
Abstract
Swine influenza virus (SIV) RNA and antigen were detected in 15 naturally infected pigs by in situ hybridization using a nonradioactive digoxigenin-labeled cDNA probe and by immunohistochemistry using an influenza virus H1N1-specific monoclonal antibody. A 582-base pair cDNA probe for viral RNA encoding the nucleocapsid protein of SIV type A H1N1 strain was generated by the reverse transcription polymerase chain reaction. In situ hybridization and immunohistochemistry gave similar results for serial sections from each of 15 lung samples. Positive cells typically exhibited a dark brown (in situ hybridization) or red (immunohistochemistry) reaction product in the nucleus and cytoplasm without background staining. A strong positive signal for both in situ hybridization and immunohistochemistry was detected mainly in the bronchial and bronchiolar epithelial cells. A less intense signal was detected in the interstitial and alveolar macrophages. Simultaneous detection of hybridization and immunohistochemical signals on serial sections provided evidence that SIV had replicated in positive cells. The in situ hybridization technique developed in this study was useful for the detection of SIV RNA in tissues taken from naturally infected pigs and may be a valuable technique for studying the pathogenesis of SIV infection.
Keywords
Swine influenza virus (SIV) infection is a commonly encountered respiratory disease of pigs throughout swine raising countries. Infections are manifested most commonly as explosive outbreaks of acute respiratory disease with fever, anorexia, weight loss, lethargy, nasal and ocular discharge, coughing, and dyspnea. High morbidity with mortality of <5% is commonly seen. 10,12 Histologically, there is widespread degeneration and necrosis of the epithelium in bronchi and bronchioli. The lumina of bronchi, bronchioli, and alveoli are filled with exudate containing desquamated cells, neutrophils, and monocytes. 2,10
Influenza virus is a member of the genus Orthomyxovirus in the family Orthomyxoviridae and has a negative single-stranded RNA molecule that contains eight segments. 18 Two subtypes of influenza A viruses have been isolated from pigs: H1N1, represented by the classical H1N1 and avian H1N1 viruses, and H3N2, represented by human H3N2 viruses. 28 The nucleocapsid protein, encoded by RNA segment 5, is the major structural protein that interacts with the RNA segments to form the ribonucleoprotein. Nucleocapsid protein is also one of the type-specific antigens that distinguishes among the influenza A, B, and C viruses. 18,20 The nucleotide sequence within the nucleocapsid gene of SIV type A is not very similar to that of other types of influenza virus (approximately 60% homology) but is highly conserved among different type A strains (approximately 90% homology), 18,20 thus, a complementary DNA (cDNA) probe transcribed from the nucleocapsid gene would allow specific detection of SIV type A.
Virus isolation is considered the standard for the diagnosis of SIV infection; however, virus culture is time consuming and expensive. Potentially infectious specimens must be handled carefully to avoid transmission of virus to humans during isolation. Human SIV infections, some of which were fatal, have been well documented in the USA and Europe. 8,9,11,17 Because of the zoonotic nature of SIV, different diagnostic methods have been employed for the detection of SIV antigen in formalin-fixed, paraffin-embedded tissues by monoclonal and polyclonal antibody–based immunohistochemical procedures. 14,19,27 Another approach for the detection of SIV is the application of in situ hybridization. In situ hybridization is a valuable adjunct to standard RNA extraction techniques for evaluating gene expression in tissues and cells. Its major advantage is the ability to determine which tissues or cells in a mixed population are expressing the RNA of interest. In situ hybridization techniques have been used successfully to detect viral nucleic acids with DNA probes complementary to viral RNA. 7,16
One objective of this study was to detect SIV nucleic acids in formalin-fixed, paraffin-embedded tissues using a digoxigenin-labeled 582–base pair (bp) single-stranded DNA probe complementary to a conserved region of the SIV nucleocapsid protein gene. The second objective was to determine the location of SIV replication in the porcine lung to better understand the pathogenesis of naturally occurring SIV infection.
Materials and Methods
Animals
Fifteen pigs approximately 56–92 days of age from 15 different herds were selected on the basis of clinical signs and characteristic lesions. All selected pigs were seropositive for SIV infection by enzyme-linked immunosorbent assay (ELISA; IDEXX Laboratories, Westbrook, ME) and negative for porcine reproductive and respiratory syndrome virus (PRRSV) and porcine respiratory coronavirus (PRCV) infection by immunohistochemistry. Samples were obtained from the necropsy cases of the Department of Veterinary Pathology, College of Veterinary Medicine, Seoul National University. All pigs were submitted alive, and samples were collected at necropsy. Positive tissue controls were provided by Dr. D. Rogers, University of Nebraska-Lincoln. Negative tissue controls were collected from a 1-day-old colostrum-deprived pig not exposed to any pathogens and from each of two pigs that had been experimentally infected with PRRSV or PRCV, respectively. 1,6 Seronegative and virus isolation–negative conventional 60-day-old and 80-day-old pigs were used as negative controls.
Tissue processing
Tissues specimens were collected from lung, tonsil, stomach, duodenum, jejunum, ileum, colon, and cecum of infected and noninfected animals, fixed in 10% (w/v) buffered formaldehyde for 24–48 hours, and embedded in paraffin according to standard laboratory procedures. Three serial sections were prepared from each of the formalin-fixed, paraffin-embedded tissues. Two sections were processed for in situ hybridization, and one was processed for immunohistochemistry. Sections were cut 3 µm thick, floated in a water bath containing diethylpyrocarbonate-treated water and mounted on Superfrost/plus slides (Fisher Scientific, Pittsburgh, PA). Sections were then subjected to optimal pressure cooking as previously described. 23
Virus isolation
Isolation of SIV from homogenates of lung was performed by inoculation of Madin Darby canine kidney (MDCK) cells as previously described. 22
RNA extraction
The SIV H1N1 (A/Seoul/20/91) strain was used as the standard strain and grown in MDCK cells. SIV-infected MDCK cells (75-cm2 flask) were harvested when 70–80% of the cells were showing cytopathic effects. Viral RNA was extracted from SIV-infected cells with Trizol LS Reagent (Gibco BRL, Grand Island, NY) according to the manufacturer's instructions.
Polymerase chain reaction
The primer from the nucleocapsid protein sequence of SIV was used in this study. The forward and reverse primers were 5′-AGTATACAGCCTAATCAGAC-3′ (nucleotides 943–962) and 5′-AGTAGAAACAAGGGTATTTTTC-3′ (nucleotides 1509–1530), respectively. The primer set resulted in amplified fragments of 582 bp.
For the first-strand cDNA synthesis, 1 µl of the SIV RNA (5 ng/µl) was supplemented in a total reaction volume of 20 µl with 1× RT buffer (50 mM Tris-HCl, 8 mM MgCl2, 30 mM KCl, 1 mM dithiothreitol, pH 8.3), 0.5 mM of each deoxynucleotide triphosphate (dNTP), 2.5 µM random hexanucleotide mixture, 20 U of RNase inhibitor, and 50 U of Moloney murine leukemia virus reverse transcriptase. After incubation for 15 minutes at 42 C, the mixture was incubated for 5 minutes at 99 C to denature the products. The mixture was then chilled on ice.
The composition of the polymerase chain reaction (PCR) mixture (150 µl) was 30 µl of cDNA (5 ng/µl), 2 µl of each primer (250 nM), 15 µl of 10× PCR buffer (10 mM Tris-HCl, 40 mM KCl, 1.5 mM MgCl2, pH 8.3), 1.2 µl of each dNTP (0.2 mM), 29 µl of 2.5 unit of Taq polymerase, and 67.2 µl of distilled water. The PCR for SIV was performed under the following conditions in a thermal cycler (Perkin-Elmer-Cetus, Norwalk, CT): one cycle of 3 minutes at 94 C and 30 cycles of denaturation at 94 C for 1 minute, annealing at 50 C for 2 minutes, and elongation at 72 C for 3 minutes.
The amplified product was visualized by standard gel electrophoresis of 10 µl of the final reaction mixture on a 2% agarose gel. Amplified DNA fragments of specific sizes were located by ultraviolet fluorescence after staining with ethidium bromide. Fragment lengths were verified by comparison with a digested lambda standard on the same gel.
To verify the specificity of the PCR, SIV H1N1 (A/Seoul/20/91), human influenza virus H3N2 (A/Johannesberg/33/95), human influenza B virus (B/Beijing/184/93), PRRSV, PRCV, transmissible gastroenteritis virus (TGEV), porcine epidemic diarrhea virus (PEDV), and porcine circovirus (PCV) types 1 and 2 were tested independently with primers.
Preparation of labeled probe
PCR products were purified using a 30-kd cutoff membrane ultrafiltration filter. The nucleotide sequences of the purified PCR products were determined by use of BigDye chemistry with the ABI Prism Sequencer (Applied Biosystems, Foster City, CA). Sequences of the purified PCR products were determined before PCR products were labeled by random priming with digoxigenin-dUTP (Boehringer Mannheim, Indianapolis, IN) according to the manufacturer's instructions. The probe for PRRSV was used as negative probe. 7
In situ hybridization
Sections were deparaffinized in xylene and rehydrated in phosphate-buffered saline (PBS; pH 7.4, 0.01 M) for 5 minutes. Deproteinization was carried out in 0.2 N HCl for 20 minutes at room temperature. Tissues were then digested at 37 C for 20 minutes in proteinase K (Gibco BRL) 200 µg/ml in PBS. One of two serial sections examined was treated with RNase A (Boehringer Mannheim) at 100 µg/ml in 10 mM Tris-HCl (pH 7.4) for 30 minutes at 37 C to remove target RNA as a specificity control. After digestion, tissues were fixed in 4% paraformaldehyde in PBS for 10 minutes. After rinsing twice with PBS, the slides were acetylated in 300 ml of 0.1 mM triethanolamine HCl buffer (pH 8.0) to which 0.75 ml of acetic anhydride (0.25%) had been added. After 5 minutes, an additional 0.75 ml of acetic anhydride was added, and 5 minutes later the slides were rinsed in 2× saline sodium citrate (SSC) (1× SSC contains 50 mM NaCl and 15 mM sodium citrate, pH 7.0). The slides were allowed to equilibrate for 60 minutes in a standard hybridization buffer that consisted of 5× SSC with 50% deionized formamide, 10× 2% buffered blocking solution (Boehringer Mannheim), 0.1% N-lauroylsarcosine, and 0.02% sodium dodecyl sulfate.
Hybridization was done overnight at 45 C. The digoxigenin-labeled probe (0.1 ng/µl) was diluted in 300 µl of the standard hybridization buffer, heated for 10 minutes at 95 C on a heating block, and quenched on ice before being applied to the tissue sections. Approximately 50 ng of the digoxigenin-labeled probe was added to the standard hybridization buffer (50 µl), which was then layered over the section. Fluid was held in place by a coverslip, and the edges were sealed with rubber cement. After overnight hybridization, sections were thoroughly washed twice in 4× SSC for 5 minutes at room temperature, twice in 2× SSC for 10 minutes at 37 C, twice in 0.2× SSC for 5 minutes at room temperature, and once in maleic acid buffer (100 mM maleic acid and 150 mM NaCl, pH 7.5) for 5 minutes at room temperature.
For detection of hybridization, sections were incubated with anti-digoxigenin conjugated with alkaline phosphatase (Boehringer Mannheim) diluted 1:250 in 0.1 M Tris HCl (pH 7.4), 0.15 M NaCl with 1% blocking reagent (Boehringer Mannheim). After three washes in buffer, substrate consisting of nitroblue tetrazolium (NBT) and 5-bromocresyl-3-indolylphosphate (BCIP) was layered over the sections. Color was allowed to develop for 5–8 hours in the dark, and the reaction was stopped by dipping slides briefly in Tris ethylenediaminetetraacetic acid (EDTA) buffer (10 mM Tris-HCl and 1 mM EDTA, pH 8.0). Sections were counterstained with 0.5% methyl green, and the slides were then washed with distilled water for 1 minute, allowed to dry completely, dipped into absolute xylene, and coverslipped with Canada balsam mounting medium (Hayashi Pure Chemical Industries Ltd., Osaka, Japan).
Immunohistochemistry
The sections were deparaffinized in xylene, rehydrated through graded alcohols, and air dried. Endogenous alkaline phosphatase was quenched with 20% glacial acetic acid solution for 2 minutes at 4 C. All slides were then incubated with normal goat serum (Sigma Chemical Co., St. Louis, MO) in PBS (0.1 M, pH 7.4) for 30 minutes at room temperature to saturate nonspecific protein-binding sites and then incubated with power block (BioGenes, San Ramon, CA) for 30 minutes. Goat anti-influenza A (H1N1) virus (Chemicon International, Inc., Temecula, CA) was diluted 1:1,000 in PBS (0.01 M, pH 7.4) containing 0.1% Tween 20. The slides were incubated with primary antibody for 1 hour at room temperature.
After three washes with 0.1% Tween 20 in PBS (0.01 M, pH 7.4), sections were flooded and incubated for 20 minutes at 36 C with biotinylated rabbit anti-goat antibody (Dako, Glostrup, Denmark) diluted 1:500 in PBS (0.01 M, pH 7.4) containing 0.1% Tween 20. After three washes with 0.1% Tween 20 in PBS, sections were flooded and incubated for 20 minutes at 36 C with alkaline phosphatase–conjugated streptavidin. The slides were then washed with 0.1% Tween 20 in PBS. Sections were then equilibrated with Tris buffer (pH 9.5) for 5 minutes at room temperature. The final reaction was produced by immersing the sections in a solution of Vector Red substrate (Vector Laboratories, Burlingame, CA) for 20 minutes at room temperature.
Results
The SIV H1N1 (A/Seoul/20/91) and human influenza virus H3N2 (A/Johannesberg/33/95) reacted with the primers of SIV. The primers did not react with human influenza B (B/Beijing/184/93), PRRSV, PRCV, TGEV, PEDV, and PCV types 1 and 2. SIV was isolated from the lung in five pigs (Nos. 2, 5, 6, 11, 14). All five influenza viruses isolated from pigs were amplified in MDCK cells, and the resulting virus RNA was reacted with primers specific for SIV by PCR (Fig. 1). PCR products from each isolate were sequenced, and their identity was confirmed as influenza virus (data not shown).
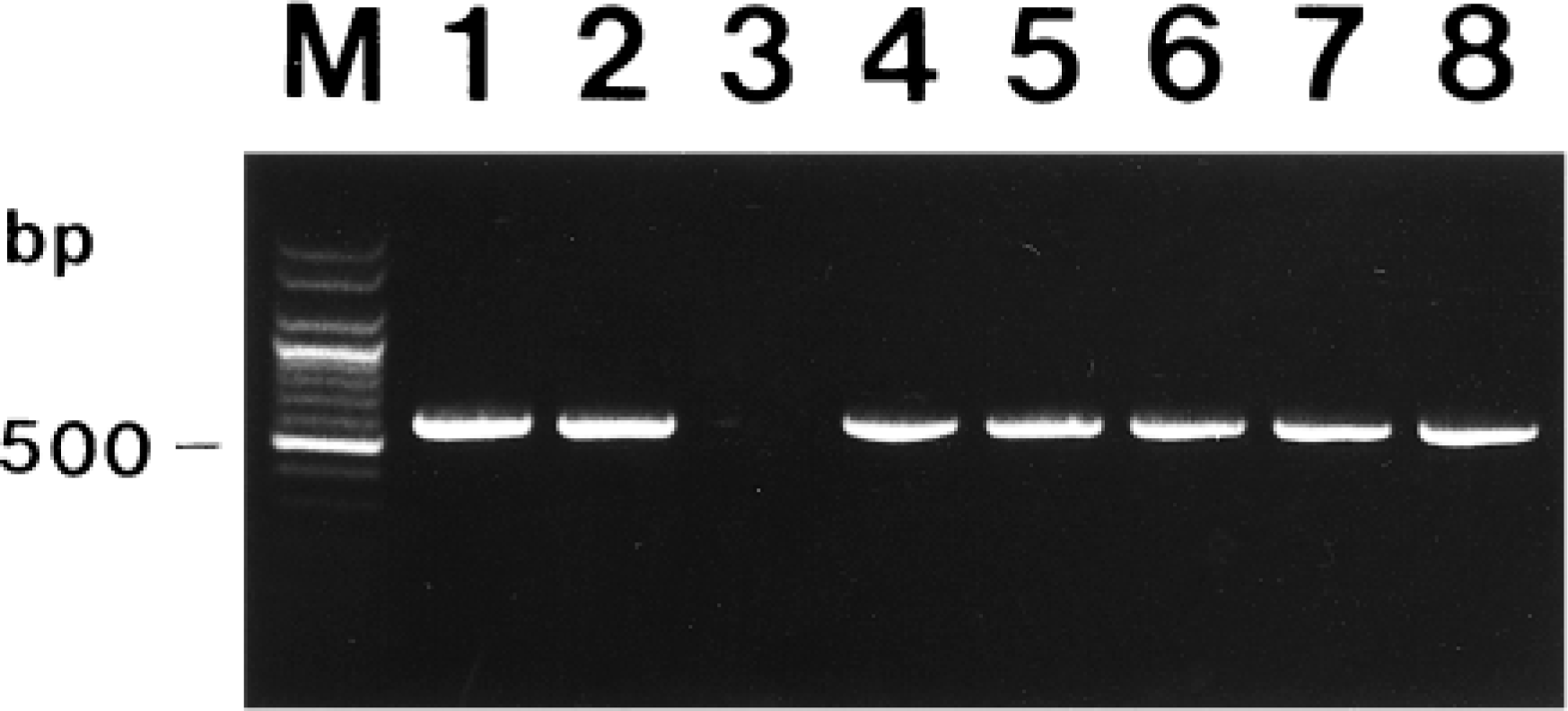
Agarose gel electrophoresis of PCR-amplified influenza virus cDNA products. From left to right: M = 100-bp DNA ladder; lane 1 = swine influenza virus (SIV) H1N1 (A/Seoul/20/91); lane 2 = human influenza virus H3N2 (A/Johannesberg/33/95); lane 3 = negative control from negative control pig; lane 4 = SIV isolated from pig No. 2; lane 5 = SIV isolated from pig No. 5; lane 6 = SIV isolated from pig No. 6; lane 7 = SIV isolated from pig No. 11; lane 8 = SIV isolated from pig No. 14.
The results of virus isolation, in situ hybridization, and immunohistochemistry are summarized in Table 1. The morphology of host cells was preserved despite the relatively high temperature required in parts of the incubation procedure. In situ hybridization and immunohistochemistry gave similar results for serial sections from each of 15 lung samples (Figs. 2, 3). Positive cells typically exhibited a dark brown (in situ hybridization) or red (immunohistochemistry) reaction product in the nucleus and cytoplasm, without background staining. Sixty percent (9/15) of the animals naturally infected with SIV had positive results by in situ hybridization and immunohistochemistry for SIV in their bronchi; most of these pigs were <80 days of age.
Virus isolation, in situ hybridization (ISH), and immunohistochemical (IHC) results in 15 pigs naturally infected with swine influenza virus.

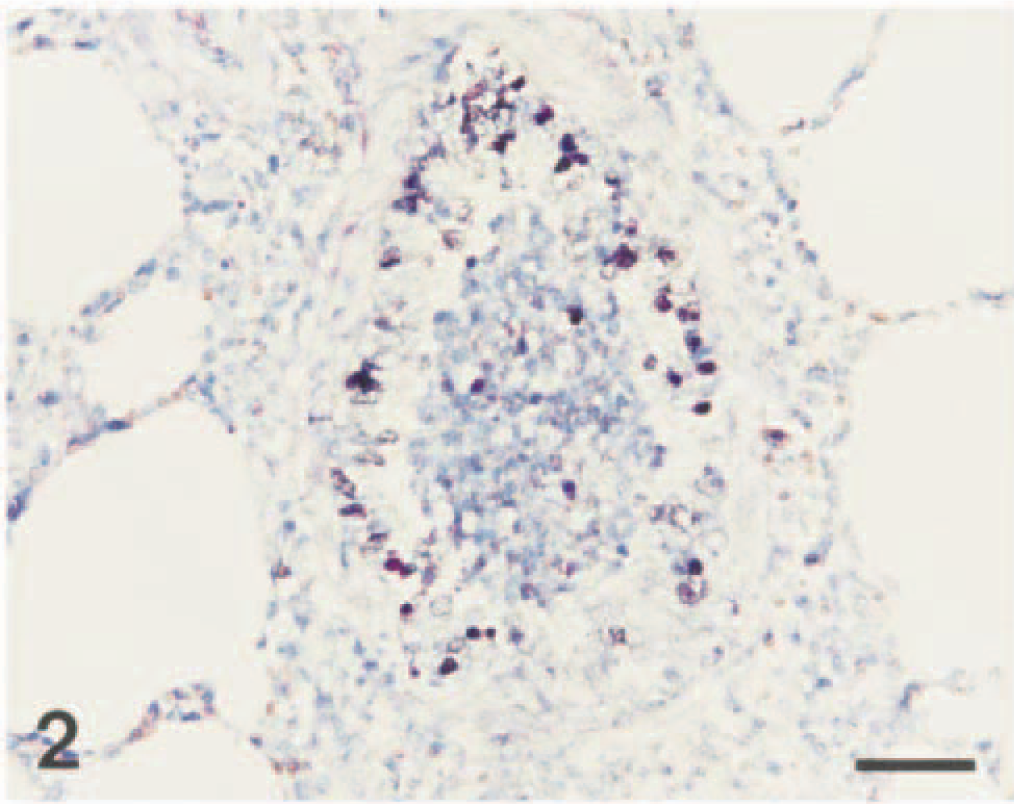
Lung; pig No. 2. Swine influenza virus RNA (dark brown reaction) is detected in the nucleus and cytoplasm of bronchiolar epithelial cells. In situ hybridization; NBT and BCIP, methyl green counterstain. Bar = 55 µm.
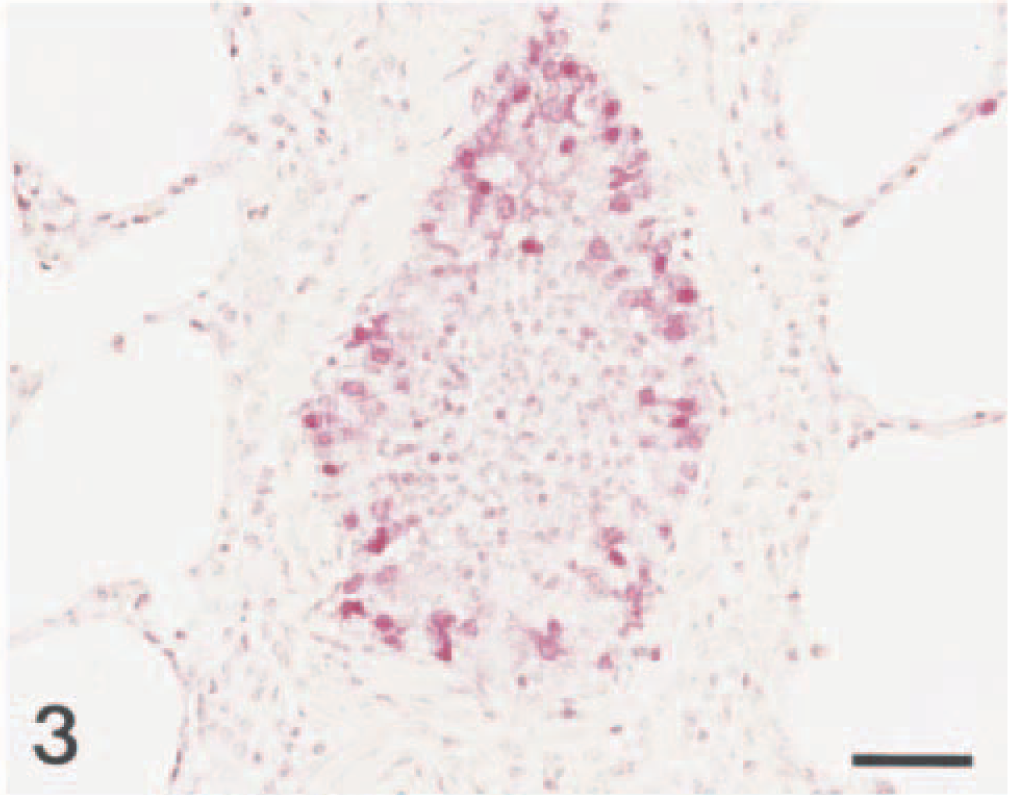
Lung; pig No. 2. Swine influenza virus antigen (red reaction) is detected in the nucleus and cytoplasm of bronchiolar epithelial cells. Immunohistochemistry; Vector Red, hematoxylin counterstain. Bar = 55 µm.
When lung tissues from the pigs with natural SIV infections were hybridized with the nonradioactive digoxigenin-labeled DNA probe or reacted with the monoclonal antibody, a strong signal was seen in bronchial and bronchiolar lining epithelial cells. The signal intensity varied within and between anatomical structures in sections and between pigs. Bronchial and bronchiolar epithelial cells (Figs. 2, 3) and alveolar and interstitial macrophages (Figs. 4, 5) had positive signals. In five pigs (Nos. 7, 9, 13–15) positive signals were detected mainly in cells randomly scattered in thickened alveolar septa and alveolar spaces. These positive cells generally had large oval nuclei and abundant cytoplasm. Comparisons of in situ hybridization and immunohistochemistry results with those of hematoxylin and eosin–stained sections from the same block indicated that many of the positive cells were interstitial macrophages and alveolar macrophages.
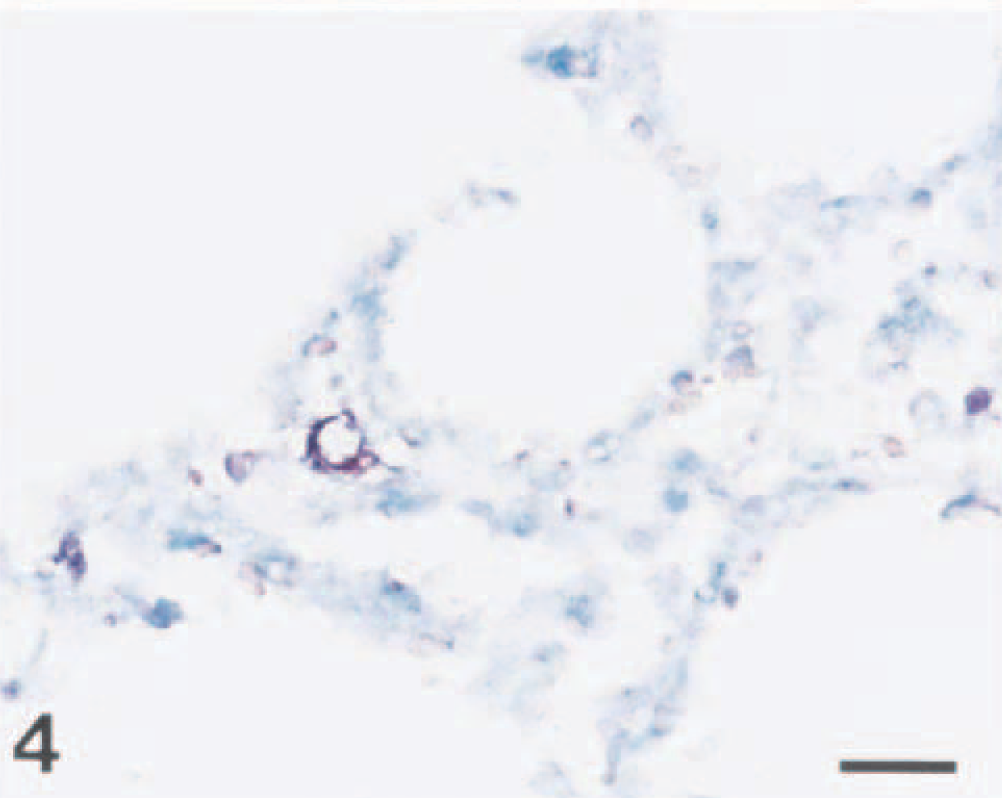
Lung; pig No. 9. Swine influenza virus RNA (dark brown reaction) is detected in the nucleus of an interstitial macrophage. In situ hybridization; NBT and BCIP, methyl green counterstain. Bar = 30 µm.
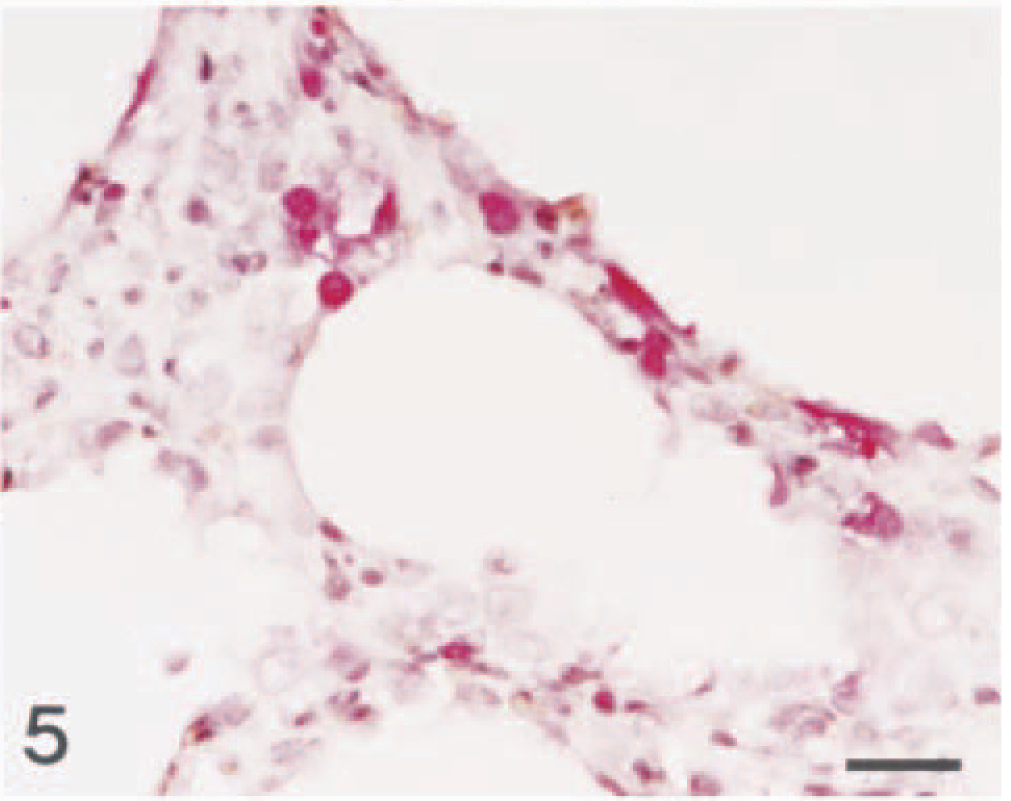
Lung; pig No. 7. Swine influenza virus antigen (red reaction) is detected in the nucleus and cytoplasm of interstitial macrophage. Immunohistochemistry; Vector Red, hematoxylin counterstain. Bar = 30 µm.
No hybridization signal was consistently seen in tissue sections treated with RNase A prior to in situ hybridization (data not shown). Sections from negative control pigs showed no hybridization signal for SIV. Probes for PRRSV gave consistently negative results in all 15 tissues tested.
Discussion
The results of this study demonstrate that SIV RNA and antigen can be detected in formalin-fixed, paraffin-embedded tissue specimens of the pig using a nonradioactively labeled cDNA probe and monoclonal antibody, respectively. Positive signal for SIV was identified in the bronchial and bronchiolar epithelium and alveolar and interstitial macrophages of pigs naturally infected with SIV. Interpretation of positive in situ hybridization and immunohistochemistry signal in cells of the macrophage lineage must involve consideration of whether the cell is truly permissive for viral infection or whether the positive signal merely represents viral nucleic acid/antigen from phagocytized infected cells. Simultaneous detection of nuclear and cytoplasmic hybridization and immunohistochemical signals in serial sections provided evidence that SIV had replicated in these positive cells.
Among RNA viruses, influenza virus is unusual in that all of its RNA synthesis, transcription, and replication takes place in the nucleus of infected cells. 18 An influenza virus binds preferentially to ribosomal RNA of cellular origin. 30 After synthesis in the cytoplasm, nucleocapsid protein molecules are transported to the nucleus. 3 Nuclear targeting of nucleocapsid protein is an intrinsic property of the protein. 18 The nucleus provides the environment for the synthesis of viral particles. Therefore, unlike other RNA viruses, SIV RNA and antigen can be detected in both the nucleus and cytoplasm of infected cells. This pattern has also been demonstrated in an ultrastructural study, in which viral particles were observed within the nucleus and cytoplasm of respiratory epithelial cells. 29
Although influenza A viruses uniformly recognize cell surface oligosaccharides with a terminal sialic acid, their receptor specificity varies. Most avian influenza viruses preferentially bind to the 2,3-N-acteylneuraminic acid–galactose linkages on sialyloligosaccharides, whereas human influenza viruses prefer the 2,6-N-acetylneuraminic acid–galactose linkages on sialyloligosaccharides. 25,26 Pigs are susceptible to infection with influenza viruses of both avian and mammalian origin because their tracheal epithelium contains sialyloligosaccharides. 15 SIV RNA and antigen were detected primarily in bronchial and bronchiolar epithelial cells, indicating that bronchial and bronchiolar epithelium may also contain sialyloligosaccharides for the influenza receptor. In an experimental study, there was extensive involvement of bronchiolar epithelium, with scattered infected cells in the lung parenchyma. 29 In situ hybridization and immunohistochemical results suggest that the major site of infection was the bronchial and bronchiolar epithelium. Infection of the mucosa of the distal portion of the respiratory tract of swine with SIV is an important initial event in the development of SIV-induced pneumonia. Further studies will be necessary to confirm the presence of influenza receptor in porcine bronchial and bronchiolar epithelium.
In situ hybridization and immunohistochemistry results suggest that SIV infects pulmonary macrophages. Destruction of pulmonary macrophages by the virus is an indication of a direct pathogenic effect of SIV. Such changes adversely affect the pulmonary host defense mechanisms and may lead to secondary viral and bacterial infections. SIV is often seen in combination with other viral and bacterial pathogens (PRRSV, PRCV, Mycoplasma hyopneumoniae, Actinobacillus pleuropneumoniae, Haemophilus spp., and Pasteurella multocida). 10,12,19 A combination of these pathogens with SIV often produces more protracted respiratory disease and higher mortality than is seen in uncomplicated cases of SIV infection. 12 In situ hybridization and immunohistochemistry are useful and applicable as diagnostic tools for the detection of SIV in combination with these other viral respiratory pathogens.
In situ hybridization and immunohistochemistry for the detection of SIV in formalin-fixed, paraffin-embedded tissues offer several advantages over other methods such as virus isolation and immunofluorescence. These advantages include direct visualization of viral nucleic acids or antigens in diseased tissue, use of ordinary light microscopy, and simultaneous observation of histopathologic changes. However, formalin fixation can denature antigens, leading to false-negative results. 13,24 In contrast to immunohistochemistry, in situ hybridization is less susceptible to structural alterations caused by fixation. 5,7 Although antigenic cross-reactivity might not be a problem with in situ hybridization, the specificity of nucleic acid hybridization has its own limitations. To avoid nonspecific hybridization of the probe, both reverse transcription and PCR must be performed under stringent conditions.
The use of high-temperature heat denaturation (pressure cooking) as a pretreatment with in situ hybridization significantly increased the intensity of hybridization signal in lung tissue. Hybridization signal was rarely seen in tissue sections that were not pretreated (data not shown). The mechanism by which high-temperature denaturation enhances in situ hybridization signal is not known but may involve heat denaturation of protein cross-links (or dimethylene bridges) caused by formalin fixation. 4 Alternatively, high-temperature denaturation may directly affect the conformation of the nucleic acid to be detected, allowing denaturation of double-stranded sequences into single strands or the unwinding of single-stranded structures that may have self-annealed. 21 This pretreatment method also reduces hybridization time and the concentration of proteolytic enzymes required. There was no evidence that heat denaturation altered morphology in any of the sections we examined, unlike tissue sections that have been overdigested by proteolytic enzymes.
Virus isolation in chicken egg inoculation was considered the gold standard for diagnosis of SIV infection. 12 Immunohistochemistry has previously been shown to have sensitivity equal to that of virus isolation. 27 In the present study, viral nucleic acid/antigen was detected in 93% (14/15) of infected tissues, but virus was isolated in only 33% (5/15) of the pigs. This discrepancy may be due to the use of cell culture instead of chicken egg inoculation.
In situ hybridization was used to detect SIV nucleic acids in formalin-fixed, paraffin-embedded tissues from naturally infected pigs. The use of nonradioactive in situ hybridization to study infectious disease is one of the most promising applications of this technique. The in situ hybridization technique developed in this study was useful for the detection of SIV RNA in tissues taken from naturally infected pigs and may be a valuable technique for studying the pathogenesis of SIV infection.
Footnotes
Acknowledgements
The research reported here was supported by the Ministry of Agriculture, Forestry and Fisheries Special Grants Research Program (MAFF SGRP) and Brain Korea 21 Project, Republic of Korea.