
Abstract
Classical swine fever (CSF) and African swine fever (ASF) are both highly contagious diseases of domestic pigs and wild boar and are clinically indistinguishable. For both diseases, antibody detection is an integral and crucial part of prevention and control measures. The purpose of our study was to develop and initially validate a duplex pen-side test for simultaneous detection and differentiation of specific antibodies against CSF virus (CSFV) and ASF virus (ASFV). The test was based on the major capsid protein VP72 of ASFV and the structural protein E2 of CSFV, both considered the most immunogenic proteins of these viruses. The performance of the pen-side test was evaluated using a panel of porcine samples consisting of experimental, reference, and field sera, with the latter collected from European farms free of both diseases. The new lateral flow assay was able to detect specific antibodies to ASFV or CSFV, showing good levels of sensitivity and specificity. These preliminary data indicate the potential of the newly developed pen-side test for rapid differential detection of antibodies found in the 2 diseases, which is of particular importance in the field and in front-line laboratories where equipment and skilled personnel are limited and control of ASF and CSF is crucial.
African swine fever (ASF) is an infectious disease of domestic and wild pigs of all breeds and ages, causing a wide range of syndromes from mild disease to lethal hemorrhagic fever.3,4 The causative agent of the infection, African swine fever virus (ASFV; family Asfarviridae, genus Asfivirus), is a large, enveloped, icosahedral double-stranded DNA virus. 7 The disease is endemic in sub-Saharan Africa and Sardinia.18,22 ASF has become endemic throughout the Caucasus and the Russian Federation since 2007, where the continued spread poses a serious threat to the swine industry worldwide.19,25 This was manifested with the introduction of the disease into the European Union member states Poland, Lithuania, Latvia, and Estonia in 2014, where the disease affected both wild boar and domestic pigs. 8 Because no vaccine against ASFV is available, the presence of antibodies against ASFV is used as an indicator of infection.
Classical swine fever (CSF) is a highly contagious disease causing major losses in swine populations almost worldwide. 17 The etiologic agent of CSF is Classical swine fever virus (CSFV; family Flaviviridae, genus Pestivirus), an enveloped, positive-stranded RNA virus. 14 Modified-live vaccines (MLVs) are used routinely in CSF endemic areas, including Asia and central-south America, and induce complete protection against virulent CSFV.5,13 Although the virus has been eradicated in many countries, the disease has been reported since 2011 in Hungary, Lithuania, Serbia, Israel, the Russian Federation, and Latvia.23,24
With the spread of ASFV from the Caucasus, the probability of areas encountering both viruses is increasing, and the presence of wild boar carrying these viruses increases the risk of their introduction into the domestic pig population. In fact, in Latvia, there are overlapping restriction zones for CSF and ASF in wild boar. For these reasons and taking into consideration that both pathogens cause diseases notifiable to the World Organization for Animal Health (OIE), early differential diagnosis is of great value for immediate implementation of control measures to prevent further spread of the diseases. However, differentiation between CSF and ASF by clinical or postmortem examination is currently not possible; therefore, pathogen verification is conducted through laboratory testing.
To date, diagnosis of ASF is based on direct identification of the virus by polymerase chain reaction (PCR) or virus isolation and detection of antibodies by enzyme-linked immunosorbent assay (ELISA), immunoblotting, or fluorescent antibody testing.1,20,21,28 Similarly, the diagnosis of CSFV is based on identification of the agent by PCR and detection of antibodies by either ELISA or a virus neutralization test (VNT).6,16 To date, multiplex PCR assays for simultaneous detection of ASFV and CSFV have been developed with good sensitivity and specificity.2,11 However, these methods are still rather time-consuming and require well-equipped laboratories and well-trained personnel, which will possibly delay disease diagnosis in remote areas. In both diseases, antibodies appear during the first to second week of infection and persist for long periods of time. Hence, in the absence of an effective vaccine, the presence of antibodies is a good marker of infection, especially in cases of subacute or chronic forms of the diseases.
We developed a rapid, 1-step, immunochromatographic test that specifically detects and distinguishes between anti-ASF and anti-CSF antibodies in serum specimens. The qualitative test is based on the use of different colored carboxyl-modified latex microspheres that are covalently linked to the specific target proteins. The glycosylated E2 structural protein of CSFV (Brescia strain) and the major capsid protein of ASFV VP72 (BA71 strain) are considered to be the most immunogenic proteins of these viruses,10,15 and thus represent appropriate target antigens for antibody detection. The VP72 protein purified from ASFV-infected cell cultures with a Vero (African green monkey kidney) cell–adapted virus was used to coat red latex particles; the recombinant affinity-purified E2 protein was linked to blue latex particles. Finally, for the control latex, green particles were used to bind biotin. Prior to protein conjugation, latex particles were activated with EDC (1-ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride) and NHS (N-hydroxysuccinimide). 12 Subsequently, the beads were coupled to the recombinant proteins at a surface concentration of 0.5 mg/m2. Finally, conjugated latex particles were diluted in Tris–HCl (10 mM, pH 8.2) and were stored at 4°C before use. The E2 and VP72 proteins were used as test line T1 and T2 capture reagents, respectively. The VP72 protein was diluted to 50 μg/mL in Tris–HCl (20 mM) buffer at pH 7.5, containing 5% sucrose and 0.095% sodium azide as preservative. The E2 protein was diluted to 150 μg/mL in the same buffer. An anti-biotin IgG monoclonal antibody (mAb) was used as the control line capture reagent, diluted to 1 mg/mL in the same buffer used for the test lines. The test and control capture reagents were dispensed in 3 parallel lines on a 25 mm × 300 mm nitrocellulose membrane d at 1 µL/cm. After drying for 5 min at 45°C, the membranes were sealed and stored at room temperature under dry conditions. To prepare the conjugate solution, the previously prepared VP72-latex, E2-latex, and biotin-latex particles were diluted at 0.2%, 0.2%, and 0.15%, respectively, in a Tris–HCl (25 mM, pH 9.5) buffer containing humidity preservatives and blocking agents (3% bovine serum albumin, 1.5% casein, 35% sucrose, 1% polysorbate 20, 0.095% NaN3). The mixture was dispensed onto the rayon conjugate pad, which was then dried for 30 min at 45°C and stored at room temperature under dry conditions. A master card was assembled as follows: on a plastic backing with adhesive, nitrocellulose membrane, conjugated pad, sample pad, and absorbent pad were pasted and covered with a protector film. The master card was then cut into strips 4.2 mm wide, which were placed individually in a plastic device.
The 2 virus-specific assays were developed individually in order to prove the validity of the reagents for the proposed test (data not shown). Then the conditions were adapted to a duplex lateral flow assay (LFA) to determine optimal settings. For this purpose, different viral protein concentrations for conjugation to the latex beads were tested, as were different running buffers and sample pads. As whole blood represented a possible biological sample, a special pad able to retain erythrocytes was used to further develop the assay. However, in the present study, only serum samples were used applying the following protocol: 10 µL of the sample (serum) are applied onto the sample pad followed by 120 µL of running buffer (Tris–HCl pH 7.5, NaCl, casein, and NaN3 as preservative), which allows the mixture to migrate through the conjugate pad and the nitrocellulose membrane by capillarity. In the presence of antibodies against ASFV or CSFV, these bind to VP72 or E2 protein–coated microparticles forming a latex antigen-antibody immune complex. As the duplex LFA was developed using a double recognition format that relies on the ability of an antibody to recognize 2 epitopes at the same time, this immune complex then reacts with the immobilized VP72/E2 protein on the membrane via the captured antibody, making the test line visible (T1/T2). Results were interpreted 10 min after adding the sample. A positive result for ASFV was recorded for samples with red and green lines visible. For samples positive to CSFV, a blue and a green line were visible, and, in the case of a negative sample, only a green line appeared (Fig. 1).
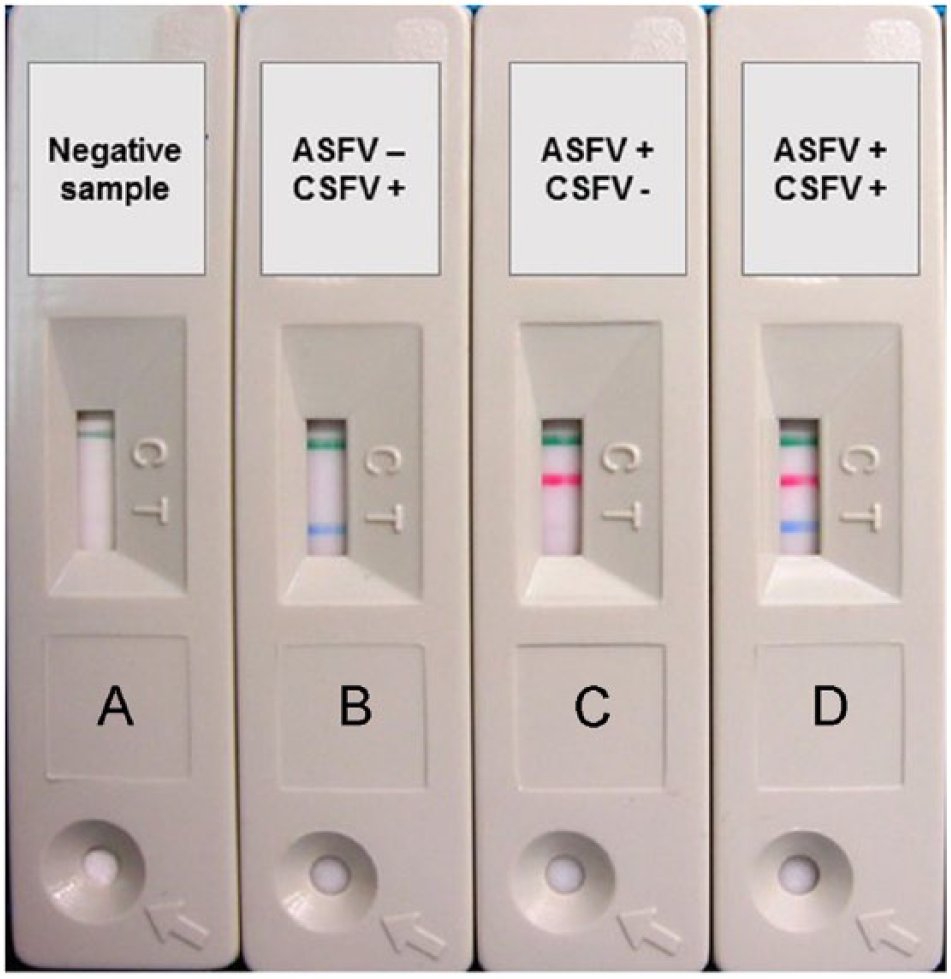
Examples of duplex lateral flow device results.
To evaluate the analytical sensitivity of the pen-side test, we used an ASFV-positive reference serum (provided by the European Union reference laboratory for ASF [EURL], Valdeolmos, Spain) and a CSFV-positive reference serum (provided by the National and FAO reference laboratory for CSF at the Friedrich-Loeffler-Institut [FLI], Isle of Riems, Germany). Using the duplex LFA, detection was possible down to the 1/3,200 dilution of the ASFV-positive serum, which had previously been characterized by the OIE ELISA against the BA71 strain, and down to the 1/64 dilution of the CSFV-positive serum, which had been characterized by VNT against CSFV strain Alfort/187 with a 50% neutralization dose (ND50) of 3,200 (data not shown). Additionally, specific mAbs to the target proteins (VP72-ASFV and E2-CSFV) were used in parallel to the reference sera to determine analytical sensitivity. 18BG3 mAb a was used to determine the sensitivity for the ASF test line, following a previously described protocol, 26 and the 18.4 mAb b was used for the CSF test line. A mixture of both antibodies diluted in running buffer at decreasing concentrations was also used to evaluate the ability of the test to detect both viruses simultaneously. Each dilution was treated as an independent sample. The test was able to detect mAbs at concentrations down to 8 ng/mL (0.96 ng/test) for 18BG3, and down to 125 ng/mL (15 ng/test) for 18.4 (Fig. 2A).
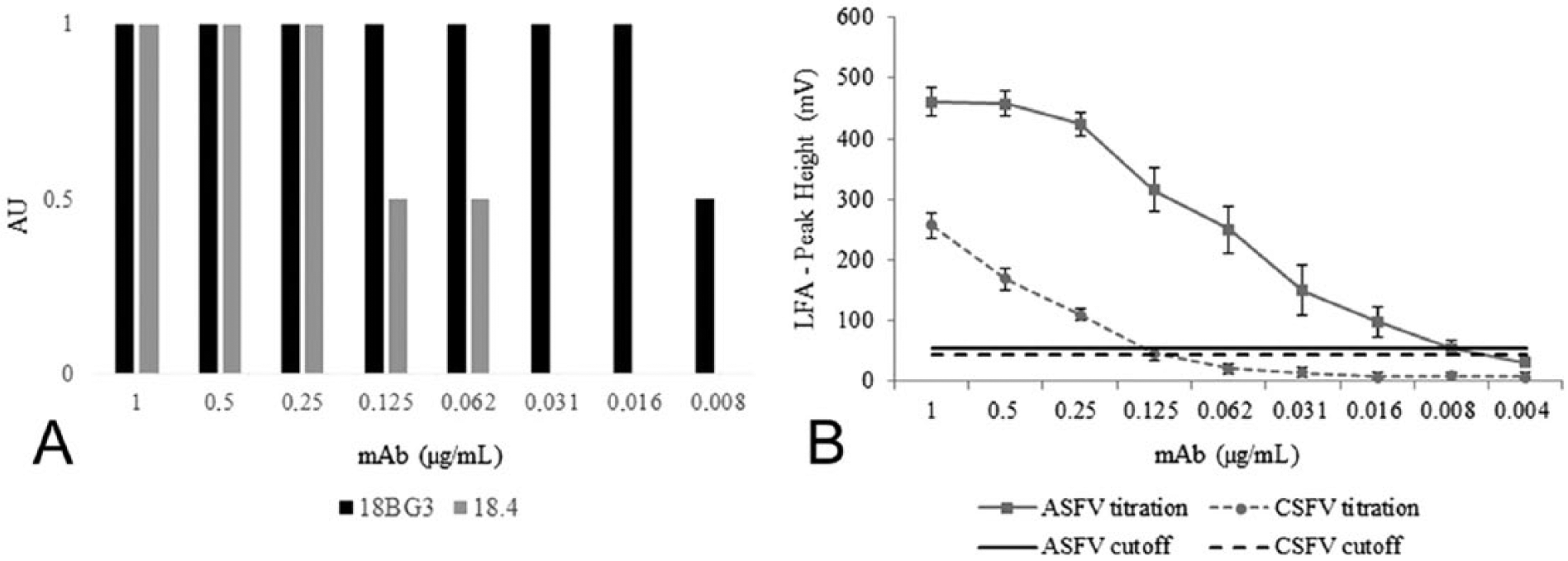
Analytical sensitivity of the duplex lateral flow assay (LFA). Serial dilutions of 2 specific monoclonal antibodies (mAbs) for Classical swine fever virus and African swine fever virus (18BG3 for ASFV, and 18.4 for CSFV) were used to determine the sensitivity of the test.
In addition to the visual evaluation, the ASFV/CSFV duplex LFA was semiquantified by a lateral flow reader e and software, following the manufacturer’s instructions. The LFA reader correlates an intensity result (in mV) with the color level of the control and test lines. By comparing the visual evaluation of each test (performed by 3 different workers) and the mV values obtained by the LFA reader, results were grouped into “positive,” “weak positive,” or “negative.” A defined range of mV values was estimated for each group of results by considering the minimum and maximum value read among all the analyzed tests. The LFA reader cutoff for each test line was established according to the minimum value of the “weak positive” group. By conducting different titration curves of mAbs for ASFV and CSFV and analyzing individual ASFV and CSFV experimental and field sera, cutoff values of 55 mV for ASFV and 43 mV for CSFV were obtained. Evaluation of the LFA reader showed that the detection limit for both mAbs was identical with the ones observed visually. In addition, semi-quantitative analysis also showed that the ASFV test line intensity reached a plateau of ~400–500 mV for antibody concentrations >0.125 µg/mL, whereas the CSFV test line intensity declined almost linearly along with the antibody concentration. No cross-reactivity was observed between the 2 test lines with any of the mAbs. The ASFV test line was more sensitive (0.96 ng/test) than the one for CSFV (15 ng/test). As described above, the concentration of protein bound to the nitrocellulose membrane was different (50 μg/mL for VP72 vs. 150 μg/mL for E2). The VP72 of ASFV showed more reactivity than the E2 of CSFV, albeit applied onto the membrane with a 3-fold lower concentration. Ideally, it was desirable for both test lines to display the same sensitivity, although the final concentration of the E2 recombinant protein obtained after the purification step did not allow a higher concentration of protein on the membrane. It is also important to mention that the VP72 protein is based on a viral antigen, whereas the E2 antigen of CSFV is a recombinant protein. The use of viral antigens has some limitations mainly related to biosecurity and standardization concerns. Although recombinant antigens are a good alternative to avoid the use of live virus, 9 the sensitivity of the test can differ. Recently, our laboratory (INGENASA, Madrid, Spain) has expressed different fragments of the VP72 protein with very good yields of expression in a baculovirus system. However, the reactivity of this recombinant protein in several immunoassays was in all cases lower than that of the viral antigen (data not published).
Finally, to determine the diagnostic sensitivity and specificity of the test, 2 panels of well-characterized swine sera were evaluated. Experimental reference sera for ASFV (n = 31) were provided by the EURL. Positive sera (22/31) were obtained from domestic pigs inoculated by the intramuscular route with 106 50% tissue culture infectious doses per milliliter (TCID50/mL) of the NH/P68 isolate (NHV), a nonhemadsorbing and low virulence ASFNHV/L60 isolate. Animals were challenged at day 29 post-inoculation with the homologous virulent ASFV strain L60. The serum samples were collected between 20 and 23 days post-challenge, and their reactivity was determined by the OIE-prescribed tests (ELISA and immunoblotting) following the protocol described in the OIE Manual of Diagnostic Tests and Vaccines for Terrestrial Animals. 27 This panel of sera comprised 22 ASFV-positive and 9 ASFV-negative sera, as determined by the OIE ELISA, which is considered the reference standard technique. The samples were also analyzed by a commercially available competition ELISA f based on mAbs to the VP72 protein showing complete concordance with the results obtained with the reference test. When these samples were tested by the duplex LFA ASFV/CSFV, 19 of 22 sera were also positive for the ASF line and 3 showed a weak positive result. The 9 negative reference sera also were reported negative in the duplex LFA in the ASF test line. None of the 31 serum samples reacted positively with the CSF test line (Table 1).
Analysis of African swine fever virus (ASFV) serum samples in the duplex lateral flow assay (LFA) and comparison with the results obtained by the OIE ELISA and a commercial competitive ELISA g (COMPAC) shown as percentage of competition.
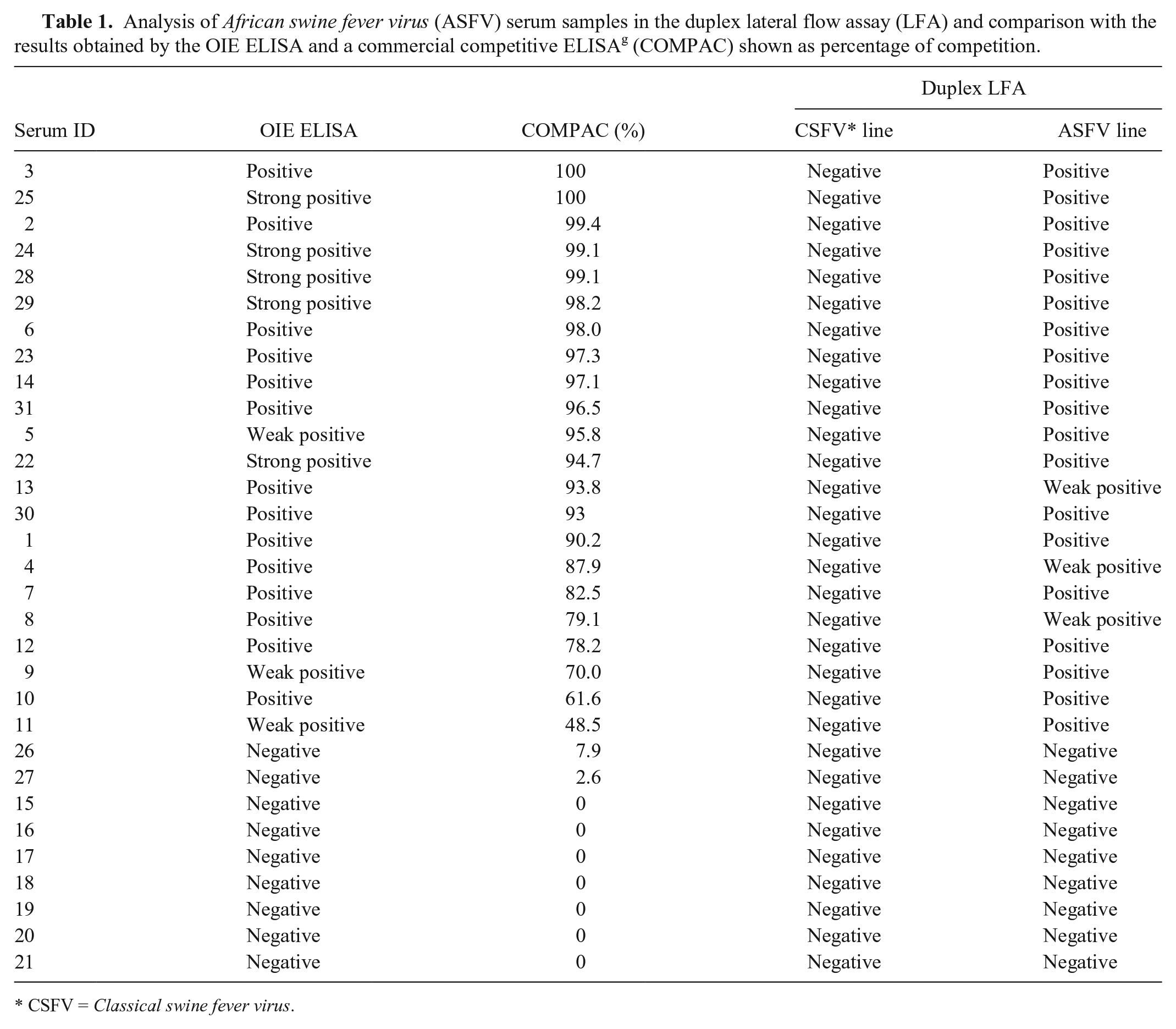
CSFV = Classical swine fever virus.
The second panel of sera used to evaluate the duplex LFA corresponded to sera for CSFV (n = 30) provided by the National and FAO reference laboratory for CSF at FLI. These sera were derived from experimentally infected pigs and characterized by VNT against CSFV strain Alfort/187 and a commercial CSFV E2 antibody ELISA. c VNT was carried out according to the recommendations of the European Commission (Commission Decision 2002/106/EC, http://goo.gl/v2VBMe). The panel was chosen to reflect different time points postinfection (early phase/convalescent phase) and different CSFV genogroups and subgenogroups (Table 2). To check for cross-reactivity, sera from pigs inoculated with ruminant pestiviruses (Bovine viral diarrhea virus and Border disease virus) were included. The early phase of infection (≤21 days postinfection) was represented by 11 sera, the convalescent phase by 13 sera, and the pestivirus samples by 6 sera. Thus, the panel followed the recommendations for CSF antibody ELISA batch release as laid down by the European Commission (Commission Decision 2002/106/EC). According to the ELISA cutoff (40% inhibition), 23 of 30 samples gave a positive result and 7 were negative. Moreover, these samples were analyzed by VNT, which is considered the gold standard technique for CSF diagnosis. In this case, according to the cutoff of the assay (10), 24 samples were positive, although it is important to point out that sample F4/Kol showed the lowest VNT titer. The 6 sera from experimental infections with other pestiviruses were positive by VNT against the homologous virus. When all of these sera were tested by LFA, 22 of 30 samples reacted positively in the CSF test line. Sample F4/Kol appeared to be negative by LFA, correlating with the result of the E2 ELISA. Sample E6/13 was also negative. The latter gave a weak positive result in the E2 ELISA, showing 41% inhibition, which is very close to the cutoff value, and also had a low VNT titer (30). All 6 negative samples were negative in the LFA. No positive results were observed for the ASF test line in any case (Table 2).
Analysis of Classical swine fever virus (CSFV) serum samples in the duplex lateral flow assay (LFA) and comparison with the results obtained by E2 ELISA and the confirmatory virus neutralization test (VNT).*
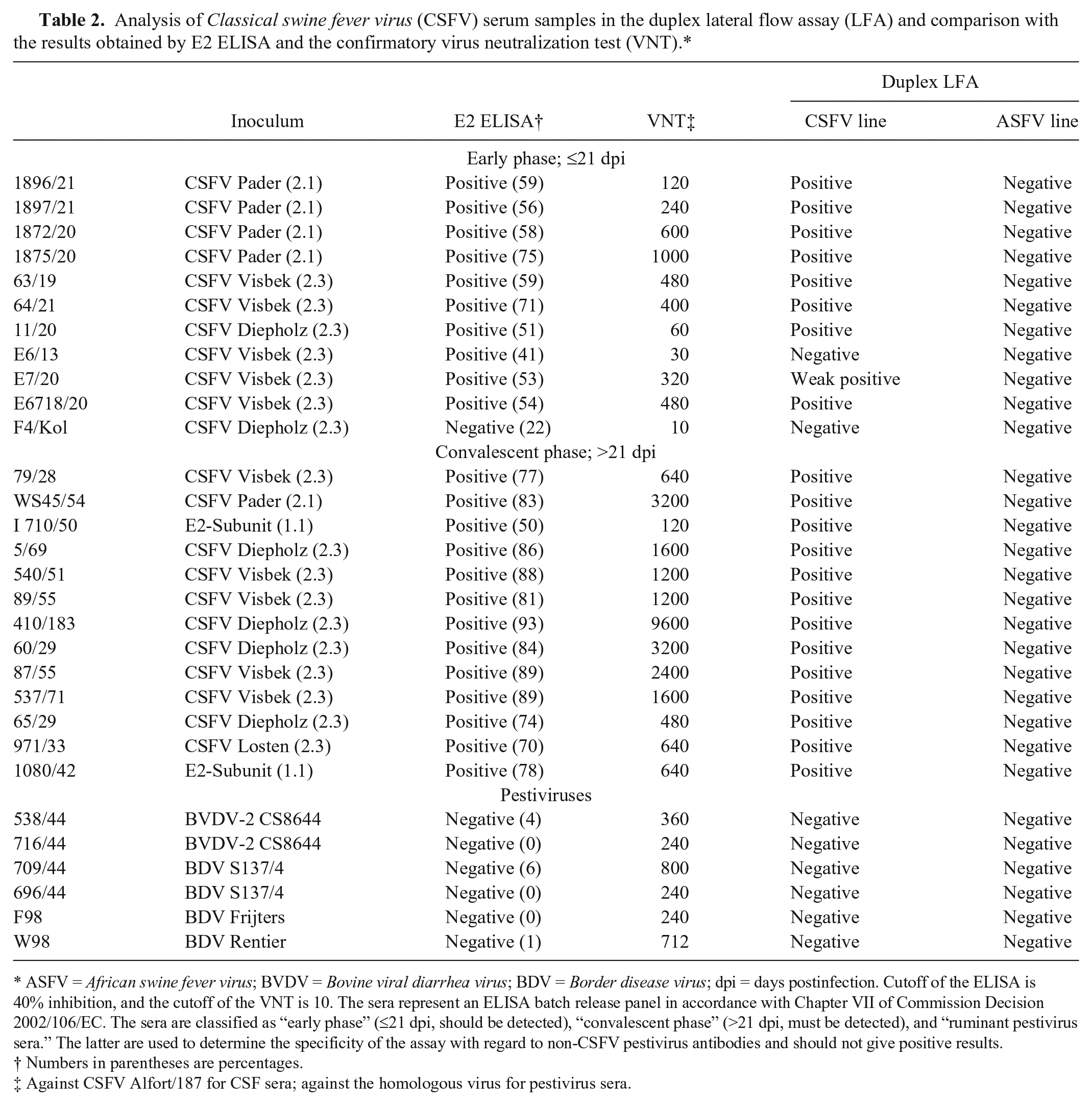
ASFV = African swine fever virus; BVDV = Bovine viral diarrhea virus; BDV = Border disease virus; dpi = days postinfection. Cutoff of the ELISA is 40% inhibition, and the cutoff of the VNT is 10. The sera represent an ELISA batch release panel in accordance with Chapter VII of Commission Decision 2002/106/EC. The sera are classified as “early phase” (≤21 dpi, should be detected), “convalescent phase” (>21 dpi, must be detected), and “ruminant pestivirus sera.” The latter are used to determine the specificity of the assay with regard to non-CSFV pestivirus antibodies and should not give positive results.
Numbers in parentheses are percentages.
Against CSFV Alfort/187 for CSF sera; against the homologous virus for pestivirus sera.
In conclusion, in the case of ASFV samples, our results revealed absolute concordance between the new LFA and different available ELISAs (Table 1), even in cases such as serum 11 with a lower percentage of competition (48.5%) and reported as a weak positive sample in the OIE ELISA, but that showed a clearly positive result in the immunochromatographic test.
Regarding CSFV samples, the results obtained by LFA were compared with the ones observed by a commercial E2 ELISA, used routinely in laboratories for large-scale screening purposes, and also with VNT, considered as the gold standard by the OIE and used as a confirmatory assay. Although concordance between the LFA and the other 2 methods was good (Table 2), the sensitivity of the newly developed test appears to be lower than the ELISA and VNT. Note that the VNT detects only neutralizing antibodies, whereas the LFA detects neutralizing and non-neutralizing antibodies; therefore, some differences could be expected.
Finally, a panel of 100 field sera from ASF- and CSF-free areas was tested. In the new duplex LFA, all samples showed a negative result for both test lines (data not shown).
Statistical analyses were performed, g and sensitivity and specificity of the LFA were evaluated by comparison with the techniques considered as reference in this study. The concordance between tests was the overall percentage agreement between the results of the 2 assays calculated using 2 × 2 contingency tables. Kappa coefficient (κ) statistics were used to evaluate the significance of the level of concordance between results beyond that expected by chance. Among the above-described panel of known status samples evaluated, the new duplex LFA ASFV/CSFV shows a diagnostic sensitivity of 100% in the case of ASFV, 96% for detection of CSFV when compared to the E2 ELISA (κ = 0.911, 95% confidence interval [CI] = 0.740–1.082), and 92% when compared with VNT (κ = 0.815, 95% CI = 0.567–1.063). The diagnostic specificity was 100% for both ASF and CSF.
This novel pen-side test offers a rapid, economical, and simple-to-use tool suitable for field application, allowing the early and specific detection of antibodies to ASFV and CSFV, thus reducing transmission of the viruses to uninfected animals and subsequent spreading of the disease. It will also allow the collection of data for epidemiologic studies from remote areas, where this has not been possible so far. Furthermore, the test has been designed to be used with either serum or blood, which makes sample processing simple and feasible even at the field level. All of these features make these devices very suitable for small field laboratories or task forces, in many cases supporting local decisions, especially in countries where laboratory infrastructure is underdeveloped or even absent. Although an antibody test may be of limited use in areas where vaccination is actively applied, as could be the case for CSF, ASF antibody detection will be very practical in areas where low-virulence viruses are circulating and the likelihood of survivors is high.
Footnotes
Acknowledgements
We thank Mercedes Montón and Tamara Ruiz for laboratory support at INGENASA. We thank Dr. John N. Barr for critical reading of the manuscript and assistance with the English language.
Authors’ contributions
P Sastre contributed to analysis and interpretation of data and drafted the manuscript. T Pérez, A Sanz, and P Rueda contributed to conception and design of the study and critically revised the manuscript. S Costa contributed to analysis and interpretation and critically revised the manuscript. X Yang, A Räber, C Gallardo, and J García contributed to acquisition of data and critically revised the manuscript. S Blome contributed to acquisition, analysis, and interpretation of data and critically revised the manuscript. KV Goller and I Tapia contributed to acquisition and analysis of data and critically revised the manuscript. All authors gave final approval and agreed to be accountable for all aspects of the work in ensuring that questions relating to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
a.
18BG3 mAb, INGENASA, Madrid, Spain.
b.
18.4 mAb, Thermo Fisher Scientific Prionics AG, Zurich, Switzerland.
c.
HerdChek CSFV Ab, IDEXX Laboratories, Westbrook, ME.
d.
HF120, HiFlow Plus nitrocellulose membrane; Merck Millipore, Darmstadt, Germany.
e.
ESEQuant, Lateral Flow Reader, Lateral Flow Studio software; Qiagen Inc., Valencia, CA.
f.
INgezim PPA COMPAC, INGENASA, Madrid, Spain.
g.
MedCalc version 10.1.7.0 for Windows, MedCalc Software, Mariakerke, Belgium.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the RAPIDIA-FIELD project (7th Framework Programme Food, Agriculture and Fisheries, and Biotechnology KBBE.2011.1.3-02 grant agreement no. 289364).