
Abstract
Three Alaskan Huskies, two females and one male, were diagnosed with GM1-gangliosidosis. Clinically, diseased animals exhibited proportional dwarfism and developed progressive neurologic impairment with signs of cerebellar dysfunction at the age of 5–7 months. Skeletal lesions characterized by retarded enchondral ossification of vertebral epiphyses were revealed by radiographs of the male dog at 5.5 months of age. Histologic examination of the central nervous system (CNS) revealed that most neurons were enlarged with a foamy to granular cytoplasm due to tightly packed vacuoles that displaced the Nissl substance. Vacuoles in paraffin-embedded sections stained positively with Luxol fast blue and Grocott's method, and in frozen sections vacuoles were periodic acid–Schiff positive. Foamy vacuolation also occurred within neurons of the autonomic ganglia. Extracerebral cells such as macrophages and peripheral lymphocytes also displayed foamy cytoplasm and vacuolation. In the CNS of diseased animals, a mild demyelination and axonal degeneration was accompanied by a significant astrogliosis (P < 0.05) in the gray matter as compared with age- and sex-matched control dogs. There was also a significant loss (P < 0.05) of oligodendrocytes in the gray and white matter of affected animals as compared with controls. Ultrastructurally, the neuronal storage material consisted of numerous circular to concentric whorls of lamellated membranes or stacks of membranes in parallel arrays. GM1-gangliosidosis in Alaskan Huskies resembles β-galactosidase deficiency in other canine breeds, and these CNS disorders may be a consequence of neuronal storage and disturbed myelin processing.
GM1-gangliosidosis is a rare inherited neurovisceral lysosomal storage disorder. 22 The term lysosomal storage disease has been applied to a group of primarily genetic diseases characterized by deficiencies of specific lysosomal hydrolases with resultant storage of uncatabolized substrates in various tissues. Mechanisms resulting in the accumulation of undegraded substrates include diminished enzyme activity due to structurally altered enzymes, lack of enzymes or activator proteins, or alterations in posttranslational processing of lysosomal enzymes. 1,25 Cellular and organ distribution and the amount of storage material determines clinical classifications such as generalized neurovisceral, visceral, or skeletal involvement. 16 GM1-gangliosidosis is caused by an acid β-galactosidase deficiency. This enzyme defect leads to lysosomal accumulation of GM1 ganglioside and other galactose-containing glycoconjugates with a nonreduced terminal β-galactosidic linkage. 38 The disease has been described in a variety of species, including cats, cattle, sheep, mice, and humans. 1–9,12,14,16,17,27–29,31,33–38 Until now, the canine form has been found in English Springer Spaniels (ESS), 3,33 Portuguese Water Dogs (PWD), 3,35,36 mixed-breed Beagles, 34 and Alaskan Huskies. 28 In humans, the disorder is classified as infantile (type 1), juvenile (type 2), and adult (type 3) forms. 31,38 Patients with infantile GM1-gangliosidosis are presented at birth or shortly thereafter with somatic and bony changes followed by severe neurologic deterioration leading to death within the first years of life. A similar type of disease in a domestic shorthair kitten included visceral, skeletal, and central nervous system (CNS) involvement. 9 Signs of juvenile GM1-gangliosidosis begin at approximately 1 year of age and consist of motor weakness, mild visceral and bone abnormalities, and slowly progressive psychomotor deterioration. Several animal models resembling the juvenile type of GM1-gangliosidosis have been described in cats, including purebred Siamese and Korat cats and outbred domestic and shorthair cats. 14,16,22 GM1-gangliosidosis in mixed-breed Beagles resembles the disease in human patients with juvenile GM1-gangliosidosis in its delayed onset of CNS dysfunction and lack of bony or craniofacial anomalies. However, the prominent morphologic and biochemical involvement of liver and kidney as observed in affected Beagles has only been described in human patients with the infantile type of GM1-gangliosidosis. 34 Skeletal dysplasia was reported in ESS and PWD, whereas only PWD displayed dwarfism and coarse facial features. 3 PWD and ESS showed different storage materials in various visceral organs. Friesian cattle with β-galactosidase deficiency expressed neurologic impairment shortly after birth, but the morphologic lesions were limited to the CNS, without accompanying visceral involvement. 17 The symptoms of adult GM1-gangliosidosis in humans consist of slowly progressive deterioration beginning in early childhood. Dystonia is the major neurologic manifestation. 25,31,38
We recently reported the biochemical findings in three Alaskan Huskies suffering from GM1-gangliosidosis. 28 Here, we describe the clinical and pathologic findings for these dogs, with special emphasis on the neuropathologic changes.
Materials and Methods
Case histories
Between 1990 and 1995, several cases of progressive neurologic impairment starting at 6 weeks of age and leading to death within the first year of life were recognized in a breeding colony of Alaskan Huskies in Germany. Using skin fibroblast cultures, researchers diagnosed the disease as GM1-gangliosidosis. 28 The dogs were descendants of phenotypically normal Alaskan Huskies from Alaska and Austria (Fig. 1). Pedigree analysis revealed a high breed consanguinity, and male and female siblings were affected. Approximately one or two offspring of each litter presented clinical signs of GM1-gangliosidosis, suggesting an autosomal recessive pattern of inheritance with variable expressivity. Based on the β-galactosidase activity of cultured primary skin fibroblasts, descendants were grouped in homozygous normal, heterozygous, and homozygous affected individuals. 28 Two dogs, one female (dog No. 2) and one male (dog No. 3), were investigated for β-galactosidase activity and expressed a loss of β-galactosidase activity of 92% as compared with controls. The β-galactosidase activity was also markedly reduced in brain, liver, spleen, and kidney of dog No. 3. 28
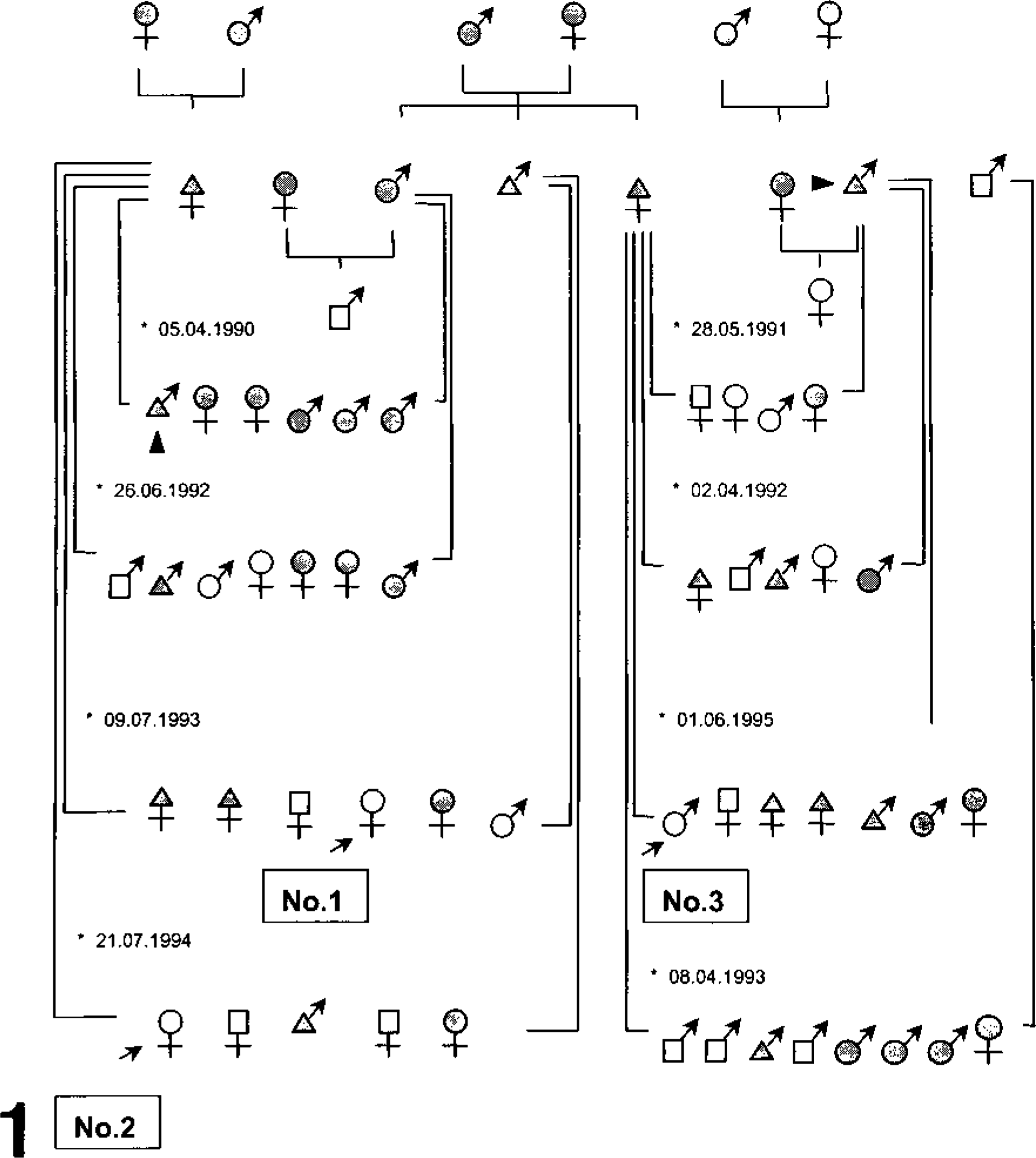
Family pedigree of Alaskan Huskies suffering from GM1-gangliosidosis. ♀ ♂, not tested for β-galactosidase activity in cultured primary skin fibroblasts; ♀ ♂, normal; ♀ ♂, heterozygous; ♀ ♂, homozygous based on β-galactosidase activity in cultured primary skin fibroblasts; 28 →, dog Nos. 1–3; ∗, date of birth; arrowhead, same animal
Clinical examination
Clinical tests of two dogs, one female (dog No. 2) and one male (dog No. 3), and control dogs (Nos. 4–9) included physical and neurologic examination, complete blood counts, plasma biochemical analyses (γ-glutamyl transferase, sorbitol dehydrogenase, aspartate aminotransferase, creatine phosphokinase, and alkaline phosphatase [ALP] activities; total bilirubin, total protein, albumin, urea nitrogen, glucose, sodium, chloride, potassium, calcium, and phosphorus concentrations), and urinalysis at 4 and 6.5 months of age in dog No. 2 and at 5.5 months of age in dog No. 3. Dog No. 1 was not available for clinical investigation. The number of vacuoles in peripheral lymphocytes of dog Nos. 2 and 3 and the six control dogs (Nos. 4–9) was counted using air-dried blood smears stained with May–Grünwald–Giemsa. Ophthalmologic examination and cerebrospinal fluid (CSF) analysis were performed and skeletal radiographs were obtained for dog Nos. 2 and 3 prior to euthanasia.
Light and electron microscopy
Affected dogs (Nos. 1–3) were euthanatized at 5.5 (dog No. 3) and 7 (dog Nos. 1, 2) months of age by a pentobarbital overdose. Corresponding age-, size-, and sex-matched dogs (Nos. 4–19) were used as controls. Samples of various tissues, including brain, spinal cord, eye, liver, kidney, urinary bladder, spleen, lymph nodes, lung, heart, intestine, pancreas, salivary gland, adrenal gland, thyroid gland, bones, bone marrow, and skin, were collected during necropsy and fixed in 10% formalin. In addition, brain tissue was embedded in OCT compound and stored frozen at −70 C. 41 Bones were cut in 4-mm-thick slices and decalcified in 10% aqueous (distilled water) ethylenediaminetetraacetic acid Na2 salt (w/v), pH 7.4, for at least 72 hours at room temperature. Paraffin-embedded sections were processed routinely for light microscopy and stained with hematoxylin and eosin (HE), Luxol fast blue–cresyl violet (LFB), periodic acid–Schiff (PAS), elastica van Gieson, cresyl-(echt)-violet, and Grocott's silver method. Frozen sections were stained with PAS. For electron microscopy, tissues including CNS and pancreas of affected animals were fixed in 2.5% glutaraldehyde in 0.166 M cacodylate buffer, postfixed in 1% osmium tetroxide, dehydrated, embedded in durcupan, sectioned at 1 µm, and stained with methylene blue. Selected sections were cut at 60–90 nm, stained with uranyl acetate and lead citrate, and examined with a EM-10 C transmission electron microscope (Carl Zeiss, Oberkochen, Germany) as previously described. 11
Immunohistochemistry
For immunohistology, CNS sections of dog Nos. 1–3 and controls (Nos. 10–19) were incubated with primary antibodies directed against glial fibrillary acidic protein (GFAP), myelin basic protein (MBP), neurofilament (NF), and major histocompatibility complex class II (MHC II) antigen employing the avidin–biotin–peroxidase complex method as previously described. 10,18,21 Sections (3 µm thick) were deparaffinized in xylene and rehydrated through graded alcohols. Endogenous peroxidase was blocked by incubation with 0.5% hydrogen peroxide in methanol at room temperature for 30 minutes. Sections were washed with Tris-buffered saline (TBS, 0.1 M Tris-HCl, 0.8% sodium chloride, pH 7.6). For demonstration of MHC II antigen, sections were treated with target unmasking fluid (Kreatech Diagnostics, Amsterdam, the Netherlands) for 10 minutes at 96 C, and for demonstration of GFAP, sections were incubated with 0.25% trypsin (pH 7.6) with 0.02% CaCl2 for 1 hour at 37 C. Following these digestion steps, sections were incubated with undiluted normal pig serum or horse serum for 10 minutes at room temperature. Thereafter, slides were incubated for 12–16 hours at 4 C with the primary antibodies: polyclonal rabbit anti-GFAP (1:500, Dako, Hamburg, Germany), monoclonal mouse anti-NF (1:400, Dako), polyclonal rabbit anti-MBP (1:1,000, Genesis, Hamburg, Germany), and monoclonal rat anti-dog MHC II antigen (1:40, kindly provided by Dr. C. Vogl). 18 Biotinylated horse anti-mouse IgG was used as link antibody for the demonstration of mouse anti-NF, and biotinylated goat anti-rabbit IgG was used for the demonstration of rabbit anti-GFAP and rabbit anti-MBP, followed by the avidin–biotin–peroxidase complex (Vector Laboratories, Burlingame, CA). For monoclonal rat antibodies, a biotinylated rabbit anti-rat antibody was applied (Vector Laboratories). Incubations were performed in a moist chamber at room temperature for 30 minutes. After each incubation step, slides were washed with TBS. Sections were incubated for 10 minutes with 0.05% 3,3′diaminobenzidine tetrahydrochloride in 0.1 M buffered imidazole/hydrogen chloride (pH 7.1) and counterstained with Papanicolaou's hematoxylin. Normal rabbit serum or cell culture supernatant were used instead of primary antibodies as negative controls. Sections of brain from control dogs served as positive controls for GFAP, MBP, and NF antigens. Sections of a cutaneous histiocytoma were the positive control for MHC II antigens.
In situ hybridization
Cerebellar and cerebral sections were investigated for proteolipid protein (PLP) mRNA in oligodendrocytes using in situ hybridization (ISH) as described previously. 18,20,30,43 A strand-specific digoxigenin-labeled mRNA probe of about 150 bases complementary to the sequence of the PLP gene was generated as previously described. 43 After proteolytic digestion, postfixation, and prehybridization, hybridization was performed overnight at 50 C. Excess labeled RNA was removed, and the sections were incubated with an anti-digoxigenin antibody conjugated with ALP (Boehringer, Mannheim, Germany). For the color reaction, nitro blue tetrazolium (NBT; Sigma, Buchs, Switzerland) and 5-bromo-4-chloro-3-indolyl phosphate (BCIP) were used.
Scoring system and statistical analysis
Using blood smears, the number of vacuoles in 50 peripheral lymphocytes of dog Nos. 2 and 3 and six control dogs (Nos. 4–9) was counted under oil immersion. GFAP and PLP mRNA-positive cells were counted in the gray and white matter of the parietal and frontal lobe of dog Nos. 1–3 and 10 age-matched control dogs (Nos. 10–19) in 10 corresponding fields using a 100-mm2 (10 × 10 mm) ocular gird (Olympus, Hamburg, Germany) at 200× magnification. Statistical analysis was carried out using BMDP/Dynamic release 7.0 software (BMDP Statistical Software, Los Angeles, CA). Data were analyzed with a t-test (BMDP3D) and with analyses of variance and covariance with repeated measures (BMDP2V). Significance was accepted at P < 0.05.
Results
Clinical findings
In all three affected dogs, clinical signs were first noticed at approximately 6–8 weeks of age and steadily progressed until the time of euthanasia. Initial signs included weight loss, moderate wide-based gait, a slight decrease in proprioception, and a fine tremor of the head. At 7 months of age, diseased animals displayed proportional dwarfism and showed clinical signs of cerebellar dysfunction including ataxia characterized by hyper- and dysmetria, particularly of the hind legs. Frequent falling and swaying was noticed, and the head tremor became more obvious. Affected dogs continued to eat and drink, although at a decreased level. A severe loss of body weight as compared with sex-matched littermates was subsequently documented. There was also a mild internal strabismus and a positional nystagmus with a fast component to the right, whereas direct and consensual pupillary reflexes were normal. However, even at that advanced stage of the disease, vision and sensory perception appeared to be normal. Dog No. 2 showed tonic-clonic convulsions in the terminal stages at 7 months of age. Dog No. 3 showed almost complete loss of proprioception with a wide sawhorse stance at 5.5 months of age. Skeletal lesions noticed in dog No. 3 at 5.5 months of age were characterized by retarded enchondral ossification of the lumbar vertebral epiphyses and abnormally wide intervertebral disc spaces. Furthermore, there was an acute fracture of the right femur and a mild periostitis of the right humerus.
Routine blood analyses revealed no abnormalities except for a moderate leukocytosis, a mild monocytosis, and a slight thrombocytosis in dog No. 3. Results of blood chemistry profiles were generally normal regardless of the clinical stage of the disease, except for a moderate increase of serum ALP in dog No. 2 at 4 months of age (406.0 U/liter; controls: <190 U/liter) and a moderate increase in serum creatine phosphokinase activity in dog No. 3 (205.0 U/liter; controls: <80 U/liter). Urinalysis revealed a mild proteinuria in both dogs (Nos. 2 and 3) and a mild hemoglobinuria in dog No. 3.
Single or multiple vacuoles were found in macrophages and lymphocytes in the CSF of both affected animals. Diseased animals demonstrated significantly more vacuolated lymphocytes in the peripheral blood (44% and 46%) as compared with controls (○ = 7.3%; range, 6–12%; P < 0.05; Fig. 2). Large vacuoles were also found within neutrophils, basophils, eosinophils, and monocytes of dog Nos. 2 and 3 but rarely in controls.
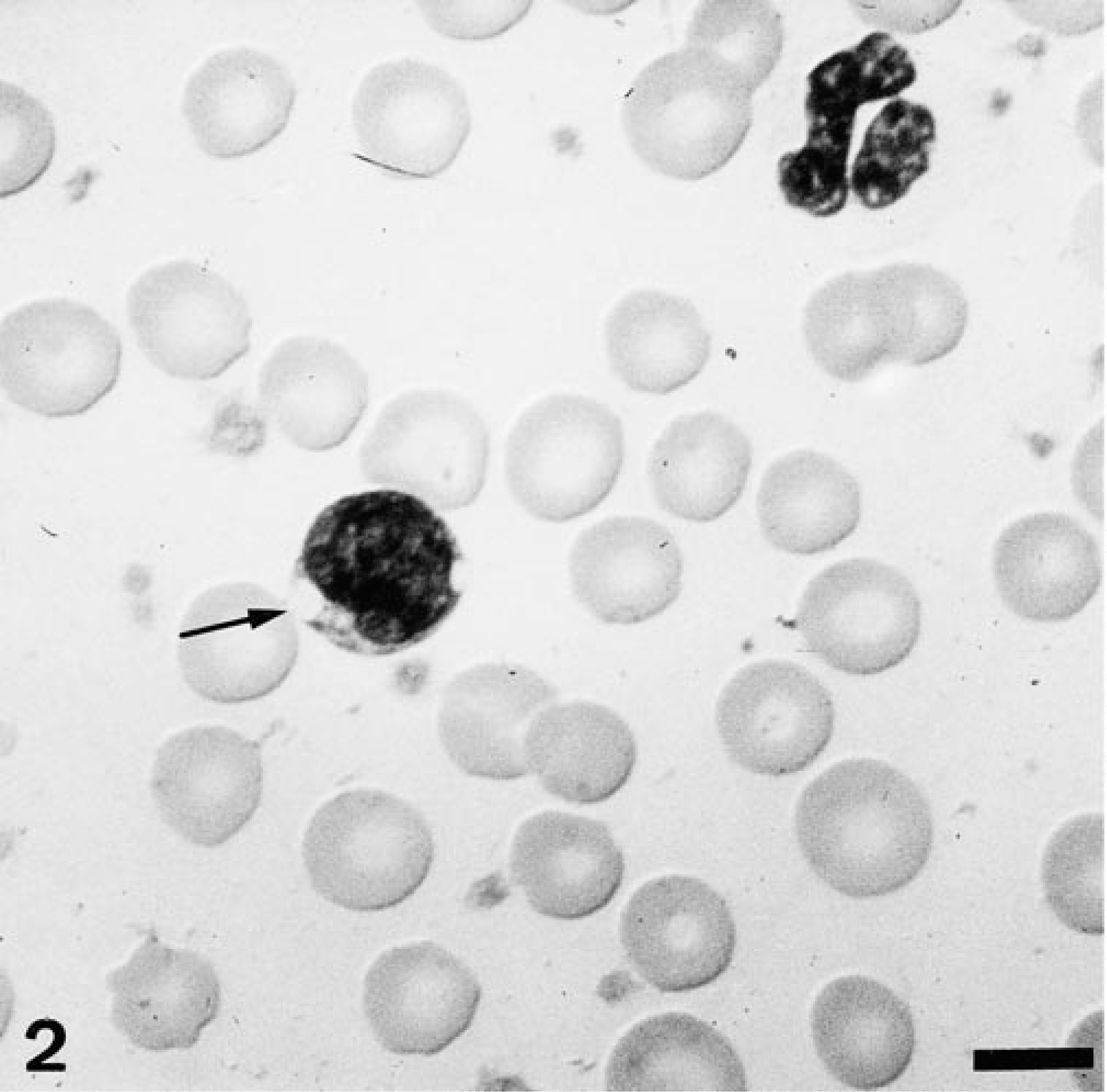
Air-dried blood smear; dog No. 2. Vacuolated cytoplasm (arrow) in a lymphocyte. May–Grünwald–Giemsa. Bar = 10 µm.
Gross pathologic findings
No significant gross lesions were observed apart from emaciation, a fracture of the right femur in dog No. 3, and irregular intervertebral disc spaces in dog Nos. 1 and 3.
Histopathology, immunohistochemistry, and ISH
By light microscopy, lesions were similar in all 3 affected animals and are therefore condensed to one description. The changes were not found in any of the control dogs. Alterations were most prominent in the central and peripheral nervous system and lymphoid tissues. Most neurons of the brain, spinal cord, and peripheral ganglia were severely affected. Neurons were enlarged by a fine to coarse granular cytoplasmic material and by vacuoles of various sizes in the cytoplasm (Fig. 3). In more severely affected neurons, the Nissl substance, as seen with cresyl violet staining, was dispersed or aggregated around the eccentrically displaced nuclei (Fig. 4). The storage material also extended into the major dendritic branches and occasionally into the axon hillocks. The storage material stained with LFB and was slightly argentophilic in paraffin-embedded tissue. In frozen sections, the inclusions stained intensely red with PAS. Similar lesions were noticed in neurons of the retinal ganglion cell layer. A few scattered cells of the inner nuclear layer of the retina were also enlarged and displayed a finely granular cytoplasm. Neurons of the ganglia of the autonomic nervous system of the intestine, urinary bladder, and adrenal glands were mildly affected. However, their vacuoles failed to react with LFB and the silver stain. The cerebral and cerebellar white matter exhibited pallor of myelin and a mild Wallerian-type degeneration characterized by myelinophagic macrophages, predominantly in the subcortical area where the U-fibers are located.
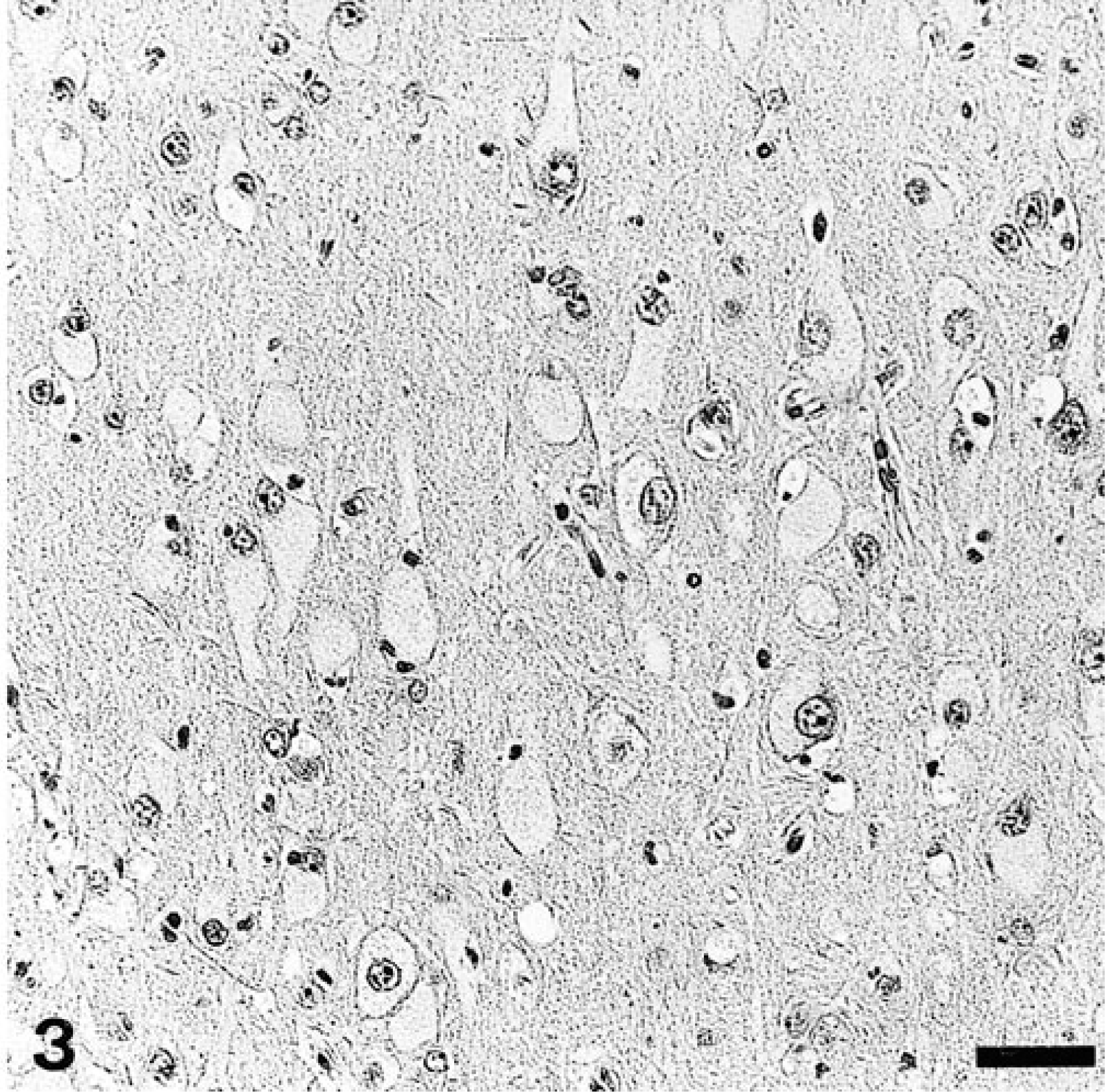
Cerebrum; dog No. 2. Neurons are enlarged and contain fine to coarse granular cytoplasmic storage material. HE. Bar = 50 µm.
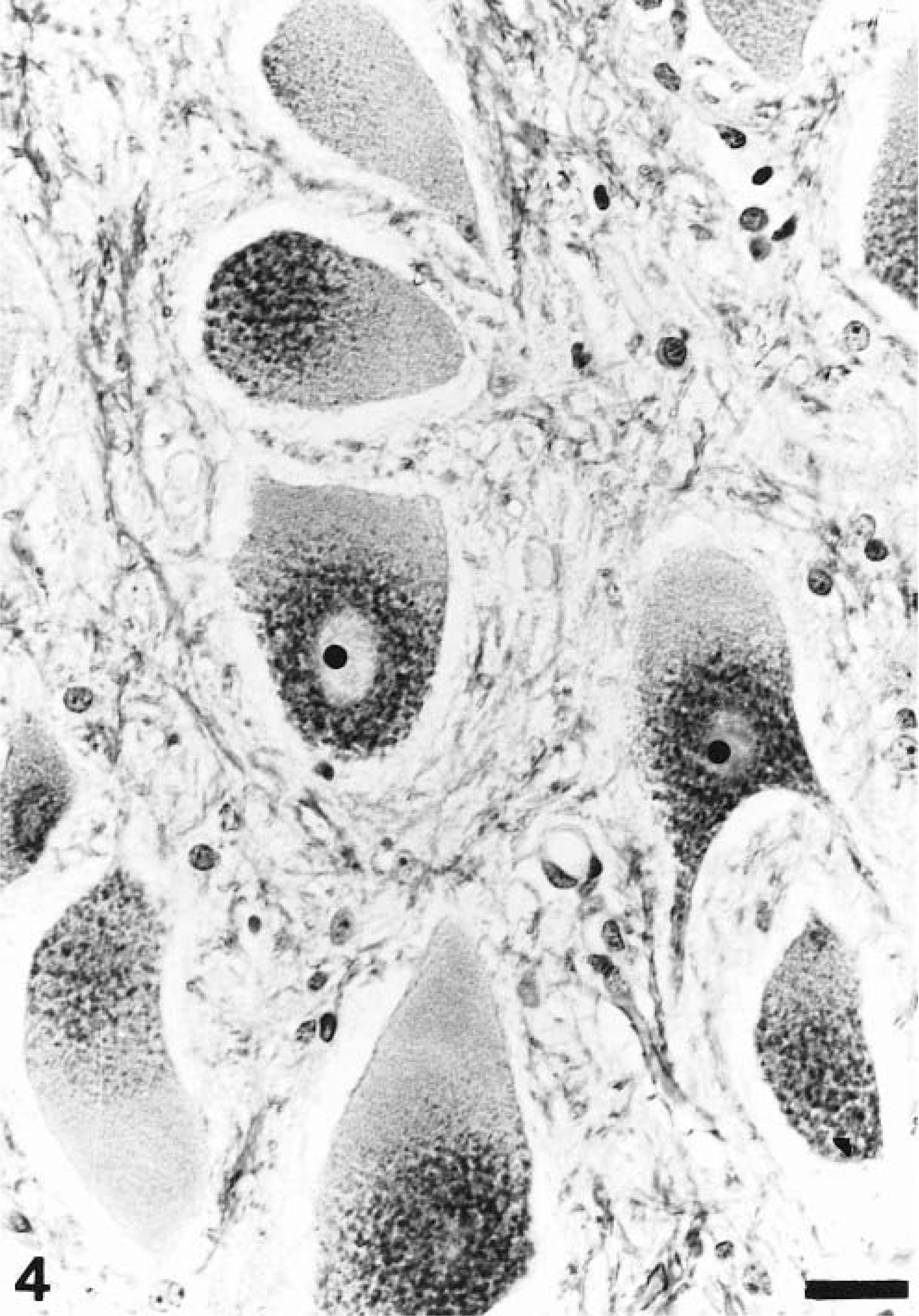
Spinal cord; dog No. 2. Neurons in the ventral gray column with marked cytoplasmic enlargement due to accumulation of LFB-positive storage material and displacement of the Nissl substance. LFB-cresyl violet. Bar = 25 µm.
Immunohistologic investigation of the CNS revealed axonal changes, including the formation of NF-positive spheroids and large torpedo-like swellings in the proximal segments of cerebellar Purkinje cells (Fig. 5). Similar axonal enlargements were also present but less prevalent in the white matter of the cerebrum and spinal cord. Additionally, there was a moderate to severe reduction of MBP expression and reduced compaction of the myelin in comparison with control tissues. The degree of myelin loss varied according to the location within the CNS and was most severe in the U-fiber region (Fig. 6a, b). A moderate to severe astrocytosis was noticed in the cerebrum and cerebellum of diseased dogs, as demonstrated by increased immunoreactivity for GFAP in the white and gray matter (Fig. 7a, b). In dog No. 2, a mild upregulation of MHC II on microglia cells was noticed in comparison to controls.
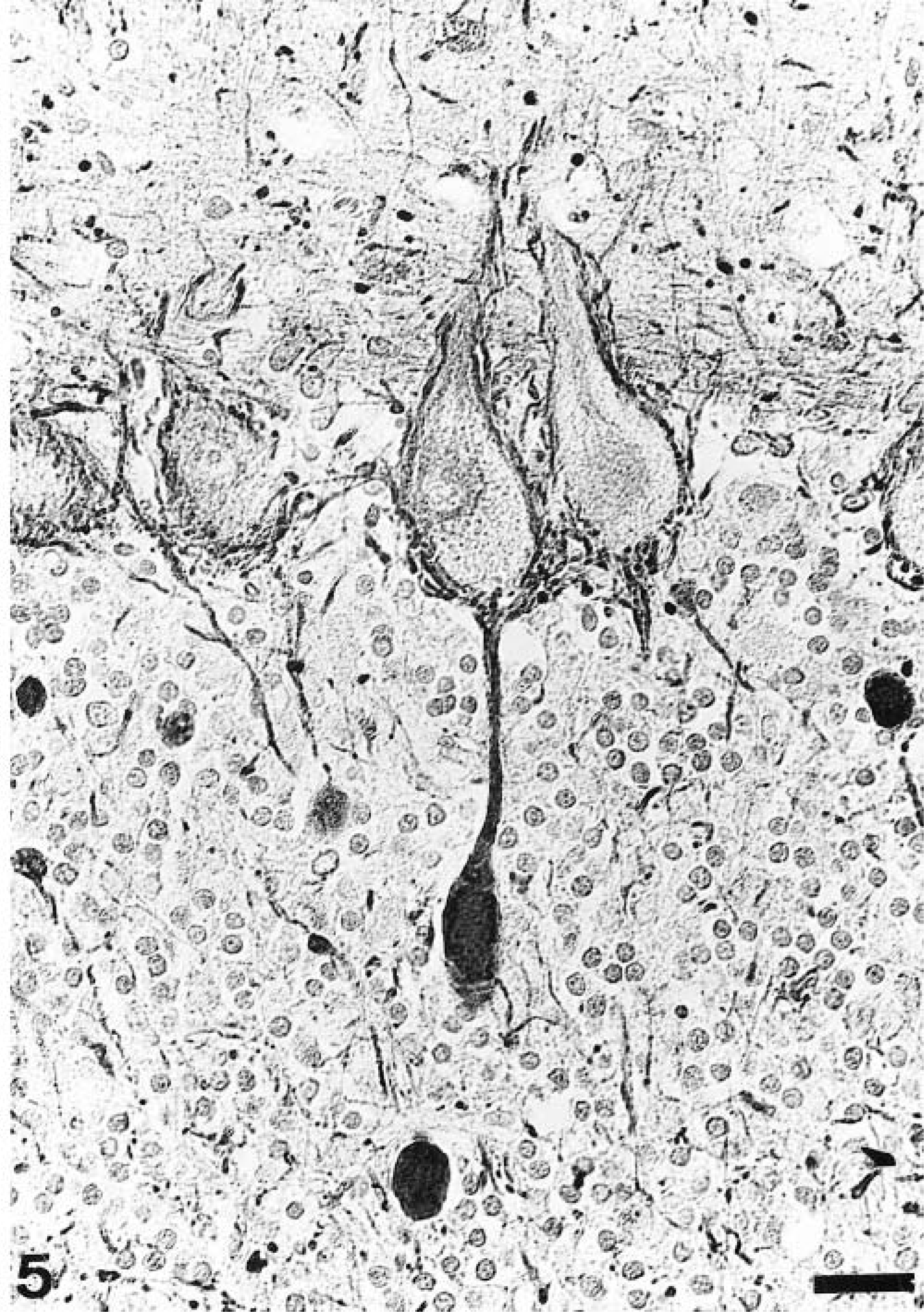
Cerebrum; dog No. 1. Large NF-positive torpedo-like swellings in the proximal axonal segment of a Purkinje cell. Monoclonal anti-NF antibody; avidin–biotin–peroxidase complex method; Papanicolaou's hematoxylin counterstain. Bar = 25 µm.

Cerebrum; dogs.Fig. 6aIn the control dog, a strong MBP immunoreactivity can be observed in the center and periphery of the cerebral white matter. Fig. 6b In the dog with GM1-gangliosidosis (dog No. 2), there is reduced expression of MBP, especially in the U-fiber region, and less compaction of the myelin in the U-fiber region (∗). wm = white matter; gm = gray matter. Polyclonal anti-MBP antibody; avidin–biotin–peroxidase complex method; slight Papanicolaou's hematoxylin counterstain. Bar = 145 µm.
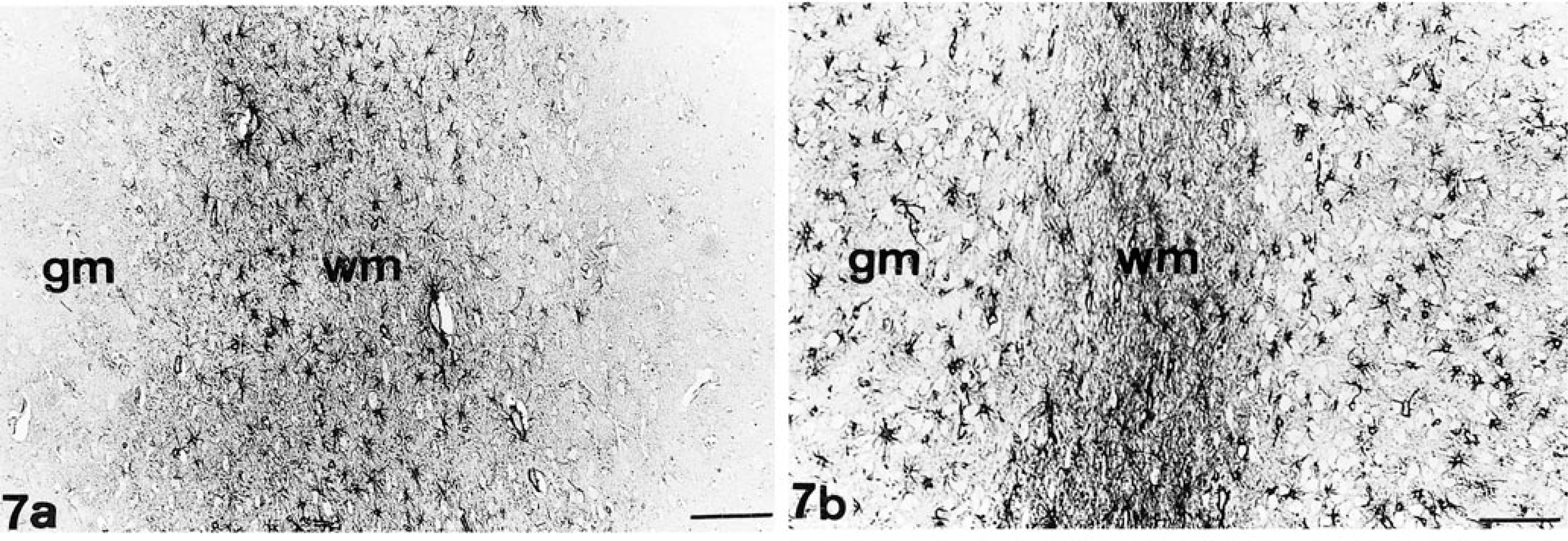
Cerebrum; dogs. Fig. 7a Control dog. Fig. 7b Dog No. 3 with GM1-gangliosidosis. Note increased GFAP immunoreactivity, especially in the gray matter. wm = white matter; gm = gray matter. Polyclonal anti-GFAP antibody; avidin–biotin–peroxidase complex method; slight Papanicolaou's hematoxylin counterstain. Bar = 145 µm.
ISH demonstrated abundant PLP mRNA–positive cells in the gray and white matter of the cerebrum of control dogs. In contrast, these brain regions showed a significant reduction of PLP mRNA–positive oligodendrocytes in affected animals. The few remaining mRNA-positive cells were located in the white matter (Fig. 8a, b). PLP mRNA–positive cells were also reduced in the granular and Purkinje cell layers of the cerebellum.
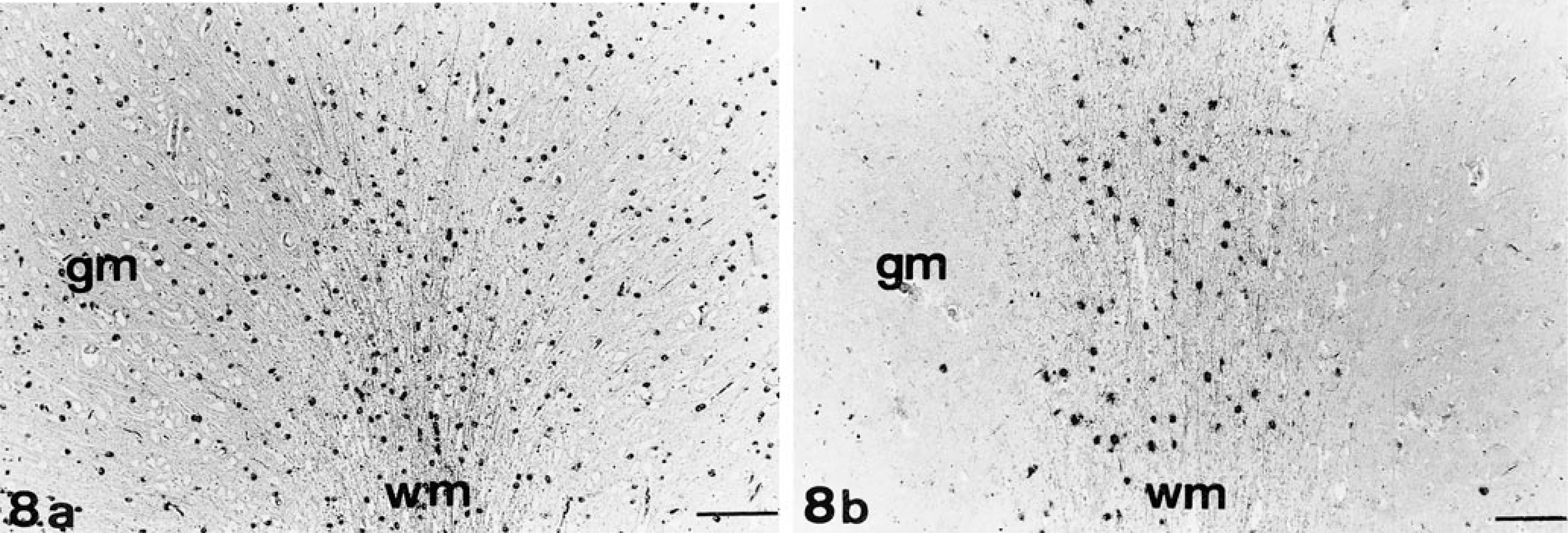
Cerebrum; dogs. Fig. 8a Control dog. Note abundant PLP mRNA–positive cells in the gray and white matter. Fig. 8b Dog No. 1 with GM1-gangliosidosis. Note reduced number of PLP mRNA–positive oligodendrocytes in the gray and white matter of the frontal lobe. wm = white matter; gm = gray matter. ISH; NBT/BCIP. Bar = 145 µm.
Statistical analysis revealed that the number of GFAP-positive cells was significantly increased in the gray matter of affected animals (○ = 140 cells/mm2; range, 116–166 cells/mm2) as compared with controls (○ = 11 cells/mm2; range, 0.4–33 cells/mm2; P < 0.05; Fig. 9), whereas the numbers were similar in the white matter. PLP mRNA–positive cells of affected animals were significantly reduced in the gray matter (○ = 1 cell/mm2; range, 0.4–2.4 cells/mm2; controls: ○ = 203 cells/mm2; range, 161–248 cells/mm2) and white matter (○ = 462 cells/mm2; range, 246–584 cells/mm2; controls: ○ = 949 cells/mm2; range, 833–1,084 cells/mm2; P < 0.05; Fig. 10).
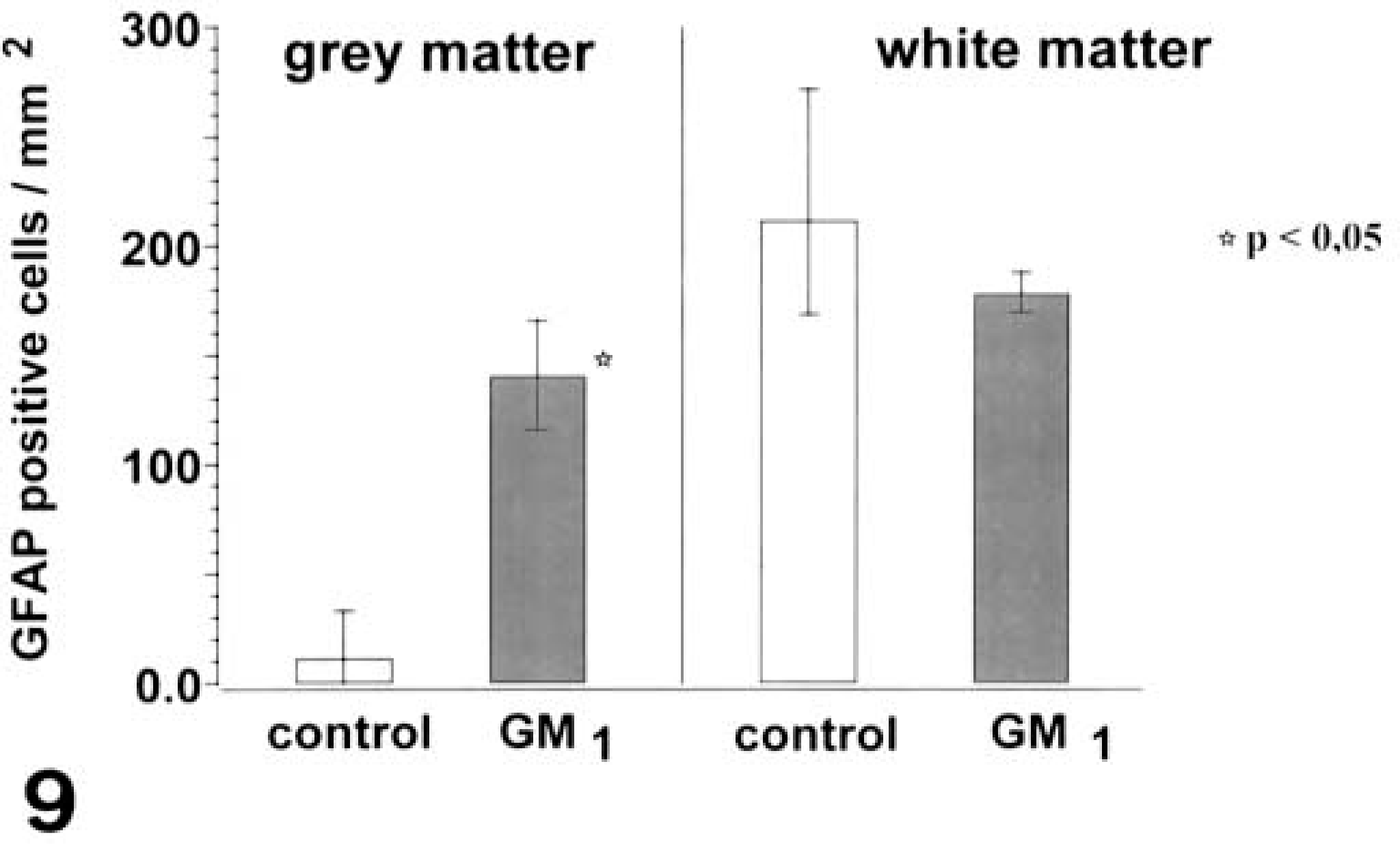
Brain; dogs. GFAP-positive cells in the parietal and frontal lobe of control dogs (n = 4) and dogs with GM1-gangliosidosis (n = 3). There is a significant increase of GFAP-positive cells in the gray matter of diseased dogs (P < 0.05; ○; range). ∗ = significantly different from controls.
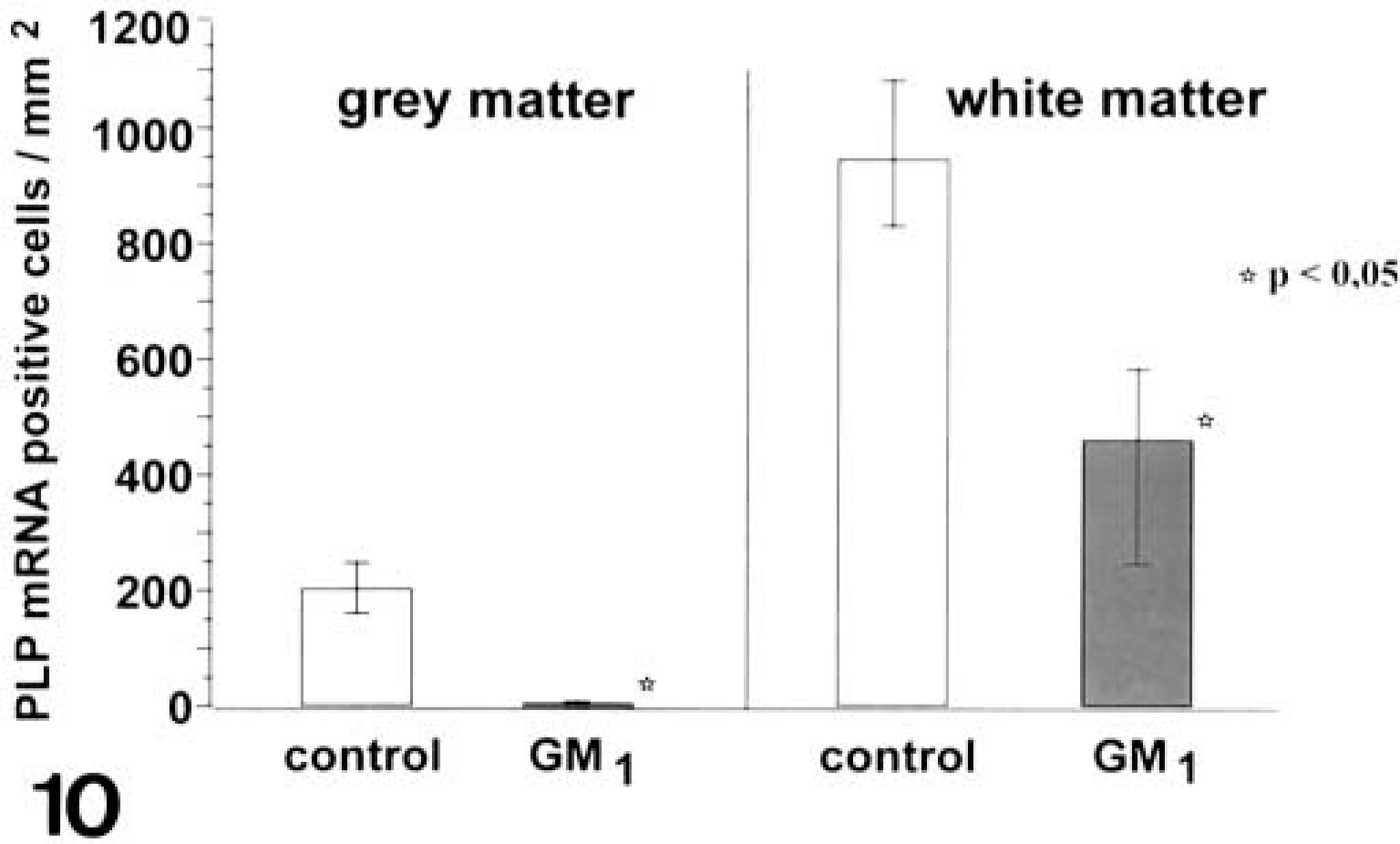
Brain; dogs. PLP mRNA-positive cells in the frontal and parietal lobe of control dogs (n = 6) and dogs with GM1-gangliosidosis (n = 3). There is a significant reduction of PLP mRNA–positive cells in the gray and white matter of diseased dogs (P < 0.05; ○; range). ∗ = significantly different from controls.
In the nonnervous tissue, alterations were present in a variety of organs and consisted of single vacuoles or foamy vacuolation within different cell types. These inclusions were not seen in controls and failed to react with LFB, PAS, and Grocott's silver stain in paraffin-embedded sections. In addition, there was an infiltration of enlarged macrophages in lymph nodes, Peyer's patches, spleen, and lung. These macrophages showed a foamy cytoplasm that was slightly argentophilic and PAS positive but failed to react with LFB. In the liver, hepatocytes with coalescent vacuoles were observed. Pancreatic acinar cells, renal tubular cells, cells of esophageal glands, and serous and mucinous cells of the salivary glands, enterocytes, and chondrocytes contained cytoplasmic vacuoles. Sections of the vertebral column revealed the formation of growth arrest lines and osteopenia. In addition, a mild focal interstitial nephritis and generalized membranous glomerulonephritis was noticed in dog No. 2. In the liver of dog No. 3, a mild periportal plasmacellular infiltration was visible.
Ultrastructural findings
Ultrastructurally, the neuronal inclusions consisted of membrane-bound enlarged lysosomes containing numerous circular to ovoid concentric whorls of lamellated membranes or stacks of membranes in parallel array (Fig. 11). Less commonly, enlarged lysosomes contained fibrillogranular or electron-dense homogeneous material. Astrocytes were ballooned and displayed mitochondrial swelling and a mild cytoplasmic vacuolation. Ultrastructural examination of the pancreatic tissue and endothelial cells revealed membrane-bound empty vacuoles, which occasionally contained amorphous granular material (Fig. 12).
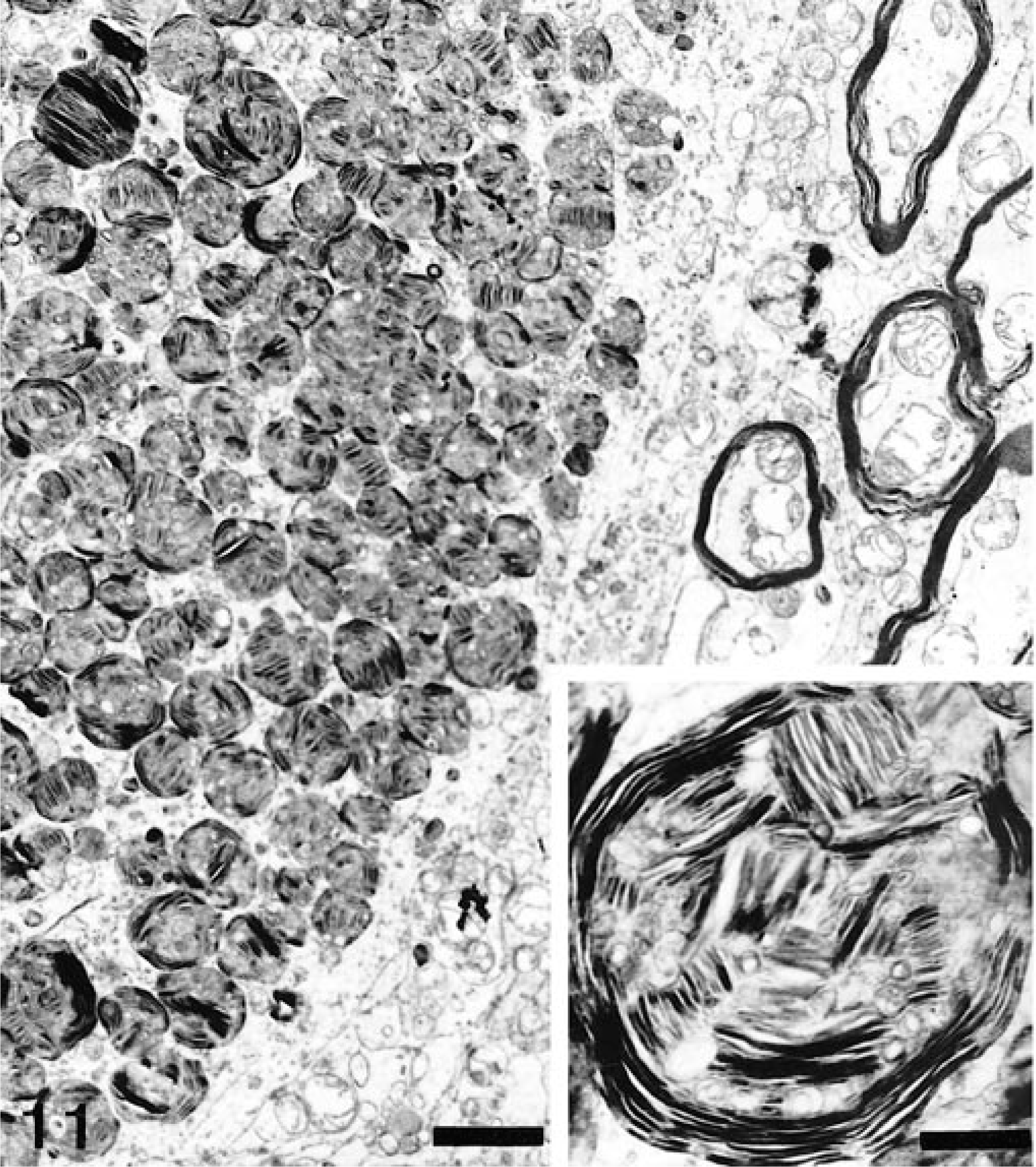
Electron micrograph. Spinal cord; dog No. 2. Neuron with cytoplasmic membranous bodies displaying concentric lamellated membranes and stacks of membranes in parallel arrays. Bar = 1.9 µm. Inset: Higher magnification of cytoplasmic membranous bodies. Uranyl acetate and lead citrate counterstain. Bar = 0.4 µm.
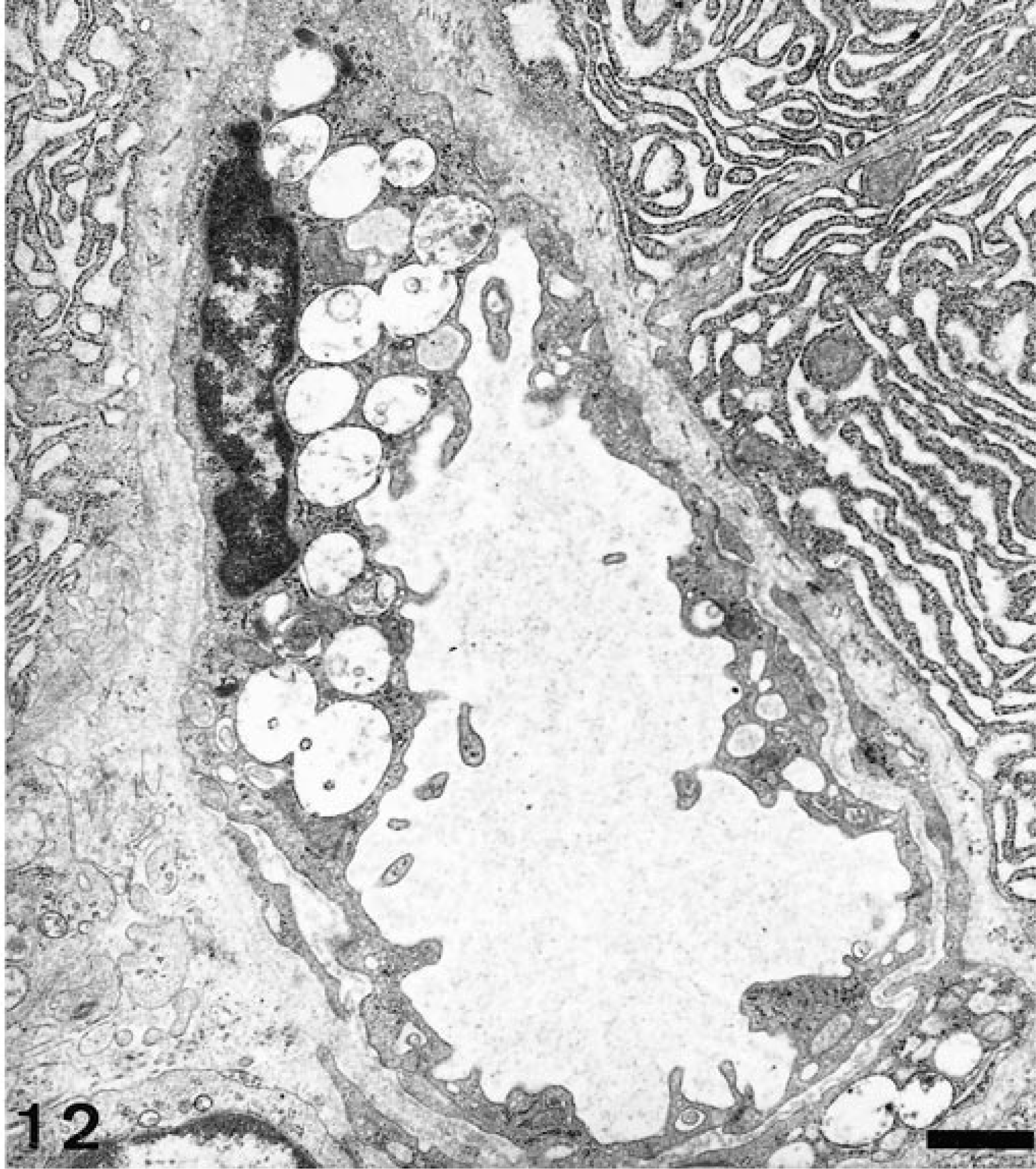
Electron micrograph. Pancreas; dog No. 2. Endothelial cell with distinct cytoplasmic membrane-bound vacuoles. Uranyl acetate and lead citrate counterstain. Bar = 1 µm.
Discussion
The clinical features of GM1-gangliosidosis in Alaskan Huskies were characterized by progressive neurologic dysfunction without visual impairment. The disease in Alaskan Huskies showed similarities to GM1-gangliosidosis in other dogs. The age of onset, neurologic signs, and rapid progression were similar to GM1-gangliosidosis of ESS, PWD, 2,3,33,35,36 and mixed-breed Beagles. 34 Proportional dwarfism has only been reported in ESS 3 and Alaskan Huskies.
The development of skeletal lesions has been described for ESS and PWD and consisted of bony dysplasia, especially of the lumbar spine, and coarse facial features in ESS. 3,32 In contrast, Alaskan Huskies showed only retarded enchondral ossification restricted to the vertebral column in one animal. Although these changes could be a consequence of lysosomal storage in bone cells, malnutrition might result in a similar growth disturbance (Dr. S. Weisbrode, Ohio State University, personal communication). There were no indications for other illnesses such as chondrodysplasia resulting in disproportionate dwarfs in this colony. Dog No. 2 showed an elevation of serum ALP activity. In humans, elevated serum ALP activity is often associated with GM1-gangliosidosis and other storage diseases with skeletal involvement, and this enzyme might represent an early biologic marker of the disease. 15 An increase of serum ALP was also detected in three young cats suffering from GM1-gangliosidosis, but no osseous changes were found. 16 The increased activity of ALP might be a physiologic age-related phenomenon unrelated to the storage disease; therefore, the significance of this observation with respect to the diagnosis of GM1-gangliosidosis in Alaskan Huskies remains unclear.
A moderate vacuolation of peripheral blood cells, most prominent in lymphocytes and similar to that in ESS 3,4 and PWD 3, was present in Alaskan Huskies. Histologically, changes associated with storage material occurred in neuronal and extraneuronal tissues. Alaskan Huskies showed prominent alterations in the gray and white matter, including enlargement of neurons, spheroid formation, and torpedo-like swellings in the proximal axonal segment of Purkinje cells. These changes resembled those observed in other species, including humans. 22,31,38 Several events such as neuronal loss, glycolipid neurotoxicity, neurotransmitter abnormalities, and changes in neuronal geometry and synaptic connectivity may lead to neurologic impairment. 39,40 Alaskan Huskies exhibited poliodystrophic and prominent leukodystrophic changes characterized by loss of myelin. There was a significant reduction of PLP mRNA–positive oligodendrocytes. These findings could be interpreted as an indicator of hypomyelination versus dysmyelination. However, there could also be a disturbed interaction between neuronal cells filled with storage material and glial cells. Gangliosides, which are anchored in the outer leaflet of the plasma membrane and represent an important membrane constituent, could play a key role in cell–cell communication. Gangliosides are presumed to be important for cell recognition and adhesion, neuronal differentiation, neuronal signal transduction and neuronal outgrowth. 42 Accordingly, neurons filled with gangliosides might be prone to disturbed signal transmission between neurons and oligodendrocytes. Neuron-derived oligodendrocytic growth factors, which could be reduced in affected neurons, represent one such pathway. 19 Consequently, the reduction of oligodendrocytes might be due to a failure in differentiation of oligodendrocyte progenitors leading to myelin loss. In this context, astrocytic proliferation might represent a reactive response. The possibility of a defection oligodendrocyte differentiation has been discussed in caprine β-mannosidosis. 13,26 Abnormalities in myelin processing have also been described in dogs suffering from GM1-gangliosidosis. 23,24 Dysmyelinogenesis in GM1-gangliosidosis of ESS and PWD was characterized by loss of myelin, reduced amounts of cerebrosides and sulfatides in the gray and white matter, and increased amounts of stage-specific embryonic antigen 1 glycolipid in the gray matter. 23,24 A paucity of oligodendrocytes associated with myelin deficiency occurred also in cats 12 and Friesian cattle 17 with GM1-gangliosidosis. Myelin rarefication has also been described in two domestic shorthair cats and one Siamese cat. 16 According to the role of gangliosides for the initiation of neuronal growth processes, 40,42 the storage of gangliosides might also be important for the modulation of axonal changes resulting in trafficking errors of signal transmission. Furthermore, the loss of myelin in Alaskan Huskies was associated with Wallerian-type degeneration reminiscent of secondary demyelination. Similarly, swollen axons and demyelinated fibers have been observed in the white matter of PWD with GM1-gangliosidosis. 35
The extraneuronal lesions in these Alaskan Huskies consisted of mild to moderate cytoplasmic vacuolation in various cells, as has been described for PWD, ESS, and mixed-breed Beagles. 3,4,34,35 However, there seemed to be some differences with respect to cell types and tissues exhibiting cytoplasmic vacuolation. It is unclear whether these differences represent actual manifestations of the underlying disease or whether they are due to unrelated metabolic disarrangement. The variable staining properties and morphologic appearance of the inclusions in the CNS and non-CNS organs likely depends on the chemical composition of different storage products, including oligosaccharides or glycoproteins. 31,38
In age of onset, temporal evolution, and a lack of hepatosplenomegaly, the disorder in the Alaskan Huskies was similar to type 2 human GM1-gangliosidosis. According to clinical, pathologic, and biochemical findings, mixed-breed Beagles showed features of both the juvenile and infantile form of GM1-gangliosidosis, 34 and ESS may represent a model for the infantile form of the disease. 4 The multiple phenotypes, particularly the variable involvement of bones in the disease process, may be due to the heterocatalytic nature of the enzyme and may depend on which natural substrate activities of the β-galactosidase are affected. 16 The phenotypic differences among various breeds or among animals from the same breed suggest distinct mutations in the β-galactosidase gene or variable expression of the same genotypic alteration. The neurologic dysfunction seemed to be a consequence of neuronal storage and disturbed myelin processing.
Footnotes
Acknowledgements
G. Müller was supported by a scholarship, Gesetz zur Förderung von Nachwuchswissenschaftlern des Landes Hessen. We thank Dr. G. Steinel for drawing our attention to the diseased dogs. We are especially grateful to T. Guth for his outstanding support regarding his Alaskan Huskies. We thank A. Artelt and U. Zeller for technical and photographic assistance. We also thank Dr. S. Weisbrode (Ohio State University, Columbus, OH) for his help and advice on the bone pathology. We are indebted to Dr. K. Failing for his help with the statistical analysis.