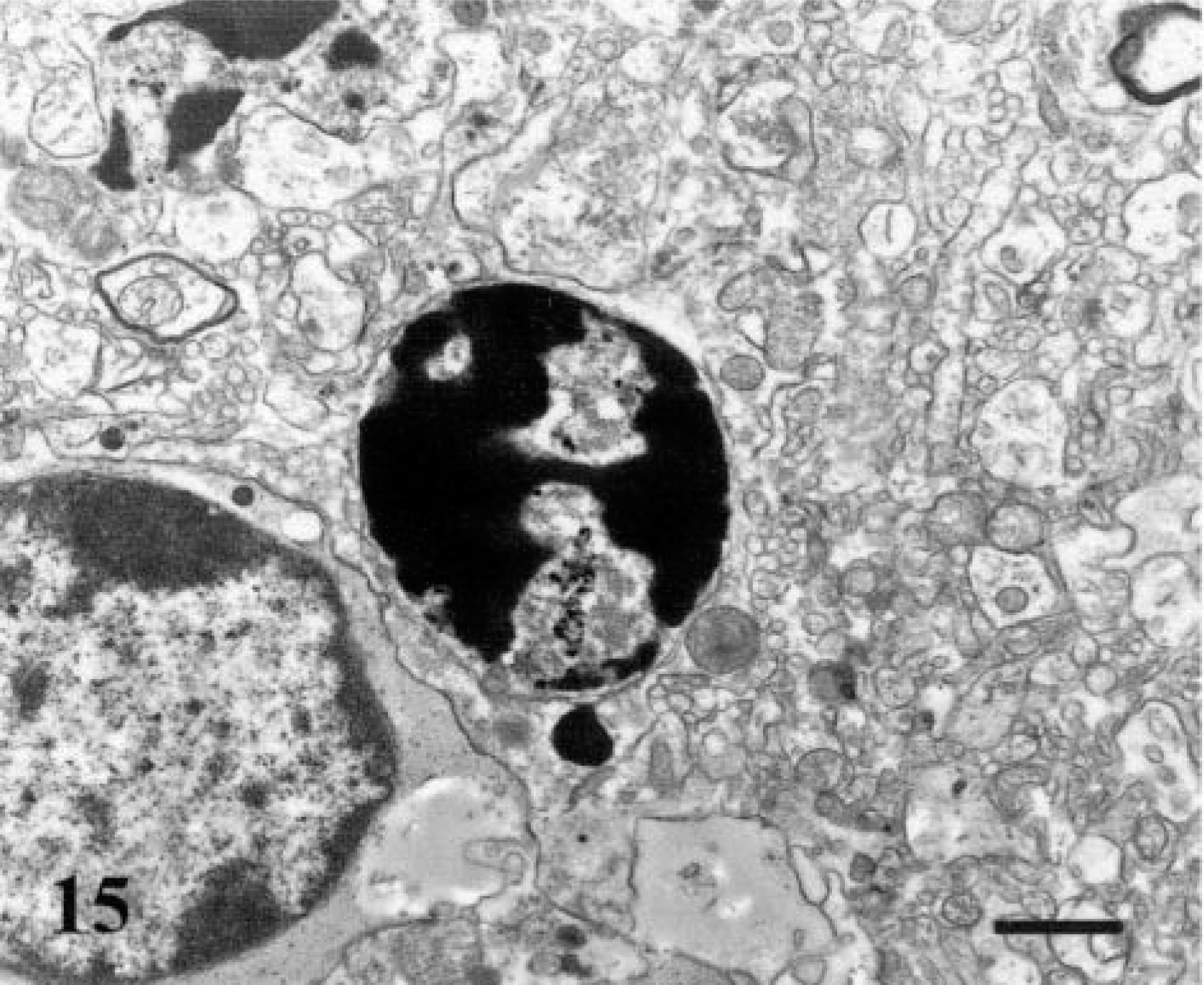
VSV-NJ–infected mice. Apoptosis was confirmed by electron micrographs in the mouse brain. Glial cell, brain stem. Condensation of chromatin accompanied by cytoplasmic shrinkage. 72 hpi. Bar = 0.7 µm.
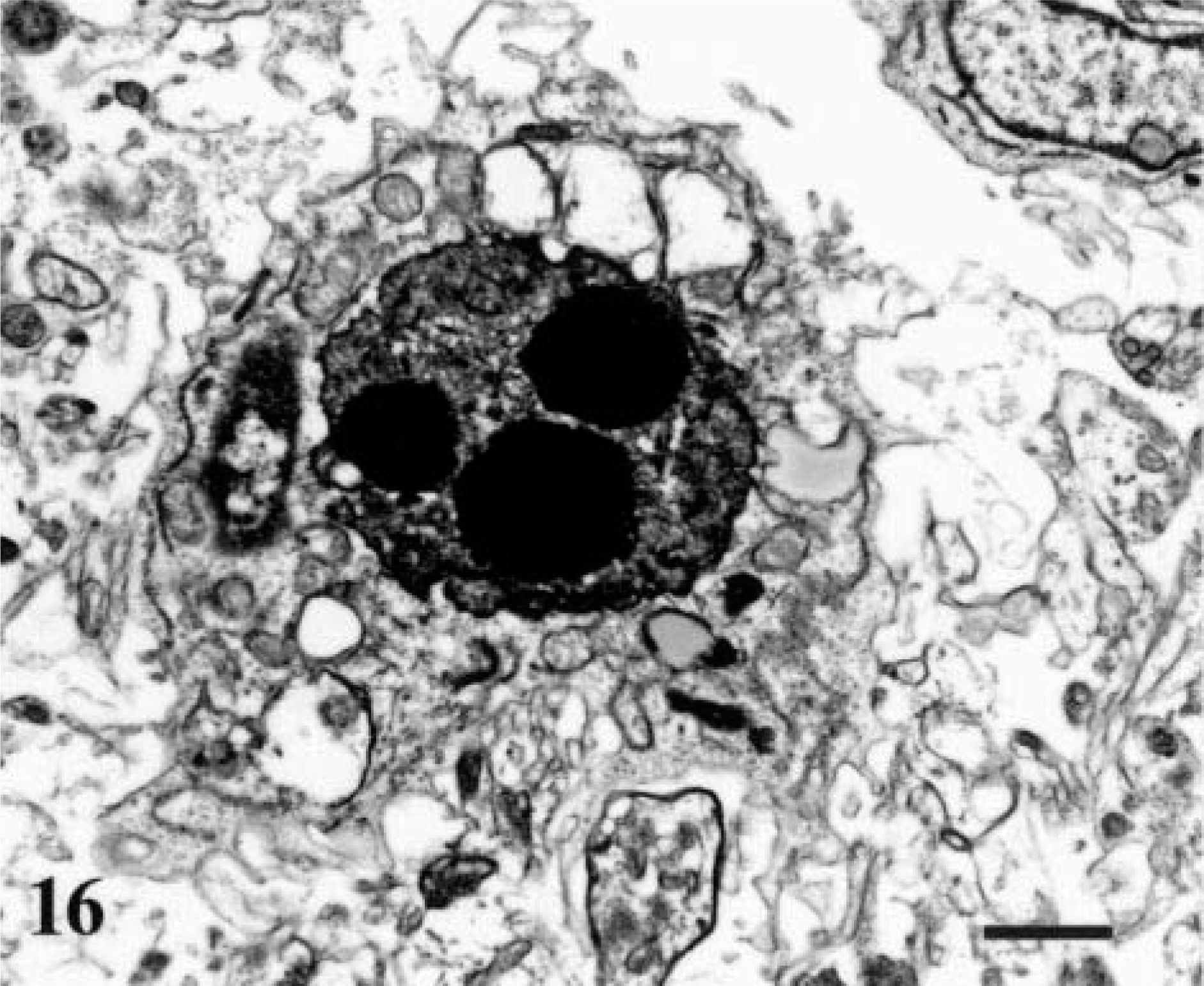
Neuron, brain stem. Condensation of chromatin accompanied by swelling and distortion of endoplasmic reticulum. 120 hpi. Bar = 0.7 µm.
Discussion
The present study examines the progression of infection and the development of brain lesions in mice after acute infection with VSV. Microscopic lesions were primarily characterized by nonsuppurative meningoencephalitis. Neuronal damage was observed in the olfactory bulb, forebrain (telencephalon), diencephalon, and brain stem (pons). The detection of VSV-RNA in mice correlated well with the location of VSV antigen and development of microscopic lesions. The patterns of lesion development and distribution are similar to those described previously with VSV infection in other species. 6,26
After intranasal inoculation, VSV infects cell bodies of the first-order olfactory receptor neurons located within the olfactory epithelium and spreads transsynaptically to higher-order neurons. Subsequent neuronal invasion probably involves replication in cell bodies and spread via axons and interneuronal synapses. 19 Previous reports have shown that VSV neurotransmission is likely to occur by retrograde transport followed by VSV dissemination via intraseptal synaptic junctions. 23 In this study, we followed VSV transneuronal passage in the mouse brain (Fig. 17). When delivered intranasally, VSV initially infected the olfactory receptor neurons in the olfactory bulb. Subsequent to replication in the olfactory bulb, VSV was detected in the telencephalon and brain stem by IHC and ISH. Viral RNA–positive cells were scattered throughout the brain, and their specific location, size, and morphology were consistent with those of neurons and glial cells.
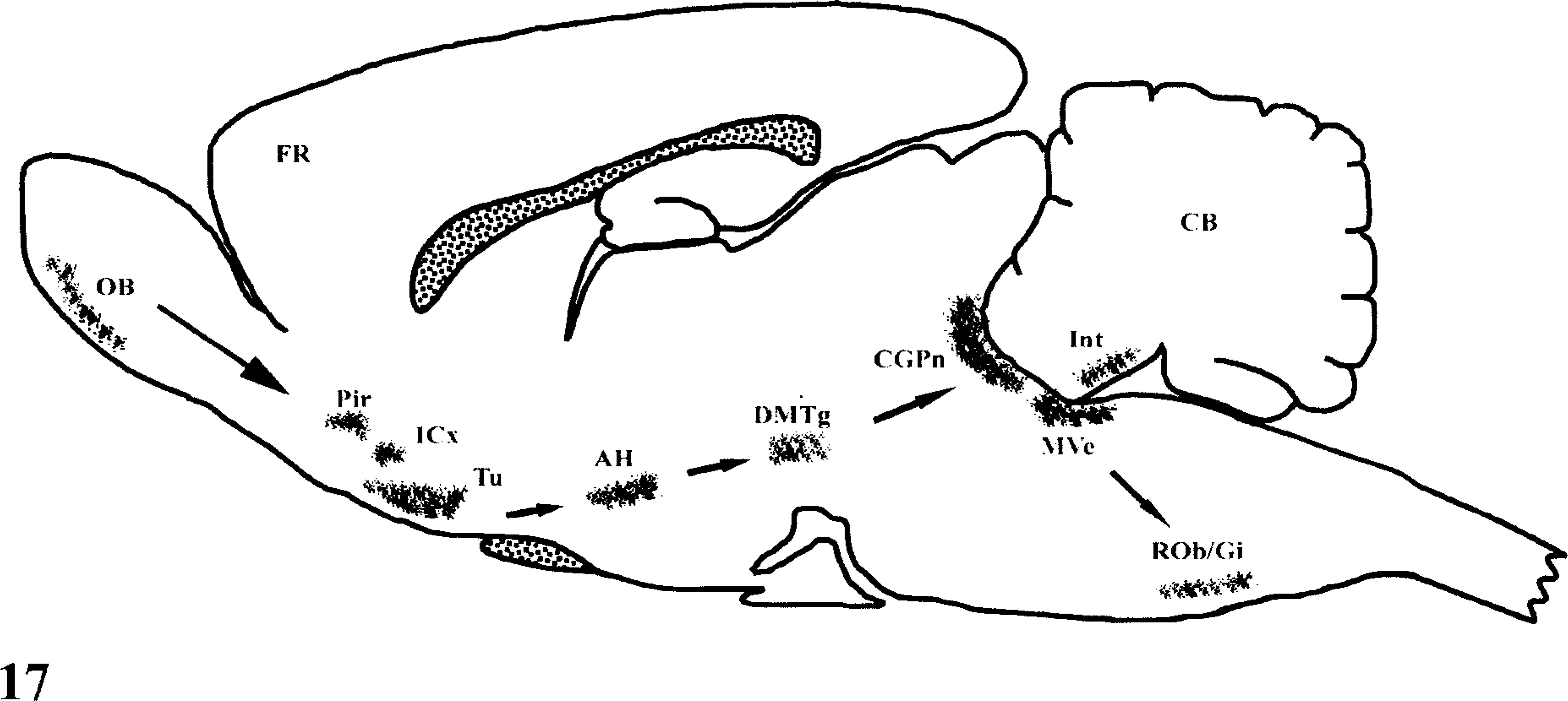
Schematic diagram illustrating the reconstruction of VSV transneuronal passage in the mouse CNS. AH, anterior hypothalamic nucleus; CB, cerebellum; CGPn, central gray, pons; DMTg, dorsomedial tegmental area; FR, forntal cerebral cortex; Icx, intermediate cortical layer; Int, interposed cerebellar nu; MVe, medial vestibular nu; OB, olfactory bulb; Pir, piriform cortex; Rob/Gi, rephe obscurus nu/gigantocellular reticular nucleus; T, thalamus; Tu, olfactory tubercule.
We believe that the infection of the olfactory bulb by VSV is important for development of efficient neuronal spread and neurovirulence in mice; however, it may not be required for initial viral entry into the trigeminal pathway. VSV viral RNA and antigen were undetectable within the SPV and SPVT in the brain stem. This result is in contrast to a previous report that indicated that VSV infection of trigeminal motor neurons occurred in experimentally infected mice. 14 This discrepancy and the difference in the regions of the brain stem shown to be infected with VSV between the two studies may have resulted from differences in the strains of VSV, duration of infection, and virulence of the VSV strain used.
Our results indicate that VSV induces apoptosis in Swiss Webster mice and is important in the development of cell injury. Although detection of single-stranded DNA-ends in nuclei of cells can occur owing to necrosis, 11,18 we resorted to detection of damaged DNA in neurons by TUNEL assay as well as ultrastructural microscopy to confirm the presence of VSV infection and apoptosis. TUNEL staining and electron microscopy indicated that VSV-infected neurons had nuclear changes characteristic of apoptosis (Figs. 14, 16). We observed that the number of VSV-RNA–positive cells was similar to the number of TUNEL-positive cells in the brain stem; however, in the Pir the number of VSV-RNA–positive cells was greater than the number of TUNEL-positive cells, suggesting that neurons, glial cells, and inflammatory cells may differ in their susceptibility to the effects of viral infection and apoptosis.
One important question is whether VSV replication and apoptosis can simultaneously occur in the same cells. Our results indicated that although many cells were labeled for either VSV RNA or apoptosis, the majority did not stain simultaneously for both. There are several possible explanations for this observation. Previous studies suggest that viral interaction with neurons and glial cells is sufficient to promote production of several cytokines, such as tumor necrosis factor, interferon, interleukin-1, and interleukin-6. Additionally, cytokines can be produced by lymphocytes, microglia, and macrophages. 13 Thus, it is possible that apoptotic cells that were negative for viral RNA in areas of VSV infection detected by ISH may have undergone apoptosis via a cytokine-dependent pathway induced by recruited inflammatory cells or reactive microglia. On the other hand, VSV RNA while undergoing extensive changes due to the ongoing infection may not reflect the terminal morphologic changes of DNA fragmentation typical of advanced terminal apoptosis. TUNEL-positive cells may have already altered or degraded cytoplasmic RNA of VSV beyond the capacity of detection by our techniques. A similar mechanism of action has been proposed for the lack of colocalization between infected and apoptotic neurons infected with murine reovirus. 22 Recent studies on viral matrix (M) protein and another viral component are responsible for induction of apoptosis in cells infected with VSV. 16 There is a possibility that M protein plays a major role in the inhibition of host-gene expression and in the induction of apoptosis, which are characteristic of VSV-infected cells. 16 Thus, these results provide a basis for exploration of the differential responsiveness among cell types to viral inducers of apoptosis and the implications of viral pathogenesis in intact hosts.
These observations suggest that neurologic damage associated with VSV infection may result from the induction of apoptosis by a combination of direct and indirect mechanisms, similar to those reported to occur with other viral infections. 9,20,25 Intranasal VSV infection of Swiss Webster mice provides an animal model for elucidation of the mechanisms by which neurovirulent viruses produce neuronal infection and induce apoptosis.
Footnotes
Acknowledgements
We thank T. Bargar of the University of Nebraska–Lincoln for technical assistance. Furthermore, we are grateful to Mrs. Kathy Apicelli and Ms. Liz Clark of the Plum Island Animal Disease Center for graphics and composite at visual information services. We also thank Dr. Gregory Mayer and Dr. Louis Rodriguez for critical review of the manuscript. This research was in part supported by a grant from Konkuk University, Kwangjin-gu, Seoul, South Korea.