
Abstract
Twenty pigs were inoculated with a virulent classical swine fever virus isolate to determine the mechanism responsible for thrombocytopenia using histopathologic, ultrastructural, and immunohistochemical (detection of viral antigens gp55 and FVIII-rag) techniques. In animals euthanatized at 2, 4, and 6 days postinoculation (dpi), clusters of granular material staining positive for FVIII-rag were observed in splenic cords, the marginal zone, hepatic sinusoids, and the perisinusoidal space. Moreover, numerous macrophages in these areas were swollen and displayed an intensely positive granular and cytoplasmic reaction. Cell alterations indicative of platelet activation and secretory and phagocytic activation of resident macrophages were also observed in these sites at 2 and 4 dpi. These results suggest that the thrombocytopenia observed in pigs is caused in the first instance by massive activation and subsequent phagocytosis of platelets secondary to the release of platelet-activating factors by activated macrophages.
Classical swine fever (CSF) is a viral disease characterized by hemorrhages associated with early intense thrombocytopenia. The onset of thrombocytopenia coincides with the appearance of fever and the start of viremia. 10,11
Over the years, a number of studies have set out to identify the pathogenic mechanism giving rise to this thrombocytopenia; various hypotheses have been advanced, some of them clearly contradictory. Although some authors have attributed thrombocytopenia to the massive degeneration of megakaryocytes observed in acute forms of the disease, 3,12 have suggested that thrombocytopenia results from a consumptive coagulopathy following onset of disseminated intravascular coagulation. 10,11 Weiss et al. 25 detected virus antigen in platelets in the acute phase of the diease and reported that thrombocytopenia was caused by the infection and subsequent destruction of these cells. Recent studies, however, have shown that the presence of virus antigen in platelets is due to the infection of a small number of megakaryocytes (2–4%), which would not account for the intensity of the thrombocytopenia observed in affected animals. 9
Numerous studies have shown that the monocyte/macrophage is the target cell of CSF virus 9,18,19,23 and that it undergoes major changes during the disease (unpublished data); analysis of these cells is thus a major relevance in the study of CSF pathogenesis, and particular attention should be paid to macrophage–platelet interactions when studying platelet alterations.
The purpose of this study was to identify both the mechanism triggering thrombocytopenia in acute forms of CSF and the possible causes of increased intensity of thrombocytopenia following disease onset.
Materials and Methods
Twenty-two Large White × Landrace pigs of both sexes were used for this study. Live weight at the start of the study was roughly 30 kg. All animals were free of infectious and parasitic disease and were housed in isolation at the Centro de Investigación en Sanidad Animal in Valdeolmos, Madrid, Spain. Two pigs were used as controls, and 20 received an intramuscular inoculation of 105 TCID50 of the virulent CSF virus isolate Quillota. This virus was titrated using the Reed and Müench method in accordance with international standards to determine the potency of the vaccine, and this strain is recognised as different from other CSF virus strains. Clinical symptoms and rectal temperature of pigs were monitored daily.
Preinoculation blood samples were taken from all pigs to obtain baseline values. Considerable care was taken in the colleciton of the blood samples to avoid hemolysis and tissue contamnation. Blood samples were obtained from the anterior vena cava with plastic syringes and disposable 18-ga needles. Blood was mixed with 1% ethylenediaminetetraacetic acid (9:1 ratio by volume) and diluted 1:200 with 1% ammonium oxalate in distilled water. A hemocytometer was used to determine the platelet count. After virus inoculation, blood samples were taken daily.
Pigs inoculated were euthanatized in groups of four at 2, 4, 6, 8, and 10 days postinoculation (dpi). For euthanasia, animals were sedated with Combelen (Pfizer, Sandwich, NJ) and then injected with T61® (American Hoechst Corp., Somerville, NJ). Spleen and liver samples were fixed by immersion in 4% formaldehyde for light microscopy and 2.5% glutaraldehyde for electron microscopy.
To corroborate the nature of viruslike particles observed in tissues of infected animals, porcine kidney cells (PK15) were infected with the Alfort strain of the CSF virus. Samples of cultures at 12, 24, and 36 hours postinoculation were fixed in 2.5% glutaraldehyde for electron microscopy.
Samples were embedded in paraffin and Epon 812 (Fluka Chemie AG, Buchs, Switzerland) following routine processing for light and transmission electron microscopy, respectively. Paraffin-embedded sections were stained with hematoxylin and eosin. The avidin–biotin–peroxidase complex (ABC) was used to demonstrate CSF virus glycoprotein 55 (gp55) and to identify platelets with anti–factor-VIII-related antigen (anti-FVIII-rag) on 3-gmm-thick paraffin-embedded sections. The primary monoclonal and polyclonal antibodies employed, dilutions, and sources were as follows: mouse anti-gp55 CSF virus (WH303) 1:20 (Central Veterinary Laboratory, Weybirdge, UK) and rabbit anti-FVIII-rag (1:t3800 (Dako, Glustrup, Denmark) and 1:500 (Dako). Phosphate-buffered saline and non-immune serum were used in place of specific primary antibodies as negative controls. Samples from two uninoculated animals also were used as controls. The specificity of these antibodies has been successfully demonstrated.
For ultrastructural study, 50-nm sections of Epon-812–embedded spleen and liver were counterstained with uranyl acetate and lead citrate and viewed through a Philips CM-10 (Eindhoven, Netherlands) transmission electron microscope.
Cell counts were performed on sections of spleen from all inoculated (2, 4, 6, 8, 10 dpi) and uninoculated animals after immunolabeling for gp55 viral antigen, FVII-rag and myeloid-histiocyte antigen. The number of macrophages engulfing platelets and the number of infected macrophages per unit area in splenic cords and the marginal zone of lymphoid follicles was determined as follows. The number of counting areas (0.3 mm2) necessary to obtain 200 cells was determined, and the distribution was expressed as the number of cells per area. The areas were chosen randomly in four diagonally positioned squares.
Data on immunopositive cell counts were assessed with an analysis of variance, followed by the Newman–Keuls method for multiple comparisons. Differences between counts for uninoculated and inoculated animals were considered significant at the P < 0.05 level; results were expressed as mean (± SD) for macrophages and mean for infected cells.
Results
Pigs infected with CSF virus showed a decrease of platelets from 475 × 103 cells µl to 342 × 103 cells µl in the first 48 hours, coinciding with pyrexia and becoming more severe over the next few days (Fig. 1).
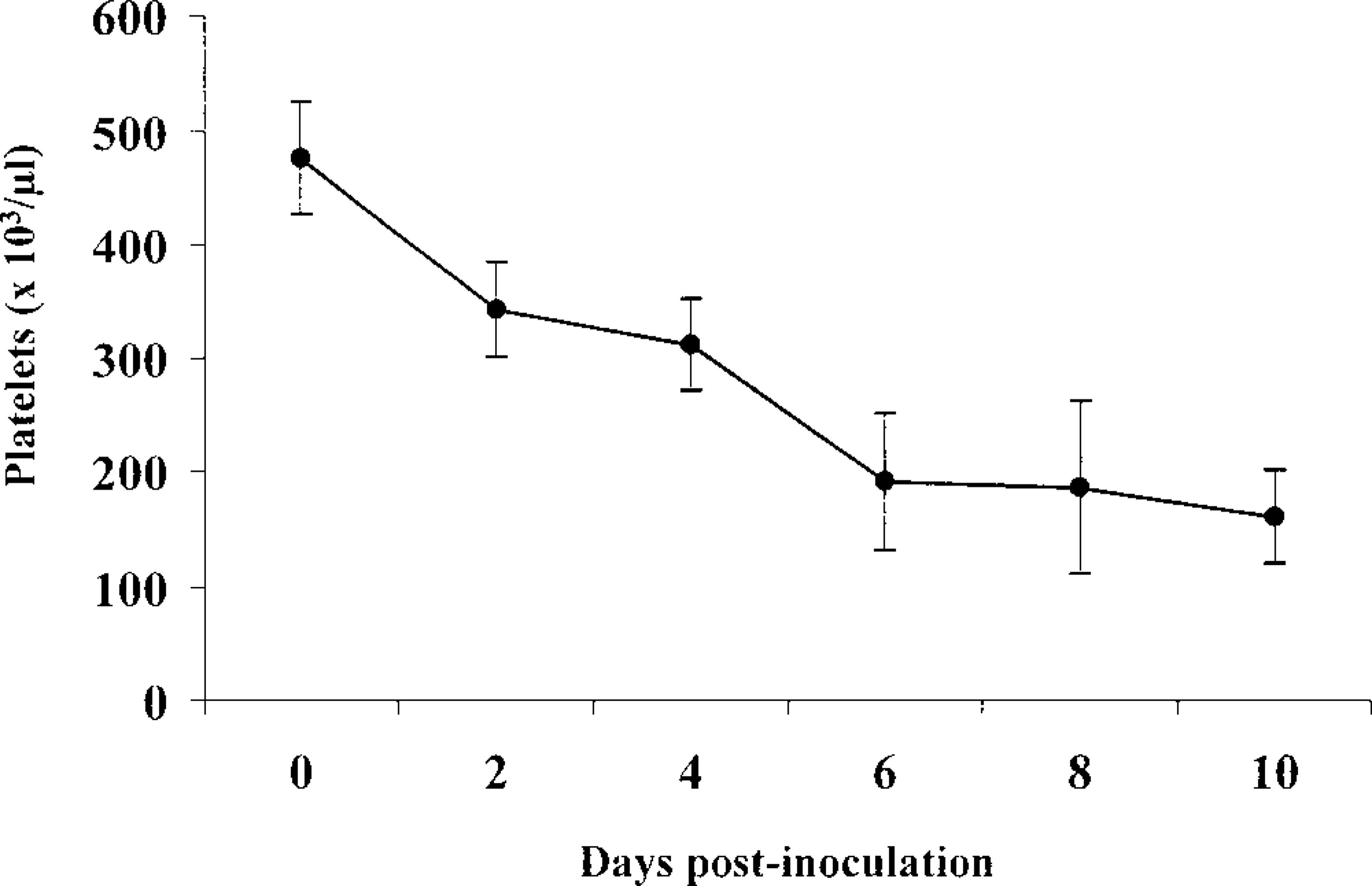
Platelet counts in control animals (0 dpi) and pigs inoculated with hog cholera virus.
Immunohistochemical detection of platelets was performed using anti-FVII-rag antibody. In controls, this antibody prompted positive granular immunostaining among sheathed capillary cells, free in splenic cords, and occasionally in macrophage cytoplasm at these sites (Fig. 2). However, in animals euthanatized at 2, 4, and 6 dpi, clusters of immunostaining granular material were observed in splenic cords and in the marginal zone. Moreover, numerous macrophages in these areas were swollen and displayed an intense positive granular and cytoplasmic reaction (Fig. 3). Subsequently, the positive reaction in macrophages and vascular lumina diminished considerably; no immune reaction was observed in periarterial macrophage sheath cells from 4 dpi onwards. Counts of macrophages engulfing platelets showed a significant increase in the number of these cells between 2 and 6 dpi (Fig. 4).
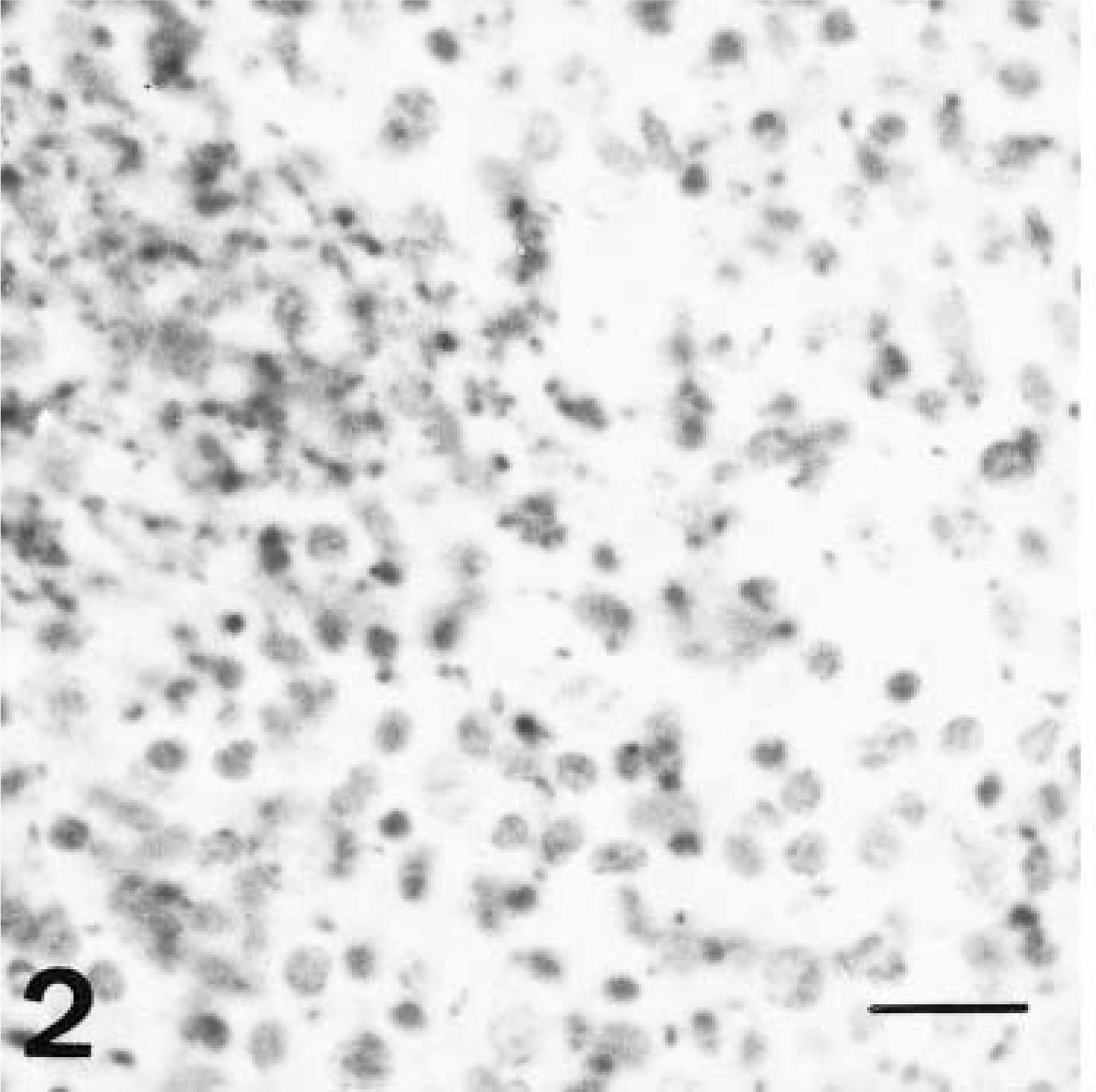
Spleen; uninoculated pig. Granular immunoreaction to FVIII-rag in splenic sheathed capillaries and in splenic cord macrophages. Intracytoplasmic reaction is occasional. ABC method, Mayer's hematoxylin counterstain. Bar = 50 µm.
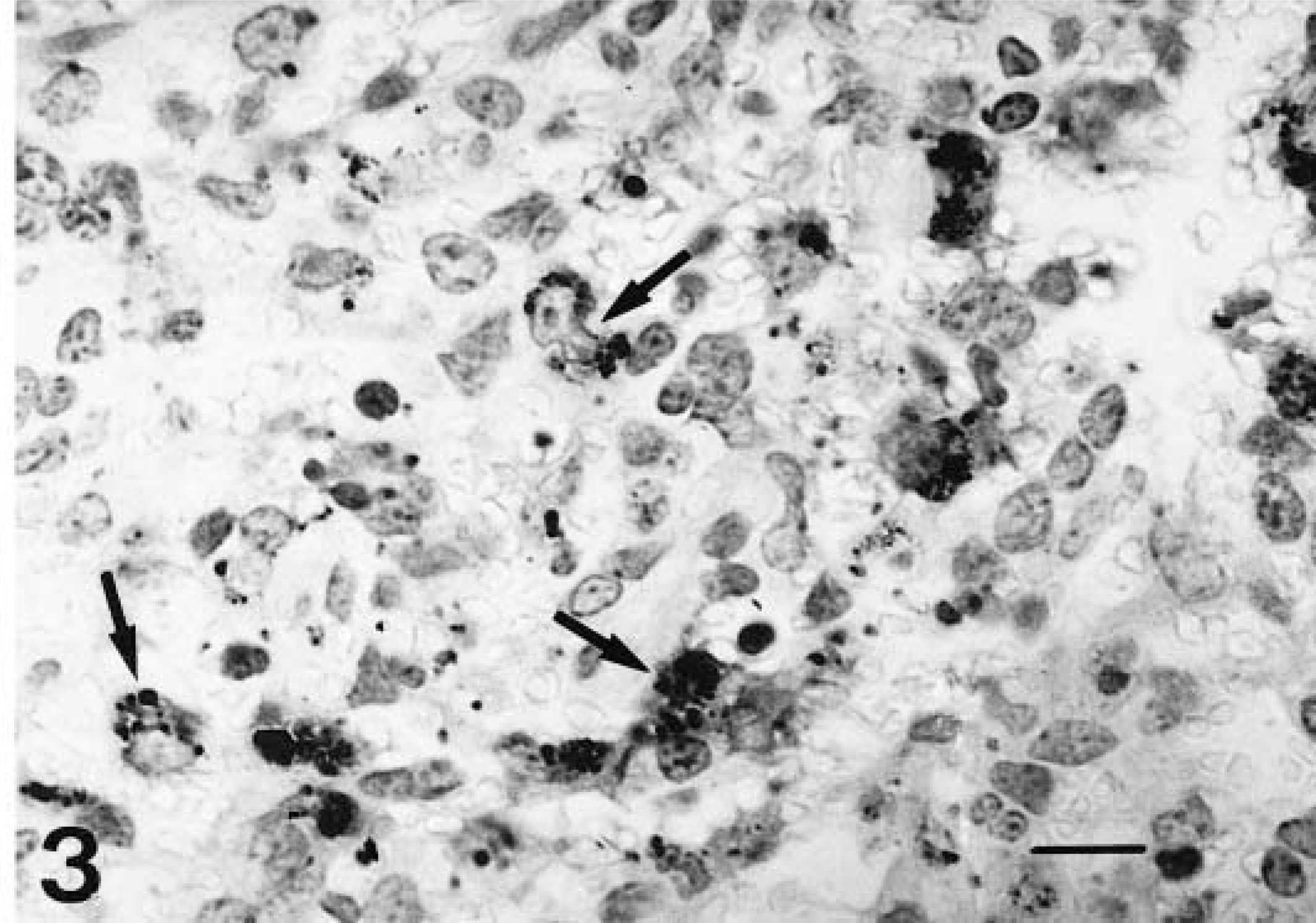
Spleen; pig, 2 dpi. Intracytoplasmic granular immunoreaction to FVIII-rag in splenic cord macrophages (arrows). ABC method, Mayer's hematoxylin counterstain. Bar = 25 µm.
In the marginal zone of lymph follicles and in splenic cords, ultrastructural changes indicative of platelet activation were observed at 2 and 4 dpi; cells were enlarged (12–15 µm) and deformed, with protrusion of one or more pseudopodia, decrease in granule numbers, and dilation of the open canalicular system. Multiple platelets formed aggregations of variable intensity (Fig. 5). Some platelet clusters retained their characteirstic morphology, and membrane fusion was evident as visible, highly electron-dense lines. Clusters contained various numbers of platelets, generally more than 10. Platelet aggregation was most manifest in the form of mosaic-like clusters comprising fully degranulated platelets with marked fusion of cytoplasmic membranes. Occasionally, these membranes completely disappeared, giving rise to a finely granular structure with low electron density, containing vestiges of platelet organelles and surrounded by a membrane layer (Fig. 6).
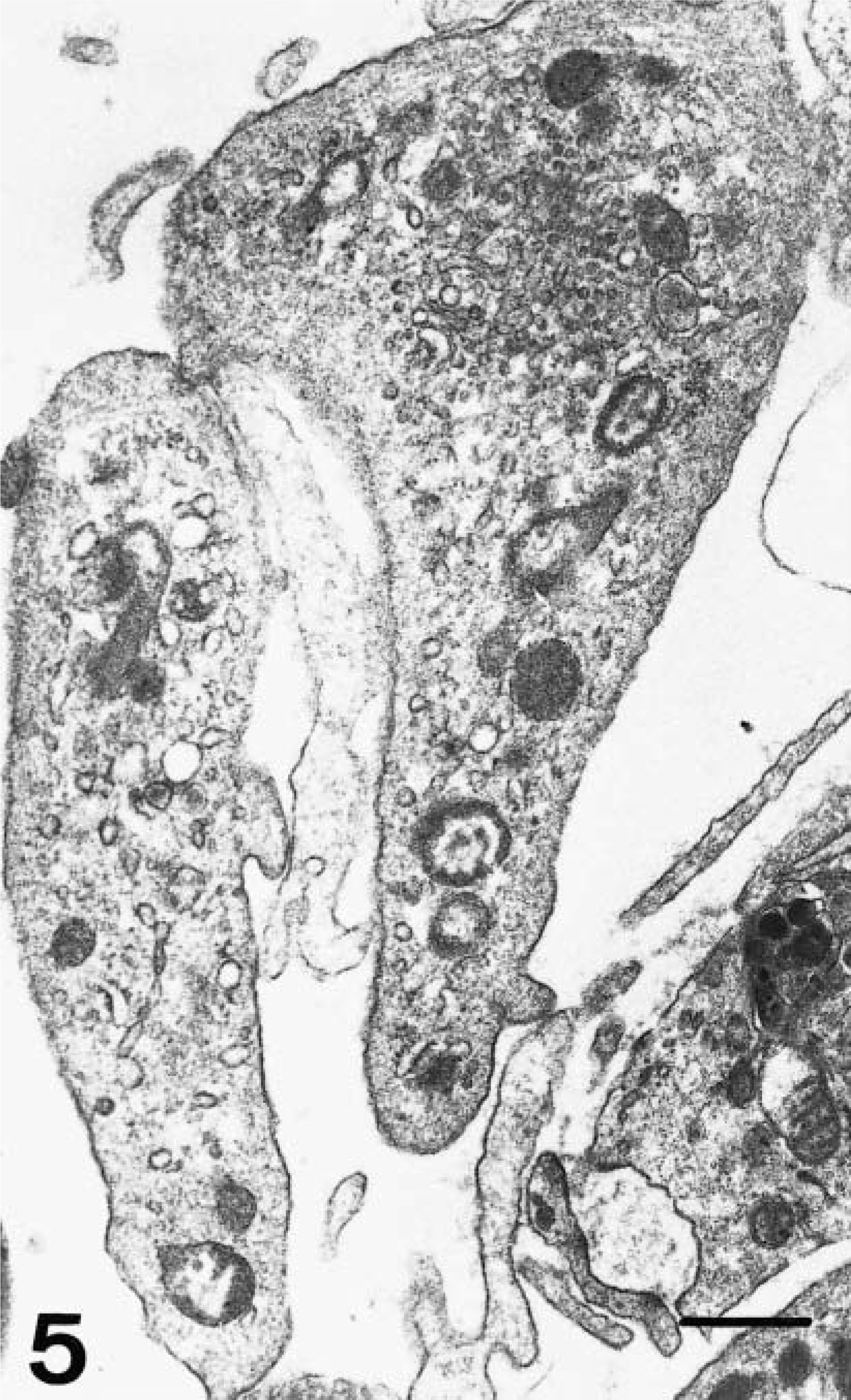
Electron micrograph. Spleen; pig, 2 dpi. Enlarged platelets in close contact and showing degranulation and dilated open canalicular system. Uranyl acetate and lead citrate. Bar = 500 nm.
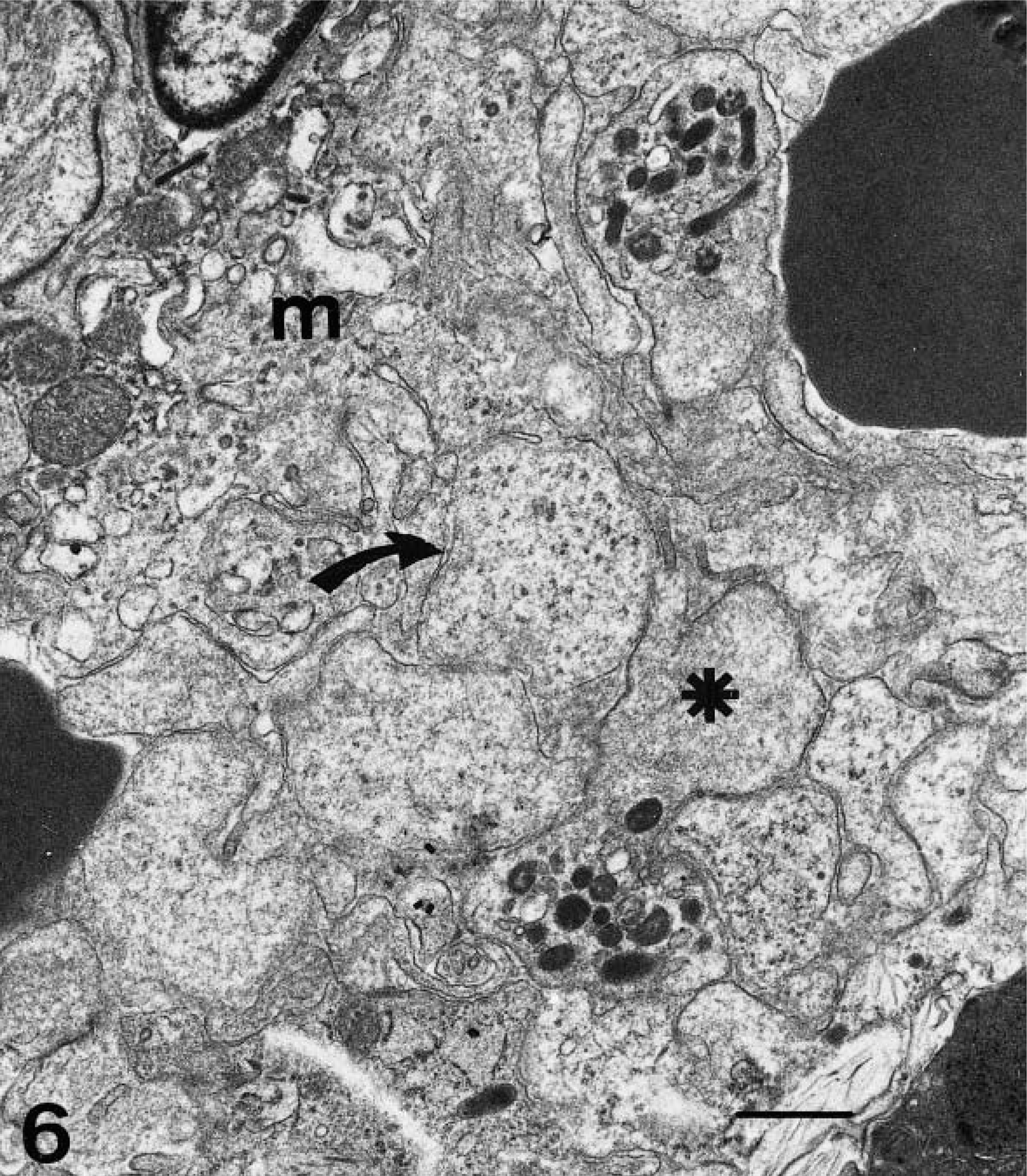
Electron micrograph. Spleen; pig, 4 dpi. Intense platelet aggregation in close contact with a fixed macrophage showing subcellular changes of activation (m). Platelets appear partially (★) or completely (arrow) degranulated. Uranyl acetate and lead citrate. Bar = 1 µm.
Over the same period (2 and 4 dpi), macrophages in the marginal zone and splenic cords displayed signs of secretory activation consisting of an increase in the size and number of rough endoplasmic reticulum cisternae and an enlargement and proliferation of Golgi complex (Fig. 7). Cell cytoplasm was swollen and rounded and displayed loss of filopodia and proliferation of primary and secondary lysosomes, subcellular changes indicative of phagocytic activation (Fig. 8). Activated platelets were in close contact with these cells (Fig. 8) and phagocyted by macrophages (Fig. 9).
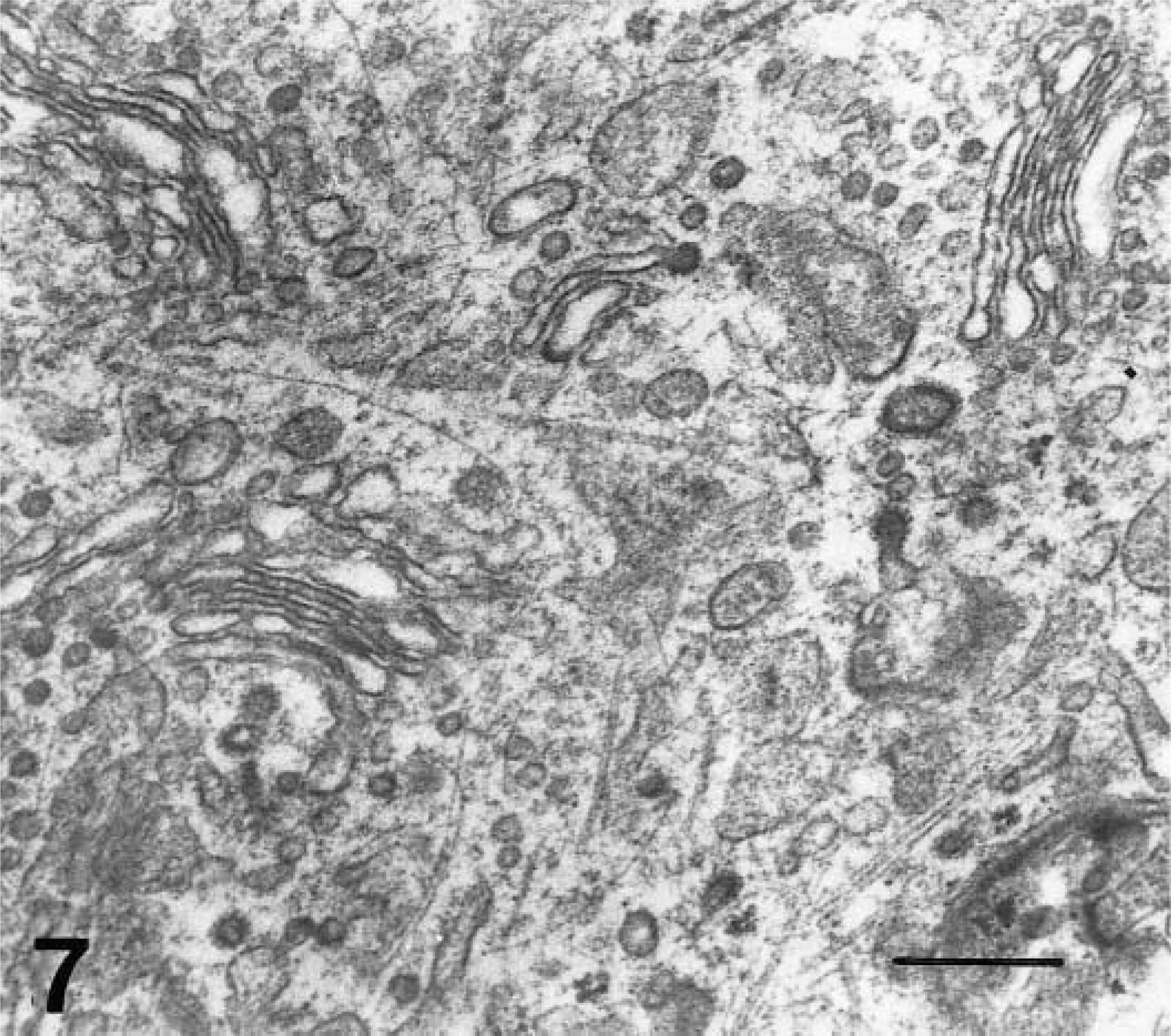
Electron micrograph. Spleen; pig, 2 dpi. Macrophage with proliferation of Golgi complex (secretory activation). Uranyl acetate and lead citrate. Bar = 500 nm.
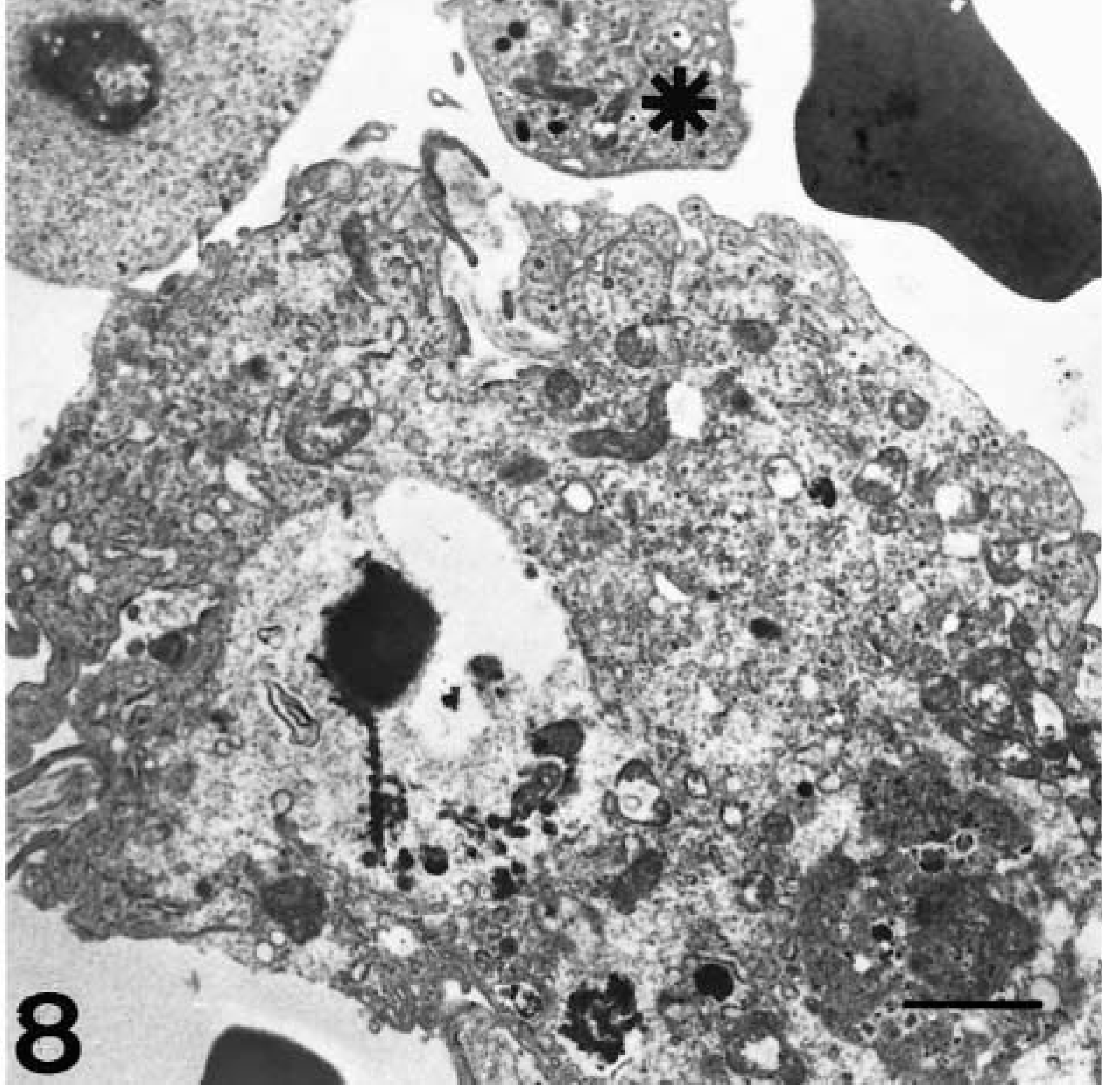
Electron micrograph. Spleen; pig, 4 dpi. Enlarged and surrounded fixed macrophage with loss of filopodia and abundant phagocytized material. Near these cells are possible activated platelets (★). Uranyl acetate and lead citrate. Bar = 2.5 µm.
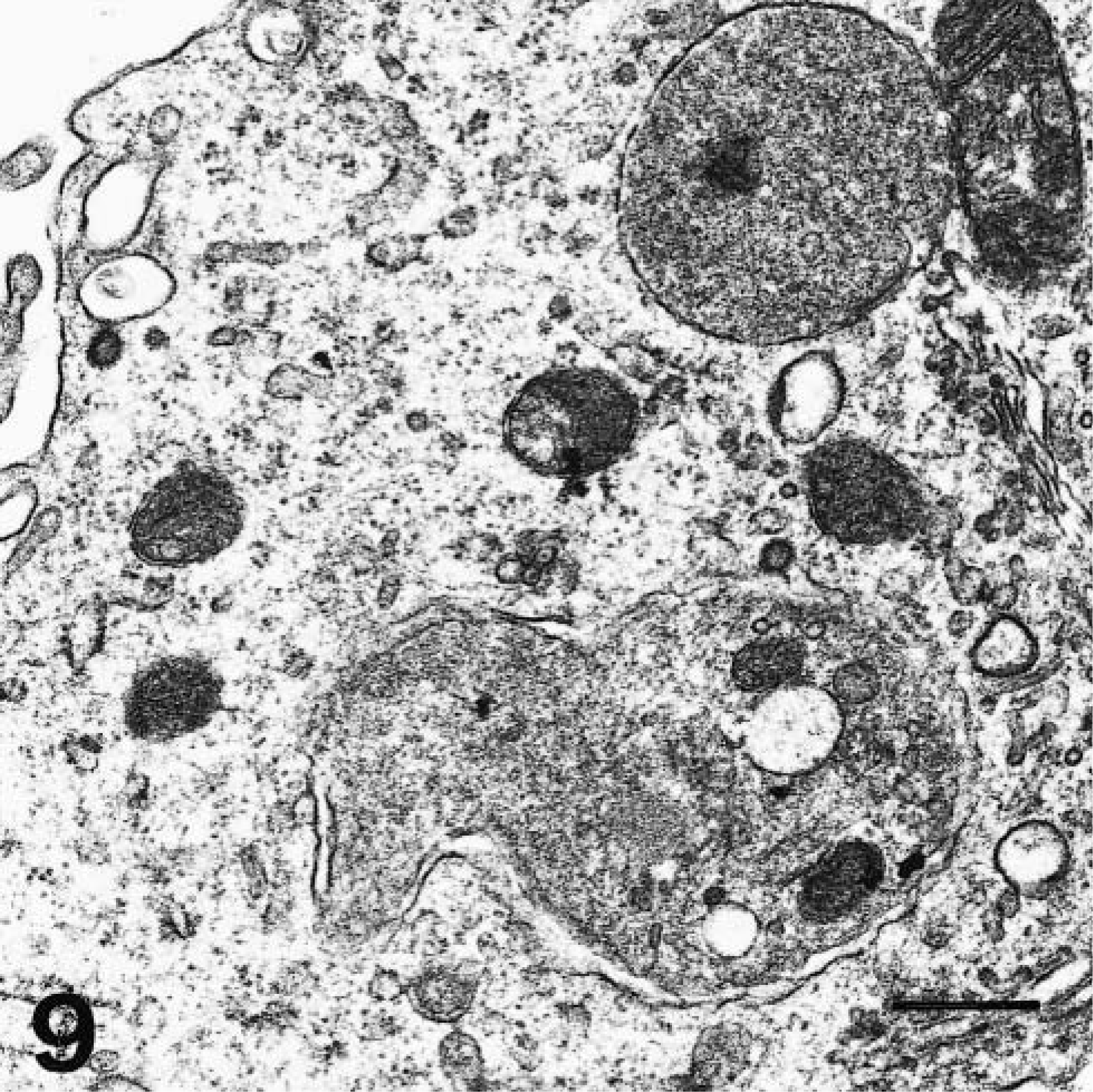
Electron micrograph. Spleen; pig, 4 dpi. Degranulated platelet phagocytized by a splenic macrophage. Uranyl acetate and lead citrate. Bar = 500 nm.
Immunohistochemical detection of viral antigen gp55 confirmed the presence of virus antigen from 2 dpi onwards in monocytes/macrophages. Ultrastructurally, these cells displayed signs of viral infection; numerous rough endoplasmic reticulum dilations and vesicles were observed, containing material of variable electron density and rounded particles both with and without nucleoids, which ranged in size from 45 to 60 nm (Fig. 10). These structures were similar to viral particles detected in PK15 cultures (Fig. 11).
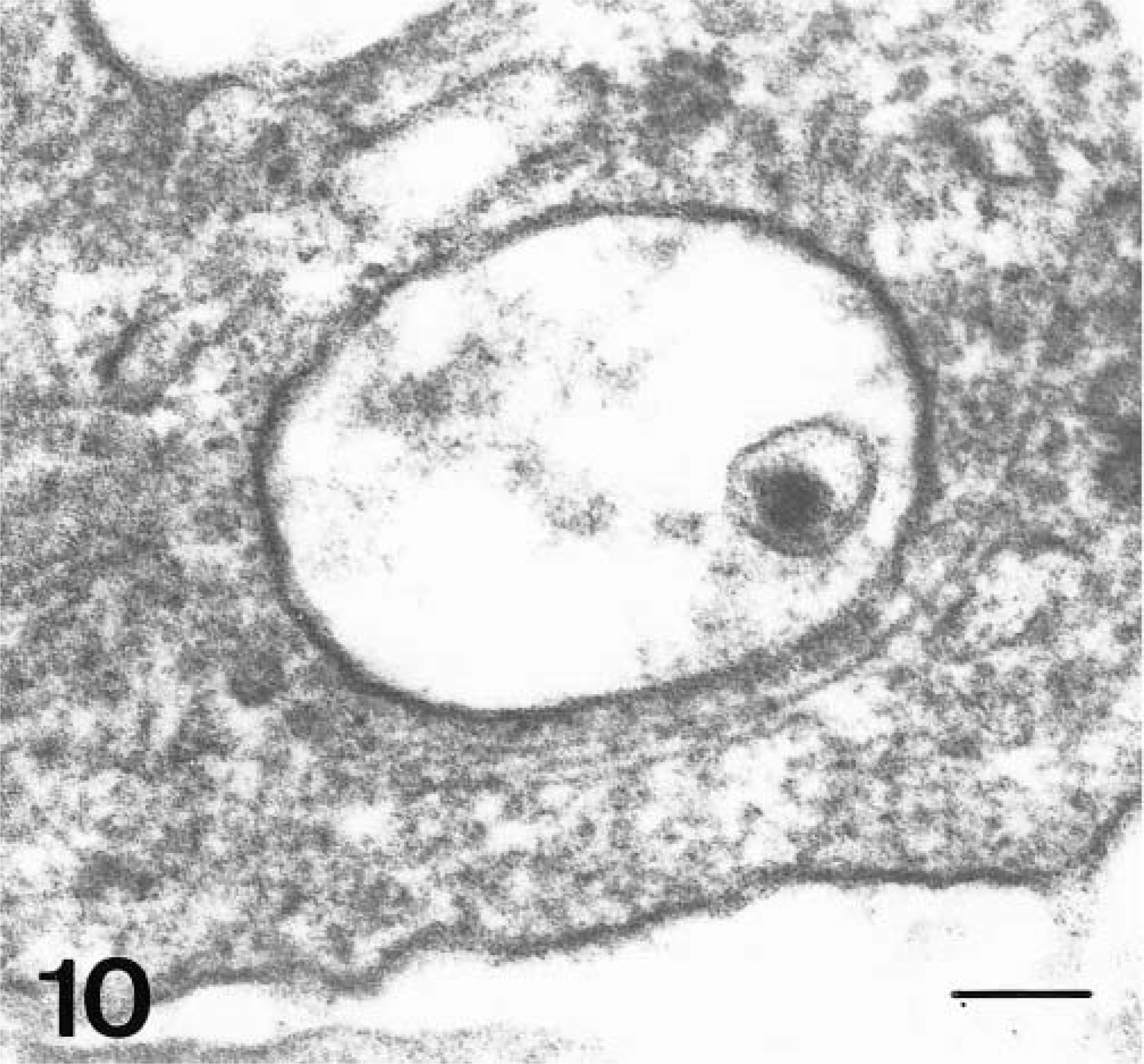
Electron micrograph. Spleen; pig, 2 dpi. Intracytoplasmatic vesicles containing virus-like particle and electrodense material. Uranyl acetate and lead citrate. Bar = 75 nm.
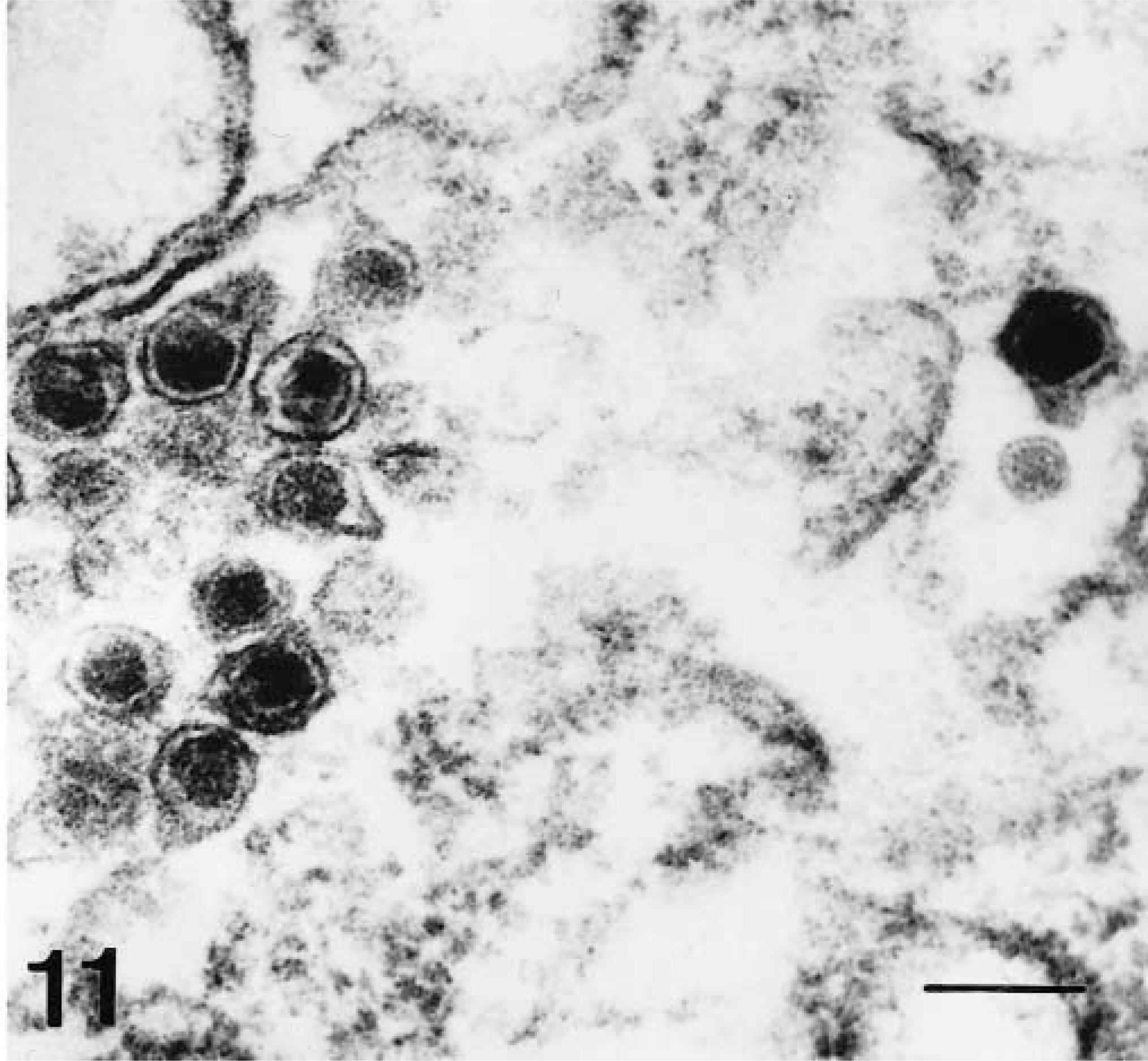
Electron micrograph. PK15 cell culture. Intracytoplasmatic vesicles containing virus particles. Uranyl acetate and lead citrate. Bar = 75 nm.
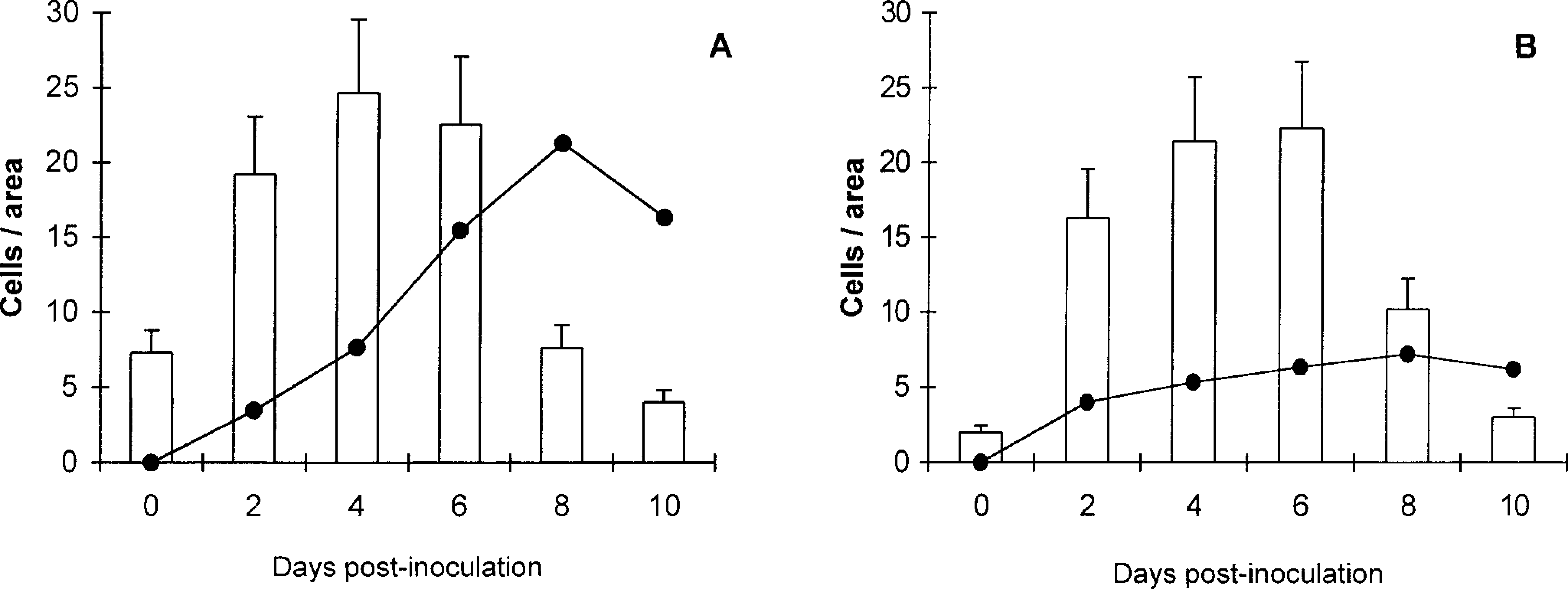
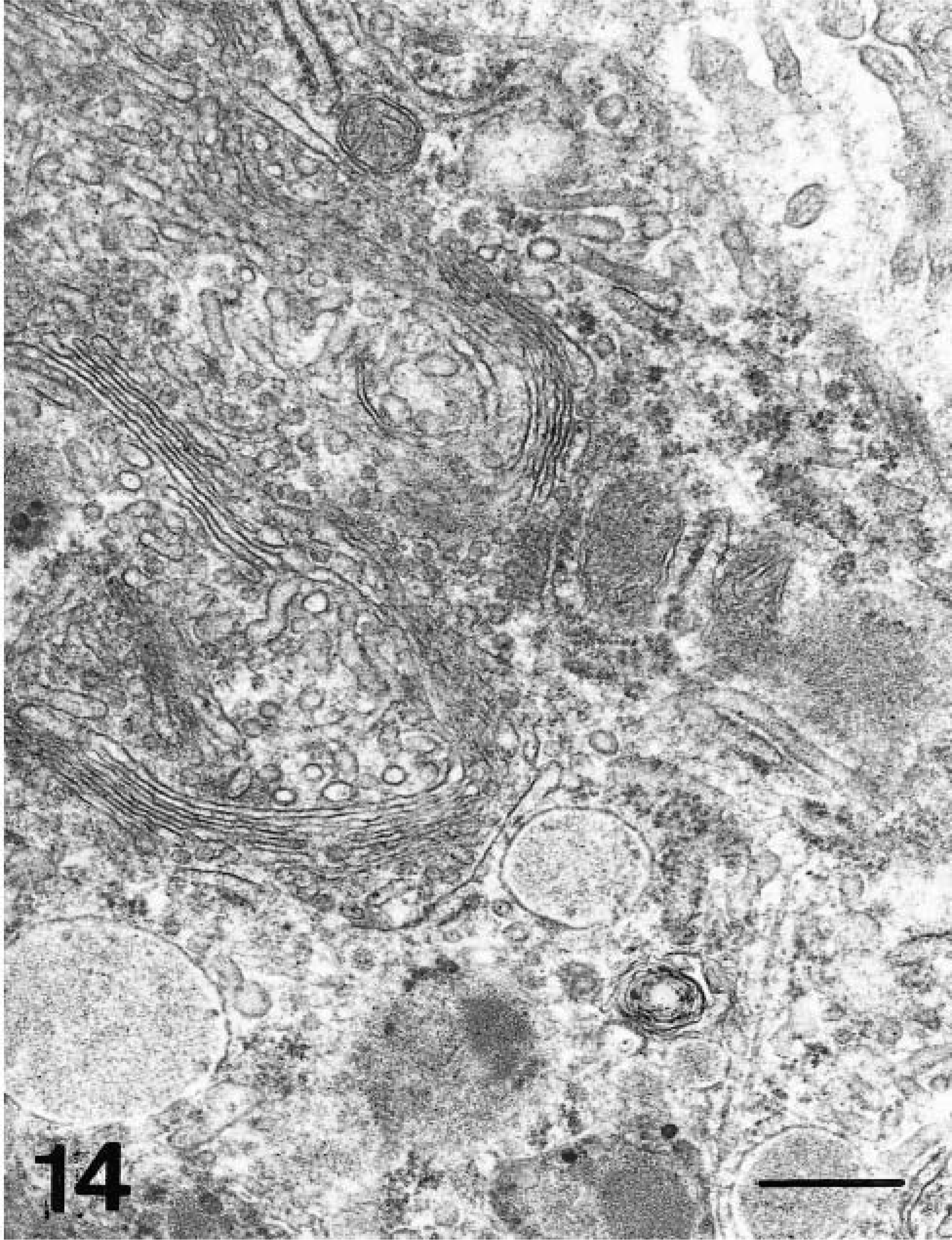
Evidence of platelet activation similar to that described above was also observed during the same period in hepatic sinusoid lumina, although clusters consisted of only two to four platelets. A few activated platelets were observed in the perisinusoidal space. Hepatic sinusoid Kupffer cells showed signs of secretory and phagocytic activation from 2 dpi, with engulfed platelets (Fig. 12) and positive reaction to FVIII-rag with significant numerical changes (Fig. 13). Kupffer cells stained positive for viral antigen gp55 from 4 dpi, and displayed ultrastructural evidence of virus infection and activation (Fig. 14).
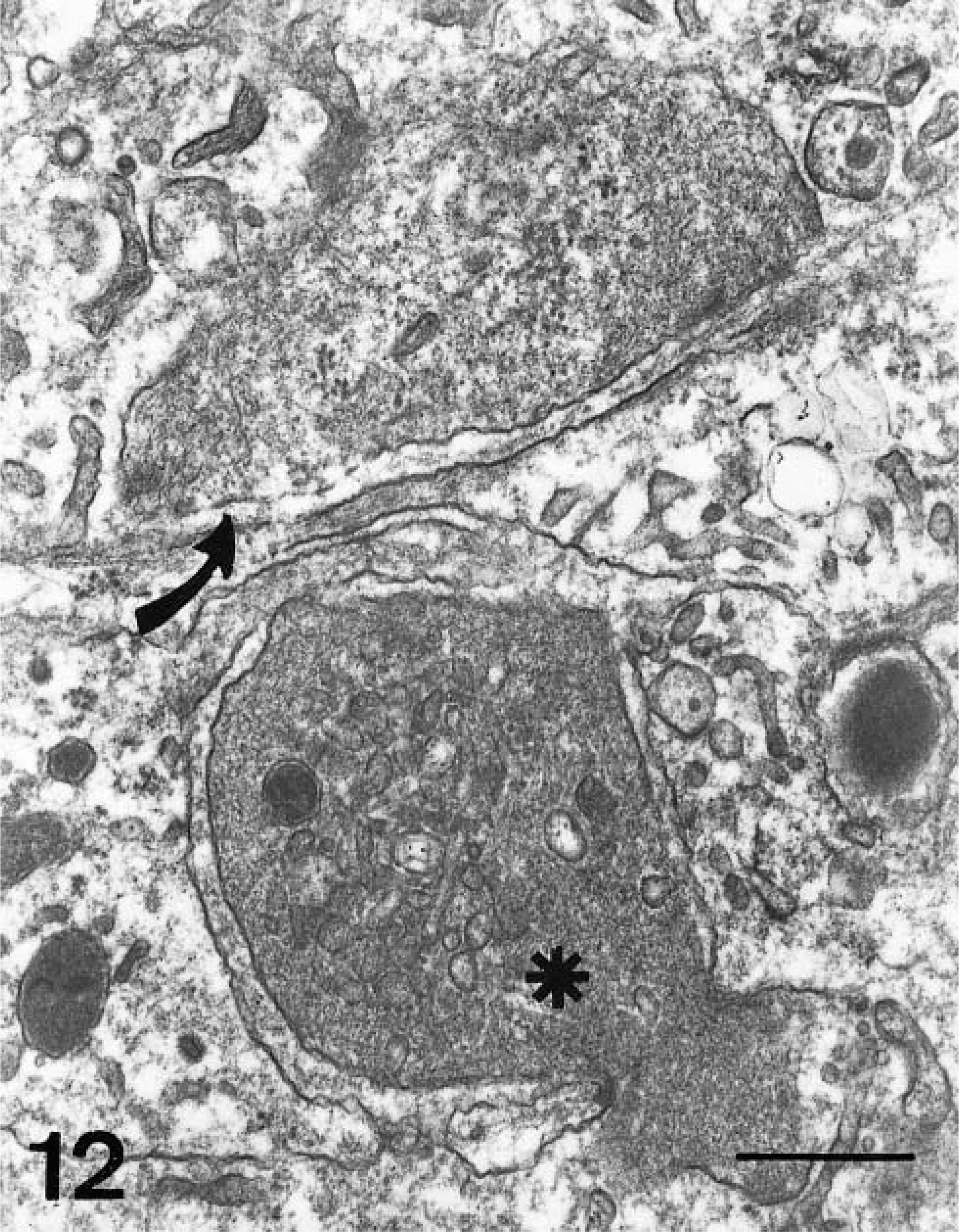
Electron micrograph. Liver; pig, 4 dpi. Partially degranulated platelet engulfed by Kupffer cell (★), and completely degranulated platelet in perisinusoidal space (arrow). Uranyl acetate and lead citrate. Bar = µm. 500 nm.
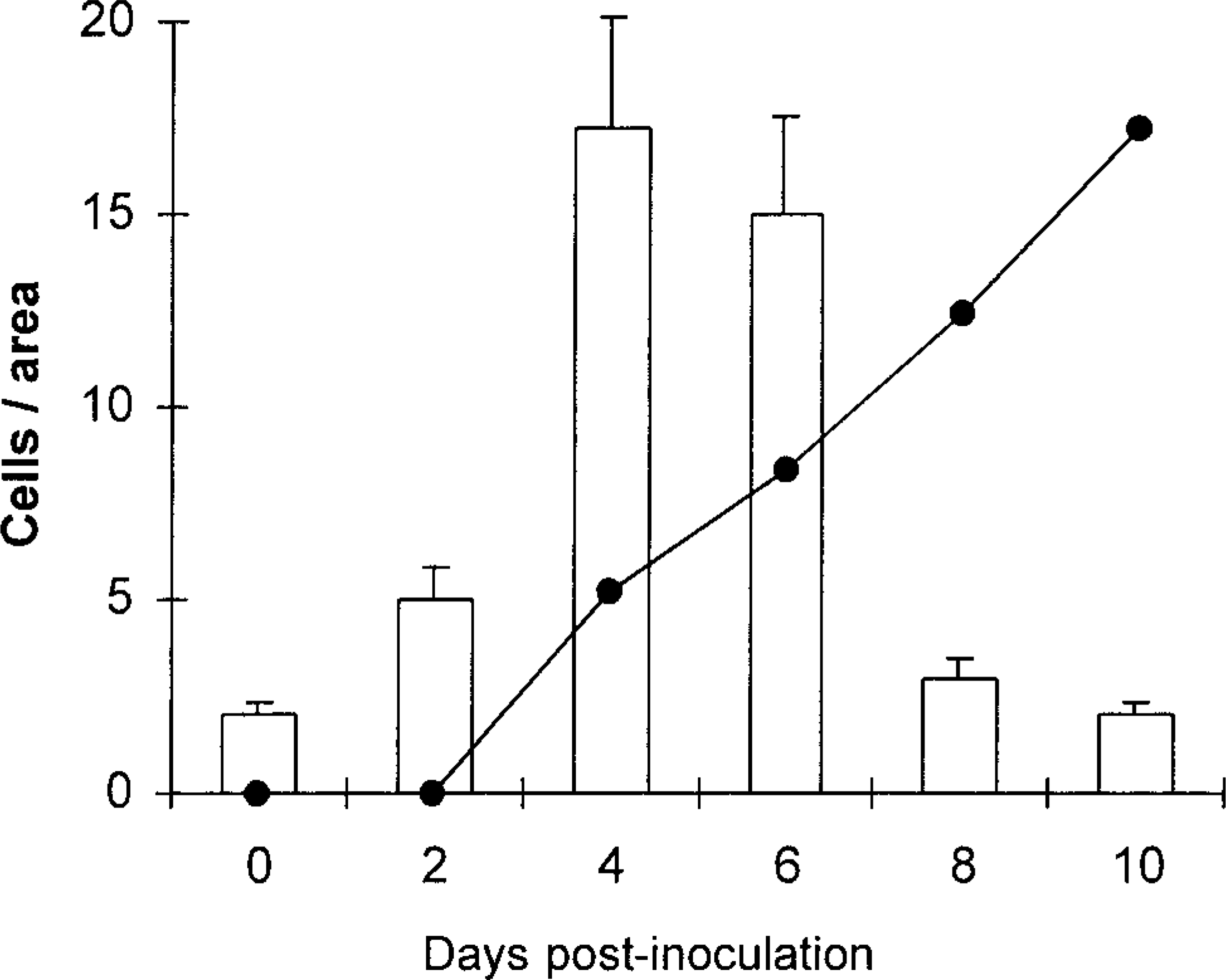
Counts of Kuffer cells immunolabeled for FVIII-rag (○ ± SD) and virus gp55 antigen (•) (means).
Electron micrograph. Liver; pig, 4 dpi. Kupffer cell showing proliferation of Golgi complex and lysosomes. Uranyl acetate and lead citrate. Bar = 500 nm.
Discussion
Results show that the thrombocytopenia observed here from 2 dpi with the CSF isolate Quillota appeared at the same time as intense platelet activation and aggregation, with subsequent phagocytosis of platelets in the spleen and to a lesser extent in the liver. These platelet changes coincided with the presence of virus antigen in liver and speeln and with phagocytic and secretory activation of the various macrophage populations.
Thrombocytopenia is a pathologic blood condition that may be caused by impaired or reduced platelet production or by increased peripheral platelet consumption due to accelerated destruction, massive utilization, or abnormal distribution. 13 True thrombocytopenia generally involves more than one of these physiopathologic mechanisms. 13
Studies by the prsent team have shown that CSF virus infection prompts significant changes in bone marrow, although these occur after the onset of thrombocytopenia. 20 Moreover, although megakaryocytes are infected by the virus, the small number of infected cells and the absence of lesions consistent with impaired thrombocytopoiesis suggest that the onset of thrombocytopenia in CSF cannot be the result of bone marrow failure. 9,20 The degenerative changes reported in megakaryocytes by some authors 3 were also encountered in this study in the form of naked megakaryocyte nuclei (unpublished data) resulting from an attempt by these cells to restore platelet numbers 1,22 ; this phenomenon must thus be viewed as a result rather than a cause of thrombocytopenia.
Peripheral platelet depletion may be due to 1) disseminated intravascular coagulation, 2) abnormal platelet distribution, or 3) phagocytosis of activated and/or damaged platelets. 13
Disseminated intravascular coagulation may be ruled out as a cause of thrombocytopenia in CSF. Although microthrombi have been observed in animals infected with the CSF virus, 12 this lesion was subsequent to the onset of thrombocytopenia and in the present study was not detected until the final phases of the disease. There also was no morphologic evidence of endothelial or tissue damage that might prompt activation of coagulation pathways, which has been indicated previously.
Massive emigation of platelets into the hepatic perisinusoidal space due to macrophage activation and cytokine release has been reported in a number of pathologic processes. 17 However, in the present study, despite morphologic changes indicative of secretory activation in Kupffer cells, the number of activated platelets observed in the perisinusoidal space was not sufficient to trigger thrombocytopenia. Consequently, thrombocytopenia cannot be attributed to abnormal platelet distribution, although such a distribution might intensify an existing thrombocytopenia.
The massive phagocytosis of platelets by macrophages observed here may be due to 1) platelet damage induced by a virus, 25 2) plasma membrane changes induced by immune-mediated mechanisms, 6,7,21 or 3) activation and aggregation triggered by the release of chemical mediators by various cells. 8 Recent studies 9 have shown that the presence of viral antigen in platelets is due to early infection of a small number of megakaryocytes. This reduced percentage of infected megakaryocytes would not account for the early intense thrombocytopenia observed in affected animals. Similarly, immune-mediated thrombocytopenia associated with the presence of immune complexes may be ruled out as the cause of platelet depletion in CSF because the onset of thrombocytopenia (2 dpi) occurs prior to the appearance of a humoral immune response. 24
The morphologic changes consistent with platelet activation observed in three pigs match those reported following administration of platelet-activating factor, 2,15 a potent mediator of platelet aggregation released mainly by macrophages. 4,5,15,16 In the present study, changes indicative of secretory activation were observed in macrophages from 2 dpi in spleen and from 4 dpi in liver. These findings suggest that this mechanism may be responsible for triggering thrombocytopenia in acute forms of CSF. Another possible inducer of platelet activation and aggregation is tissue factor, which initiates the extrinsic coagulation cascade. 14 However, the absence of fibrin strands in affected organs at the onset of thrombocytopenia enables this factor to be ruled out as a cause of platelet depletion.
Footnotes
Acknowledgements
This work was supported financially by grants from the Plan Andaluz de Investigatión (AGR 137), DGESIC PB98-1033 and FONDECYT-CONICYT (98152008-1.0, 98152009-1.2).