
Abstract
Nitric oxide synthase 2 (NOS2) and tumor necrosis factor α (TNF-α) were detected and localized in 15 pigs with naturally occurring pleuropneumonia by use of in situ hybridization with a nonradioactive digoxigenin-labeled cDNA probe. Two cDNA probes 491 and 219 base pairs for NOS2 and TNF-α, respectively, were generated by reverse transcription polymerase chain reaction. All 15 pigs infected with Actinobacillus pleuropneumoniae had distinct positive hybridization signals for NOS2 and TNF-α. Strong hybridization signals for both NOS2 and TNF-α were evident in degenerate alveolar leukocytes bordering zones of coagulative necrosis and in alveolar spaces. NOS2 nucleic acids were detected in neutrophils and macrophages. In situ hybridization of serial sections of lung tissue revealed numerous cells positive for NOS2 and TNF-α, suggesting that NOS2 and TNF-α expression may play a role in the pathophysiology of pleuropneumonia.
Keywords
Actinobacillus pleuropneumoniae causes severe, often fatal fibrinohemorrhagic necrotizing pleuropneumonia in pigs. A. pleuropneumoniae is detected primarily within neutrophils and alveolar macrophages at the periphery of foci of acute coagulative necrosis and at the edge of granulation tissue in chronic lesions. 23 Several bacterial components, including lipopolysaccharide (LPS), Apx toxins, and polysaccharide capsule, likely contribute to the cause of disease. 12
Nitric oxide (NO) is a molecule with various biologic properties such as neurotransmission, immunologic functions mediated by macrophages, and regulation of the vasotonus.
15,24,29
NO is synthesized from
In contrast to the constitutive isoforms, NOS2 is present following stimulation of the host cells with bacteria or bacterial products such as LPS and/or inflammatory cytokines such as tumor necrosis factor α (TNF-α) and interleukin (IL)-1. 20,24–26,30 The LPS elaborated by A. pleuropneumoniae appears to be associated with the early inflammatory response. 1,3,4,10 The inflammatory cytokines IL-1, TNF-α, IL-6, and IL-8 are produced mainly by monocytes/macrophages in response to A. pleuropneumoniae LPS. 1,9 Intense and consistent expression of these proinflammatory cytokines was demonstrated in lesions caused by A. pleuropneumoniae. 7 The objectives of this study were to investigate the expression of NOS2 by in situ hybridization in naturally occurring pleuropneumonia and to determine the localization of NOS2 and TNF-α in infected porcine lungs.
Materials and Methods
Animals
Samples were obtained at necropsy from pigs submitted to the Department of Veterinary Pathology of Seoul National University from January 1994 to December 1997. Fifteen pigs (Nos. 1–15) approximately 87–140 days of age from 15 different herds were selected on the basis of clinical signs, characteristic lesions, bacterial isolation, and serotype. All 15 pigs were negative for Mycoplasma hyopneumoniae, Pasteurella multocida, and porcine reproductive and respiratory syndrome virus by culture and in situ hybridization. 6,19
Positive control sections were from pigs that were naturally infected with A. pleuropneumoniae and expressed inflammatory cytokines. 7,23 Negative control sections were prepared from 1-day-old colostrum-deprived pigs that had not been exposed to any viral and bacterial pathogens. Conventional 60-day-old and 80-day-old pigs that were negative for bacterial isolation also were used as negative controls.
Tissue processing
Samples of lung, liver, kidney, tonsil, lymph node, and large and small intestine from infected and noninfected animals were fixed in 10% (w/v) neutral buffered formalin for 24–48 hours, embedded in paraffin, sectioned at 4 µm, floated on a water bath containing diethylpyrocarbonate-treated water, and mounted on Superfrost/plus slides (Fisher Scientific, Pittsburgh, PA).
RNA extraction
The Concanavalin A–stimulated porcine splenocytes, LPS-stimulated peripheral blood mononuclear cells, and pleuropneumonic lung specimens from 15 pigs were used for extraction of NOS2 and TNF-α RNA as previously described. 27,31 RNA was extracted from these cells and lungs with Trizol LS reagent (Gibco BRL, Grand Island, NY) according to the manufacturer's instructions.
Primer
The primers were designed based on the porcine NOS2 cDNA sequence (GenBank accession no. U59390), corresponding to exons 7–10 of the human NOS2 gene. The forward and reverse primers were 5′-CGTTATGCCACCAACAAT-3′ and 5′-ACTTCCTCCAGGATGTTGTA-3′, respectively. These primers amplified a 491-base pair (bp) cDNA fragment. The primers for TNF-α were used as previously described. 31 The forward and reverse primers were 5′-GCTGTACCTCATCTACTCCC-3′ and 5′-TAGACCTGCCCAGATTCAGC-3′, respectively. These primers amplified a 291-bp cDNA fragment.
Reverse transcription polymerase chain reaction
For the first-strand cDNA synthesis, 1 µl of the NOS2 RNA (5 ng/µl) was supplemented in a total reaction volume of 20 µl with 1× RT buffer (50 mM Tris-HCl, 8 mM MgCl2, 30 mM KCl, 1 mM dithiothreitol, pH 8.3), 0.5 mM (each) deoxynucleotide triphosphates (dNTPs), 2.5 µM random hexanucleotide mixture, 20 U of RNase inhibitor, and 50 U of Moloney murine leukemia virus reverse transcriptase. After incubation for 15 minutes at 42 C, the mixture was incubated for 5 minutes at 99 C to denature the products. The mixture was then chilled on ice.
The composition of the polymerase chain reaction (PCR) mixture (150 µl) was 30 µl of cDNA (5 ng/µl), 2 µl of each primer (250 nM), 15 µl of 10× PCR buffer (10 mM Tris-HCl, 40 mM KCl, 1.5 mM MgCl2, pH 8.3), 1.2 µl of each dNTP (0.2 mM), 2.5 unit of Taq polymerase (Bioneer Corp., Cheungwon, Korea) in a volume of 29 µl, and 67.2 µl of distilled water. The PCR for NOS2 was performed under the following conditions in a thermal cycler (Perkin-Elmer-Cetus, Norwalk, CT): 35 cycles of denaturation at 94 C for 30 seconds, annealing at 56 C for 30 seconds, and elongation at 72 C for 30 seconds and 1 cycle of 2 minutes at 94 C, 2 minutes at 58C, and 2 minutes at 72 C. Reverse transcription (RT)-PCR of TNF-α was performed as previously described. 31
Preparation of labeled probe
RT-PCR products of NOS2 and TNF-α were purified using a 30-kd cutoff ultrafiltration filter membrane. The nucleotide sequences of the purified RT-PCR products were determined by use of BigDye chemistry with the ABI Prism Sequencer (Applied Biosystems, Foster City, CA). Sequencing of the purified RT-PCR products was performed before RT-PCR products were labeled by random priming with digoxigenin-dUTP (Boehringer Mannheim, Indianapolis, IN) according to the manufacturer's instructions.
In situ hybridization
Sections were deparaffinized in xylene and rehydrated in PBS (pH 7.4, 0.01 M) for 5 minutes. Deproteinization was carried out in 0.2 N HCl for 20 minutes at room temperature. Tissues were then digested at 37 C for 20 minutes in 100 µg/ml proteinase K (Gibco BRL) in PBS (pH 7.4, 0.01 M). Serial sections of each tissue section examined were treated with RNase A (Boehringer Mannheim) at 100 µg/ml in 10 mM Tris-HCl (pH 7.4) for 30 minutes at 37 C to remove target RNA as a specificity control. After digestion, tissues were fixed in 4% paraformaldehyde in PBS for 10 minutes. After rinsing twice with PBS, the slides were acetylated in 300 ml of 0.1 mM triethanolamine-HCl buffer (pH 8.0) to which 0.75 ml of acetic anhydride (0.25%) had been added. After 3 minutes, an additional 0.75 ml of acetic anhydride was added, and 3 minutes later the slides were rinsed in 2× saline sodium citrate (SSC; 1× SSC contains 50 mM NaCl and 15 mM sodium citrate, pH 7.0). The slides were allowed to equilibrate for 60 minutes in a standard hybridization buffer that consisted of 5× SSC with 50% deionized formamide, 10× 2% buffered blocking solution (Boehringer Mannheim), 0.1% N-lauroylsarcosine, and 0.02% sodium dodecyl sulfate.
Hybridization was performed overnight at 45 C for NOS2 and TNF-α. The digoxigenin-labeled probe (0.1 ng/µl) was diluted in 300 µl of the standard hybridization buffer, heated for 10 minutes at 95 C on a heating block, and quenched on ice before being applied to the tissue sections. Approximately 50 ng of the digoxigenin-labeled probe was added to the standard hybridization buffer (50 µl), which was then layered over the section. Fluid was held in place by a coverslip, and the edges were sealed with rubber cement. After overnight hybridization, sections were thoroughly washed twice in 4× SSC for 5 minutes at room temperature, twice in 2× SSC for 10 minutes at 37 C, twice in 0.2× SSC for 5 minutes at room temperature, and once in maleic acid buffer (100 mM maleic acid and 150 mM NaCl, pH 7.5) for 5 minutes at room temperature.
For detection of hybridization, sections were incubated with anti-digoxigenin conjugated with alkaline phosphatase (Boehringer Mannheim) diluted 1:250 in 0.1 M Tris-HCl (pH 7.4), 0.15 M NaCl with 1% blocking reagent (Boehringer Mannheim). After three washes in buffer, substrate consisting of nitroblue tetrazolium (NBT) and 5-bromocresyl-3-indolylphosphate (BCIP) was layered over the sections. Color was allowed to develop for 5–8 hours in the dark, and the reaction was stopped by dipping slides briefly in tri-ethylenediaminetetraacetic acid (EDTA) buffer (10 mM Tris-HCl and 1 mM EDTA, pH 8.0). Sections were counterstained with 0.5% methyl green, and the slides were then washed with distilled water for 1 minute, allowed to dry completely, dipped into the absolute xylene, and coverslipped with Canada balsam mounting medium (Hayashi Pure Chemical Industries Ltd., Osaka, Japan).
Results
Amplification of template cDNA with primers for NOS2 or TNF-α resulted in amplified products corresponding to those of the predicted size, i.e., 491 bp (NOS2) and 291 bp (TNF-α). PCR products were sequenced, and their identities were confirmed as NOS2 and TNF-α. To determine expression of NOS2 and TNF-α, RT-PCR analyses were performed using RNA extracted from lung tissues of pigs that had been infected with A. pleuropneumoniae. NOS2 and TNF-α were consistently detected in lung tissues of all 15 pigs with naturally occurring pleuropneumonia (Figs. 1, 2). In contrast, NOS2 and TNF-α were not detected in mock-infected alveolar macrophages from negative control pigs.
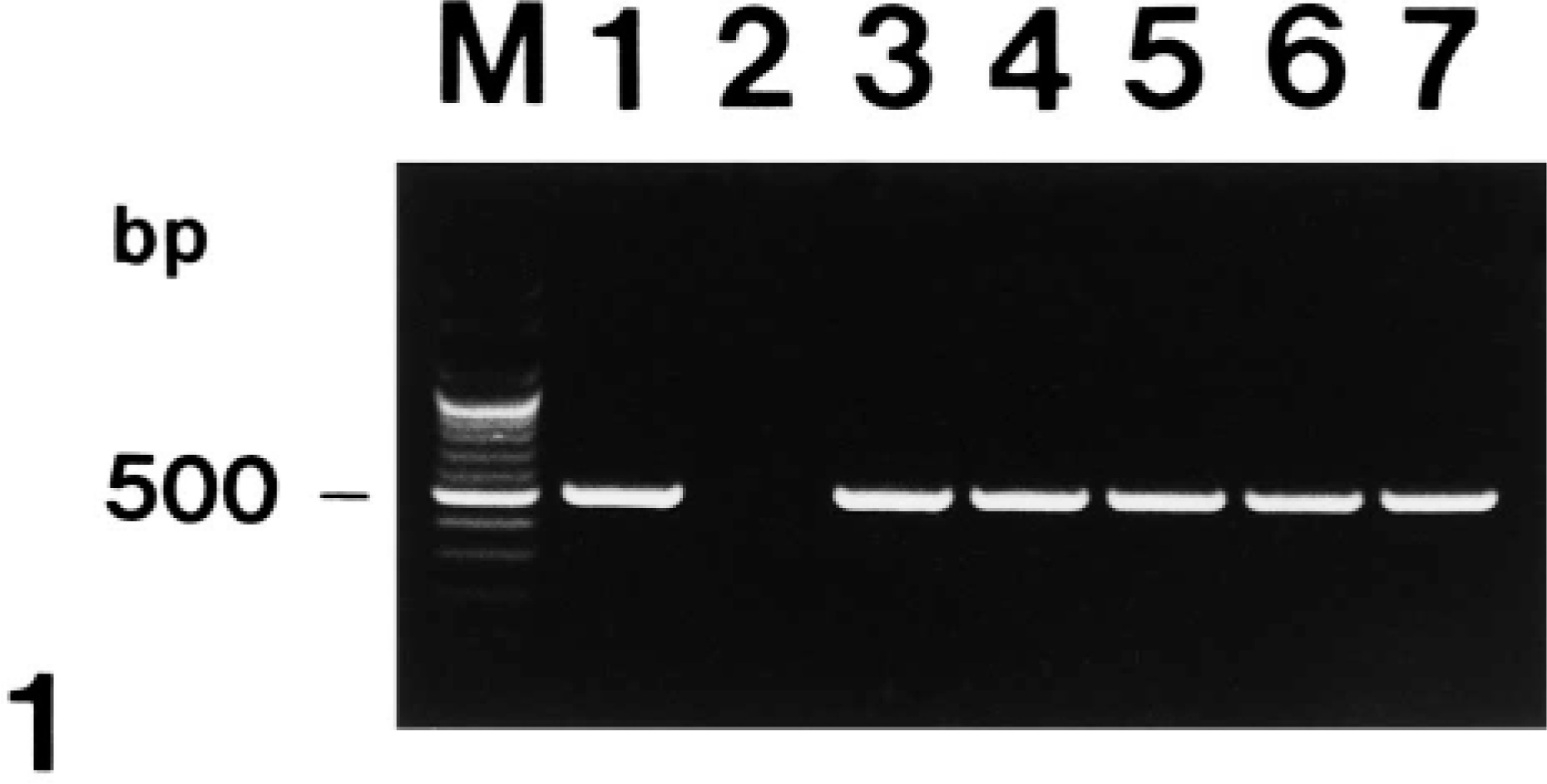
Agarose gel electrophoresis of polymerase chain reaction-amplified NOS2 cDNA products. From left to right: M = 100-bp DNA ladder; lane 1 = positive control from lipopolysaccharide-treated macrophages; lane 2 = negative control from normal macrophages; lane 3 = positive NOS2 cDNA from pig No. 2; lane 4 = positive NOS2 cDNA from pig No. 5; lane 5 = positive NOS2 cDNA from pig No. 8; lane 6 = positive NOS2 cDNA from pig No. 10; lane 7 = positive NOS2 cDNA from pig No. 12; lane 8 = positive NOS2 cDNA from pig No. 15.
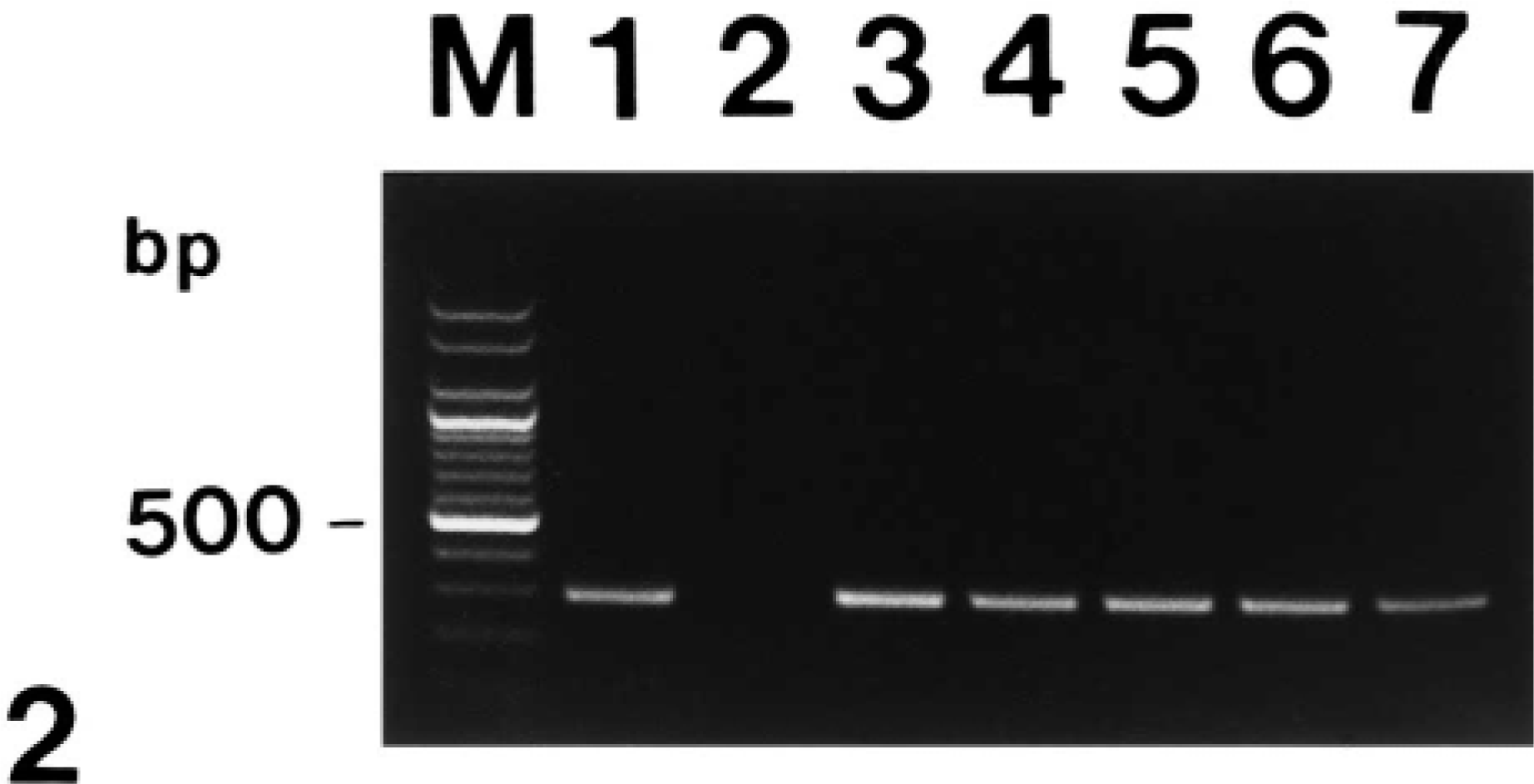
Agarose gel electrophoresis of polymerase chain reaction-amplified TNF-α cDNA products. From left to right: M = 100-bp DNA ladder; lane 1 = positive control from lipopolysaccharide-treated macrophages; lane 2 = negative control from normal macrophages; lane 3 = positive TNF-α cDNA from pig No. 2; lane 4 = positive TNF-α cDNA from pig No. 5; lane 5 = positive TNF-α cDNA from pig No. 8; lane 6 = positive TNF-α cDNA from pig No. 10; lane 7 = positive TNF-α cDNA from pig No. 12; lane 8 = positive TNF-α cDNA from pig No. 15.
All 15 pigs infected with A. pleuropneumoniae had distinct and positive hybridization signals for NOS2 and TNF-α. The morphology of host cells was preserved despite the relatively high temperature required during the incubation procedure. Signal intensity varied within and between anatomic structures within sections and between pigs. Positive cells were identified as having a dark brown or black reaction product without background staining.
A strong hybridization signal for NOS2 was seen in degenerate alveolar leukocytes bordering zones of coagulative necrosis and alveolar spaces. Hybridization signal was consistently detected in neutrophils and macrophages within the alveolar spaces but not inside blood vessels. Identification of cell types expressing NOS2 genes was occasionally difficult, but examination of serial sections stained with hematoxylin and eosin confirmed that NOS2 genes were present in neutrophils (Fig. 3) and macrophages (Fig. 4).
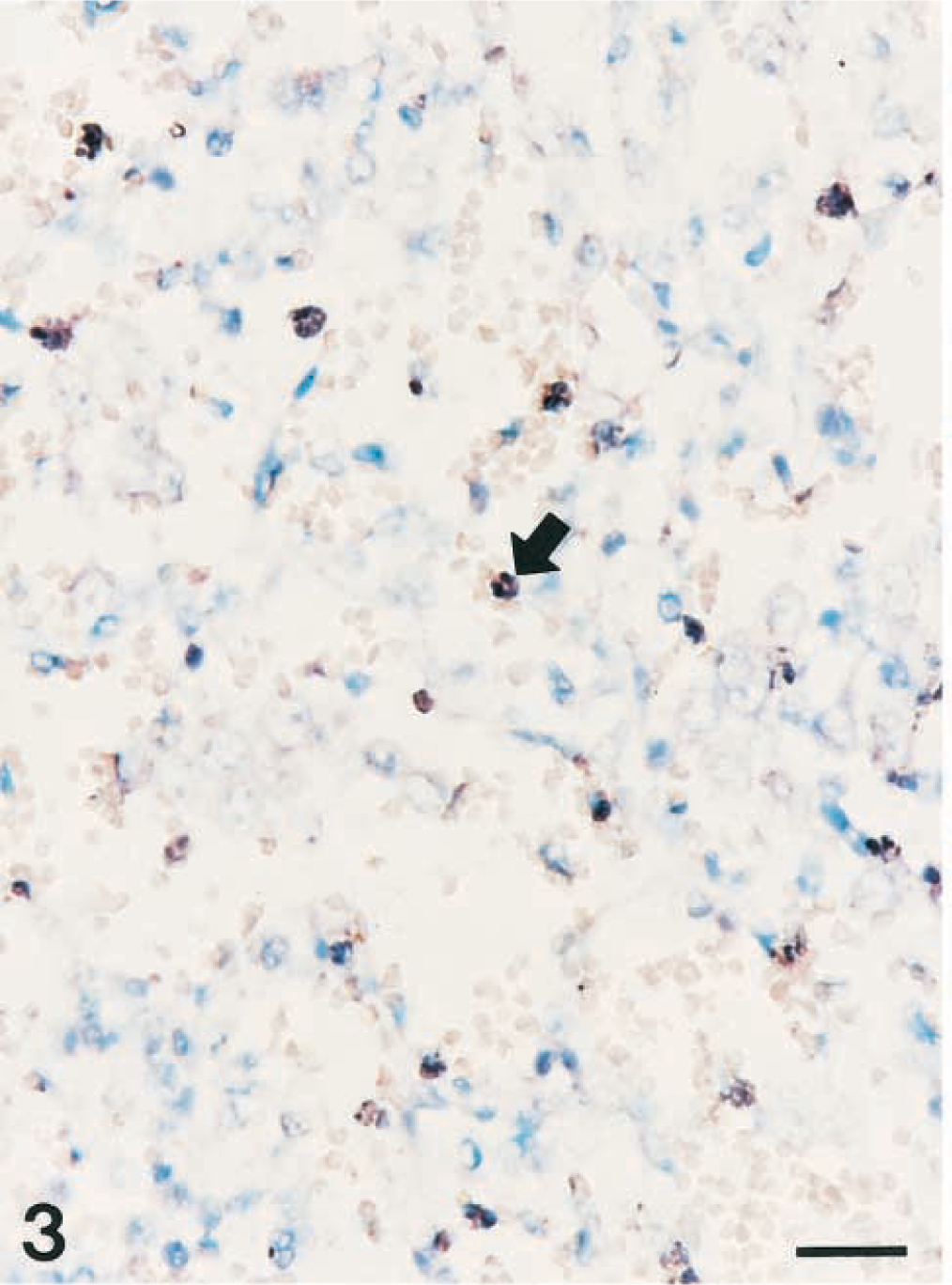
Lung; pig No. 2 naturally infected with Actinobacillus pleuropneumoniae serotype 2. Nitric oxide synthase 2 RNA (dark brown reaction) was detected in neutrophils (arrow) of alveolar spaces. In situ hybridization, cDNA probe, NBT/BCIP, methyl green counterstain. Bar = 30 µm.
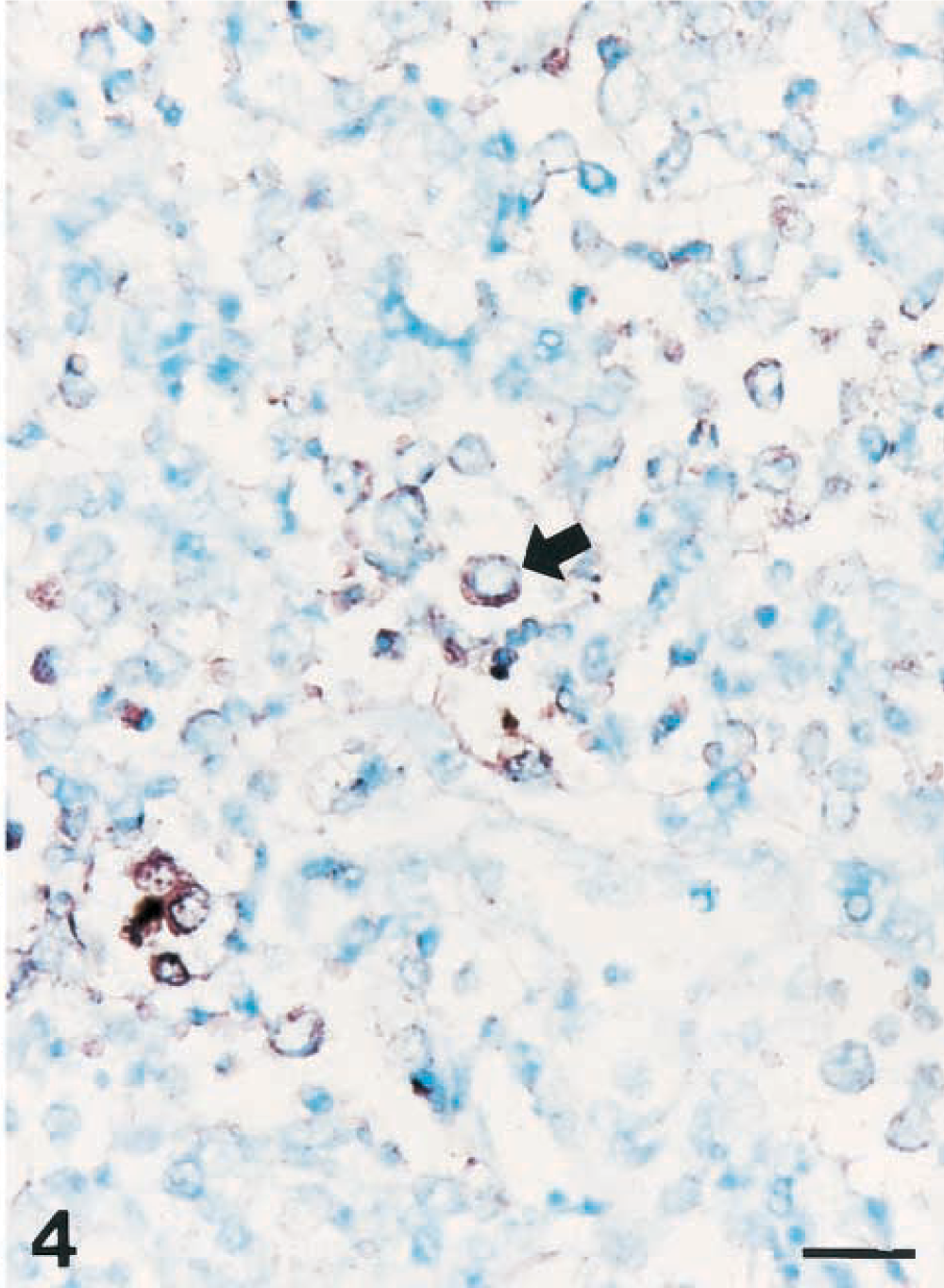
Lung; pig No. 6 naturally infected with Actinobacillus pleuropneumoniae serotype 5. Nitric oxide synthase 2 RNA (dark brown reaction) was detected in macrophages (arrow) of alveolar spaces. In situ hybridization, cDNA probe, NBT/BCIP, methyl green counterstain. Bar = 30 µm.
TNF-α expression was particularly intense in the clustered leukocytes with streaming nuclear chromatin that are a characteristic histologic feature of porcine pleuropneumonia. Neutrophils and macrophages in alveolar spaces frequently showed a strong signal. Expression of NOS2 and TNF-α was always present within inflammationary lung lesions but was minimal in nonaffected lung of A. pleuropneumoniae-infected pigs.
No hybridization signals for NOS2 and TNF-α were seen in sections incubated with a solution of RNase A prior to the performance of in situ hybridization. Sections from negative control pigs showed no hybridization signal for NOS2 and TNF-α.
Discussion
In this study, in situ hybridization with nonradioactive digoxigenin-labeled probes was successfully applied to A. pleuropneumoniae infected lung to detect NOS2 and TNF-α and to identify the sites at which these molecules were expressed. Intense and consistent expression of NOS2 and TNF-α was demonstrated in lesions of pleuropneumonia caused by A. pleuropneumoniae. In contrast, expression of inflammatory cytokines was minimal in noninflammed lung of infected pigs and in normal lung from control pigs. The results strongly suggest that NOS2 and TNF-α may be involved in lesion development and/or progression during acute pleuropneumonia. However, the detection of NOS2 and TNF-α nucleic acid does not necessarily indicate functional activity of NO and TNF-α production in vivo.
In situ hybridization confirmed increased expression of NOS2 by inflammatory cells in A. pleuropneumoniae-induced pleuropneumonia. To date, the induction of NOS2 had been shown in numerous cell types during inflammation, including neutrophils, macrophages, epithelial cells, and endothelial cells. 5,11,13,17 Macrophages produce NO and NOS2 under specific conditions, 28 and alveolar macrophages in inflamed lung show NOS2 staining. 16 Induction of NOS2 in macrophages and other cell types results in the release of large quantities of NO from these cells. 25,26 NOS2 is constitutively expressed in tissues, and increased expression is a normal response to invading organisms. In the lung, NOS2 is only induced in inflammatory cells after transmigration out of the vasculature into the interstitium and alveolar space, 22 consistent with our observations of the appearance of NOS2 in macrophages and neutrophils within the alveolar spaces but not in the vasculature. Our data suggest that epithelial and endothelial cells did not express NOS2 or did not express it to an extent that was detectable. However, there was limited epithelial regeneration in the tissues studied, which may have limited our ability to detect NOS2 in these cells.
Expression of NOS2 was greater in inflammatory lesions than in lung tissue showing no lesions. Therefore, it appears that induction of NOS2 is a direct response to A. pleuropneumoniae after stimulation of cells by LPS. Alternatively, expression of NOS2 may be a secondary effect of cytokine produced in response to A. pleuropneumoniae. NOS2 is present after stimulating the host cells with proinflammatory cytokines (e.g., TNF-α and IL-1). 25,26 Expression of TNF-α and IL-1 are detected in pigs with pleuropneumonia by use of in situ hybridization. 1,7 The combination of two or more of the stimulating cytokines is required to achieve optimal induction of NOS2 and maximal synthesis of its product, NO. 25,26 In situ hybridization of serial sections of lung indicates that the majority of areas containing numerous NOS2-positive cells also have numerous TNF-α–positive cells. Although the exact manner in which these molecules interact is not completely understood, TNF-α and IL-1 may act synergistically with A. pleuropneumoniae LPS to induce NOS2 expression by macrophages and other inflammatory cells in infected lungs.
Antimicrobicidal activity of NO has been demonstrated for several pathogens, including viruses. 21 There is substantial evidence that NO can downregulate mucosal inflammation and injury. NOS2-derived NO is a potent inhibitor of leukocyte adhesion to activated endothelial cells in vitro and in vivo. 2,18 Thus, NOS2 could have both proinflammatory and anti-inflammatory effects on the lungs. This study suggests that NOS2 and TNF-α expression may play a role in the pathophysiologic processes in pleuropneumonia.
Footnotes
Acknowledgements
The research reported here was supported by the Ministry of Agriculture, Forestry and Fisheries Special Grants Research Program (MAFF-SGRP) and by Brain Korea 21 Project, Republic of Korea.