
Abstract
Mucopolysaccharidoses are inherited metabolic disorders that result from a deficiency of lysosomal enzymes required for the catabolism of glycosaminoglycans. Lysosomal glycosaminoglycan accumulation results in cell and organ dysfunction. This study characterized the phenotype and genotype of mucopolysaccharidosis VI in a Great Dane puppy with clinical signs of stunted growth, facial dysmorphia, skeletal deformities, corneal opacities, and increased respiratory sounds. Clinical and pathologic evaluations, urine glycosaminoglycan analyses, lysosomal enzyme assays, and ARSB sequencing were performed. The urine mucopolysaccharide spot test was strongly positive predominantly due to the accumulation of dermatan sulfate. Enzyme assays in leukocytes and tissues indicated a deficiency of arylsulfatase B (ARSB) activity. Histologic examination revealed cytoplasmic vacuoles in many tissues. Analysis of the exonic ARSB DNA sequences from the affected puppy compared to the published canine genome sequence revealed a homozygous nonsense mutation (c.295C>T) in exon 1, replacing glutamine with a premature stop codon (p.Gln99*), predicting no enzyme synthesis. A polymerase chain reaction–based restriction fragment length polymorphism test was established to assist with the clinical diagnosis and breeding of Great Danes. This genotyping test revealed that the clinically healthy parents and some other relatives of the puppy were heterozygous for the mutant allele, but all 200 clinically healthy dogs screened including 15 Great Danes were homozygous for the normal allele. This ARSB mutation is the fourth identified genetic variant causing canine mucopolysaccharidosis VI. Mucopolysaccharidosis VI is the first lysosomal storage disorder described in Great Danes but does not appear to be widespread in this breed.
Keywords
Mucopolysaccharidoses are a group of inherited lysosomal storage disorders in which a specific lysosomal enzyme required for the step-wise catabolism of glycosaminoglycans (GAGs, originally called mucopolysaccharides) is deficient or dysfunctional. Cartilage, bone, skin, blood vessels, tendon, and cornea require GAGs for maintaining the structure and function of these tissues. The physiological concentration of GAGs is controlled by enzymatically regulated metabolic pathways. If the activity of any catabolic lysosomal enzyme is severely reduced, lysosomal GAG accumulation occurs in tissues, causing progressive and irreversible cellular damage that affects growth, morphology, mobility, organ function, and sometimes neurological development. 21 To complete degradation of GAGs, 11 different lysosomal enzymes are required. Deficiency of any 1 of them will result in a specific type of mucopolysaccharidosis (MPS). 16
Mucopolysaccharidosis type VI (Maroteaux-Lamy syndrome, Online Mendelian Inheritance in Man #253200) is an autosomal recessive lysosomal storage disorder caused by a deficiency of N-acetylgalactosamine 4-sulfatase (also called arylsulfatase B [ARSB], EC 3.1.6.12). This enzyme plays a critical role in the catabolism of dermatan sulfate. The typical clinical features of MPS VI include progressive abnormal cartilage and bone development leading to short stature, dysostosis multiplex, degenerative joint disease, growth retardation, physical disability, facial malformations, and corneal clouding. Despite GAG storage being systemic and commonly involving the central nervous system, patients with MPS VI usually do not have primary neurological signs but rather severe skeletal and ocular signs. 27
Currently, more than 150 ARSB gene mutations have been documented in humans with MPS VI (The Human Gene Mutation Database, http://www.hgmd.org), despite representing an orphan disorder. Furthermore, MPS VI has been described in several animal species. 14,15,17,18,22,23,32
Canine MPS I, III, VI, and VII have been previously described and share close similarities in clinical disease presentation with other species. With the exception of MPS IIIB (Sanfilippo syndrome B), which is predominantly an adult onset neuropathy, the other MPS disorders largely affect the joints, bones, and eyes. The clinical manifestations include growth retardation, facial dysmorphia, pectus excavatum, hip and other joint laxities, umbilical hernia, corneal clouding, hepatomegaly, and cardiovascular dysfunction. These dogs die or are humanely euthanized before they reach adulthood. Dogs with MPS serve as naturally occurring models to understand the disease and assess therapeutics for humans and animals. 14
Here, we report the clinicopathologic, histopathologic, biochemical, and molecular genetic characteristics of a Great Dane puppy with MPS VI and its family.
Materials and Methods
Sample Collection and Preparation
A 4-month-old male Great Dane puppy (proband) with the clinical signs of a possible inborn error of metabolism was referred to the Pediatrics, Genetics and Reproduction Clinic and PennGen Laboratory Services at the School of Veterinary Medicine, University of Pennsylvania, Philadelphia, for diagnosis and management. Urine, serum, and ethylenediaminetetraacetic acid (EDTA)–anticoagulated blood samples were collected. The proband was euthanized at 6 months of age and representative tissues (no spine and brain) were obtained by the Quality Pet Care clinic in Monticello, New York, for histopathologic and biochemical analyses. Furthermore, EDTA-anticoagulated blood or buccal swab samples from the proband’s relatives were obtained, and archived DNA samples from clinically healthy dogs, including Great Danes, were genotyped. The investigations were approved by the Institutional Animal Care and Use Committee at the University of Pennsylvania.
Leukocytes were purified from fresh EDTA-anticoagulated blood using a dextran density gradient separation technique. 12 Fresh liver and spleen samples (20 mg) were homogenized in 1.0 ml of 0.9% saline with 0.2% Triton X-100, and the extracts of the lysates were separated by centrifugation. Protein concentrations were determined by the Bradford-dye protein assay kit (Bio-Rad Laboratories, Hercules, CA, USA). Genomic DNA was extracted from EDTA blood and buccal swabs using the Qiagen DNA purification kit (Qiagen, Valencia, CA, USA). These samples were stored at –80°C for further study.
Representative tissues were fixed in 10% neutral buffered formalin, routinely processed, embedded in paraffin, and sectioned at 5 μm. Histologic sections were stained with hematoxylin and eosin for routine examination, periodic acid–Schiff to detect polysaccharides including GAGs, and toluidine blue to specifically detect GAG storage, as previously described. 26
Urinary GAG Analysis
The toluidine blue mucopolysaccharide spot test 2 was performed on the urine from the proband and a normal juvenile dog. Urine (5 µl) was applied on filter paper (Whatman™ 3MM), stained with 0.2% toluidine blue O for 45 seconds, and then destained with 0.6% acetic acid. The occurrence of a purple (metachromatic) spot indicated the presence of GAGs in urine. The type of GAG was analyzed by thin layer chromatography (Biocontrol, Ingelheim, Germany).
Lysosomal Enzyme Assays
Using 4-methylumbelliferyl (4-MU)–based substrates, the activities of 3 lysosomal enzymes, ARSB (for MPS VI), α-L-iduronidase (EC 3.2.1.76 for MPS I), and β-D-glucuronidase (EC 3.2.1.31 for MPS VII), were examined in leukocytes, liver, and spleen of the proband and a healthy control juvenile dog. Activities of α-L-iduronidase and β-D-glucuronidase were assayed by previously established fluorescence methods. 11 The ARSB activity was measured by a slightly modified protocol 7 : 10 µl of tissue extract containing 10 µg protein was incubated with 5.0 mM fluorescent substrate 4-MU sulfate potassium salt (4-MUS; Glycosynth, Warrington, UK) prepared with acetate buffer (50 mM, pH 5.6) containing 3.0 mM lead acetate and 0.3 mM silver nitrate (both from Sigma-Aldrich, St Louis, MO, USA) in a final volume of 200 µl for 2 hours at 37°C. The reaction was terminated with 2 ml of stopping buffer (0.32 M glycine, 0.2 M sodium carbonate, and 1 mM EDTA, pH 10). The substrate fluorescence signal was measured with a fluorometer (VersaFluor™; Bio-Rad Laboratories) at an excitation and emission wavelength of 365 nm and 450 nm, respectively. The results of the specific enzyme activities were calculated from a standard curve of 4-MU and expressed as nM of 4-MU released per mg of protein per hour.
ARSB Gene Sequencing
The 8 exons of the ARSB gene were sequenced from genomic DNA of the proband, its sire, and an unrelated healthy dog using previously established hot-start polymerase chain reaction (PCR) and automated Sanger sequencing techniques. 28 The PCR primers surrounding the exons and conditions are listed in Supplemental Table 1. The sequencing results were analyzed using Lasergene software (DNASTAR Inc., Madison, WI, USA) and compared to the published canine genome sequence (NCBI Genbank, Gene ID: 610364, RefSeq #NM_001048133.1, http://www.ncbi.nlm.gov) and Ensembl: Transcript ID ENSCAFT00000014585.3, CanFam3.1 canine reference genome assembly (http://www.ensembl.org) as well as to the sequencing results of the sire and the control dog.
Lysosomal Enzyme Activities in Leukocytes, Liver, and Spleen of the Mucopolysaccharidosis (MPS) VI Affected Dog (Proband), 1 Clinically Healthy Dog (Control), and Normal Reference Range (Reference).
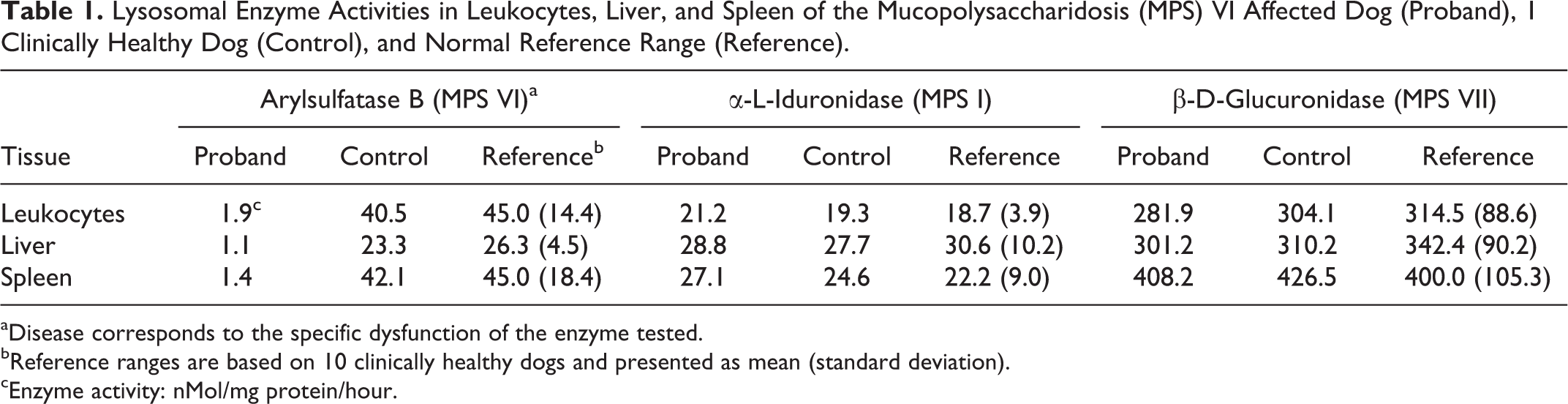
aDisease corresponds to the specific dysfunction of the enzyme tested.
bReference ranges are based on 10 clinically healthy dogs and presented as mean (standard deviation).
cEnzyme activity: nMol/mg protein/hour.
Genotyping Test
Once the ARSB mutation was discovered, a genotyping test using a restriction fragment length polymorphism (RFLP) allele specific analysis was developed for screening EDTA blood and buccal swab samples of the proband, its family members, and other samples. The DNA fragment containing the mutation was amplified using the PCR primers listed in Supplemental Table 1. The BmtI restriction endonuclease recognizes the bases 293-298 of ARSB exon 1 (5’…GCTAG∧C…3’), which is present in the mutant but not in the wild-type/normal allele. Thus, the uncut fragment consists of 318 base pairs (bp), whereas the cut fragments are 278 and 40 bp long. Ten µl of the digestion mixture containing BmtI (10 000 units/ml), 10X NEBuffer 3.1 (both were from New England Biolabs, Inc., Ipswich, MA, USA), and dH2O at a ratio of 0.5:2.0:7.5 was mixed with 10 µl of the PCR product and digested at 37°C for 2 hours. The PCR fragments were separated on a 2% agarose gel by electrophoresis and stained with ethidium bromide for visualization.
Results
Clinical Findings
A 4-month-old male Great Dane puppy was presented to the primary care clinician for its third booster vaccinations. At the time, he was also noted to have hind limb splaying with progressive weakness over the previous 2 weeks. The puppy was purchased from a breeder but no pedigree information was made available. The breeder did not recall any similar manifestations in the remainder of the litter or any of the puppies previously produced in the kennel.
The puppy was then referred to a specialist clinic where it appeared disproportionally stunted and had a body condition score of 2/9 at a weight of 16 kg, with normal temperature, heart rate, and respiratory rate and slightly increased lung and upper airway sounds. Other abnormalities noted included firm swelling of the paws, valgus deformities of both pelvic limbs, various deformities of both forelimbs, and prominent pelvic bones with bilateral swelling perianally, likely representing pressure point seromas. It was ambulatory with normal cranial nerve responses and conscious proprioception but had severe neck pain. Results of an instrumental complete blood count, chemistry screen, and urinalysis were within normal reference ranges for the patient’s age.
Radiographs revealed severe generalized skeletal abnormalities including irregularity of the vertebral endplates of the entire spine, mild soft tissue swelling of the elbows, bilateral cranial bowing of antebrachii, bowing of metacarpal bones, soft tissue swelling within the metacarpophalangeal joints, abnormal angulation of proximal femurs, and generally decreased bone density of hind limbs, with bilaterally increased intramedullary density of the mid tibial diaphysis and irregular margins of the femoral condyles. In addition, increased opacities in the larynx region were noted, likely responsible for the loud upper respiratory sounds.
Results of synovial fluid analyses from several joint taps showed turbid, light red to light orange fluid with a specific gravity of 1.017 and containing 20 000 to 50 000 red blood cells/µl and 120 to 150 mononuclear cells/µl including macrophages, synoviocytes, and small mature lymphocytes, but neither neoplastic cells nor infectious organisms were noted. Bacterial culture of the synovial fluid was negative.
The proband (Fig. 1) was referred to the Pediatrics, Genetics and Reproduction Clinic at the School of Veterinary Medicine of the University of Pennsylvania. In addition to the previously noted clinicopathologic and imaging manifestations, mild corneal opacities, facial dysmorphia, and muzzle sloping, with a prominence of the rostral ventral mandible, prognathism, and clear serous nasal discharge and laryngeal sensitivity were observed. Spinal palpation was unremarkable, but pain was elicited on dorsal neck extension.

Mucopolysaccharidosis type VI Great Dane puppy. 4 months of age. The puppy has stunted growth and swollen joints and paws.
Routine automated complete blood count, chemistry screen, and urinalysis of the proband were again within normal reference ranges. In a blood smear stained with Wright-Giemsa, granulocytes, lymphocytes, and monocytes contained moderate to abundant, fine to coarse dark purple cytoplasmic granules, suggestive of a storage disease (Fig. 2).
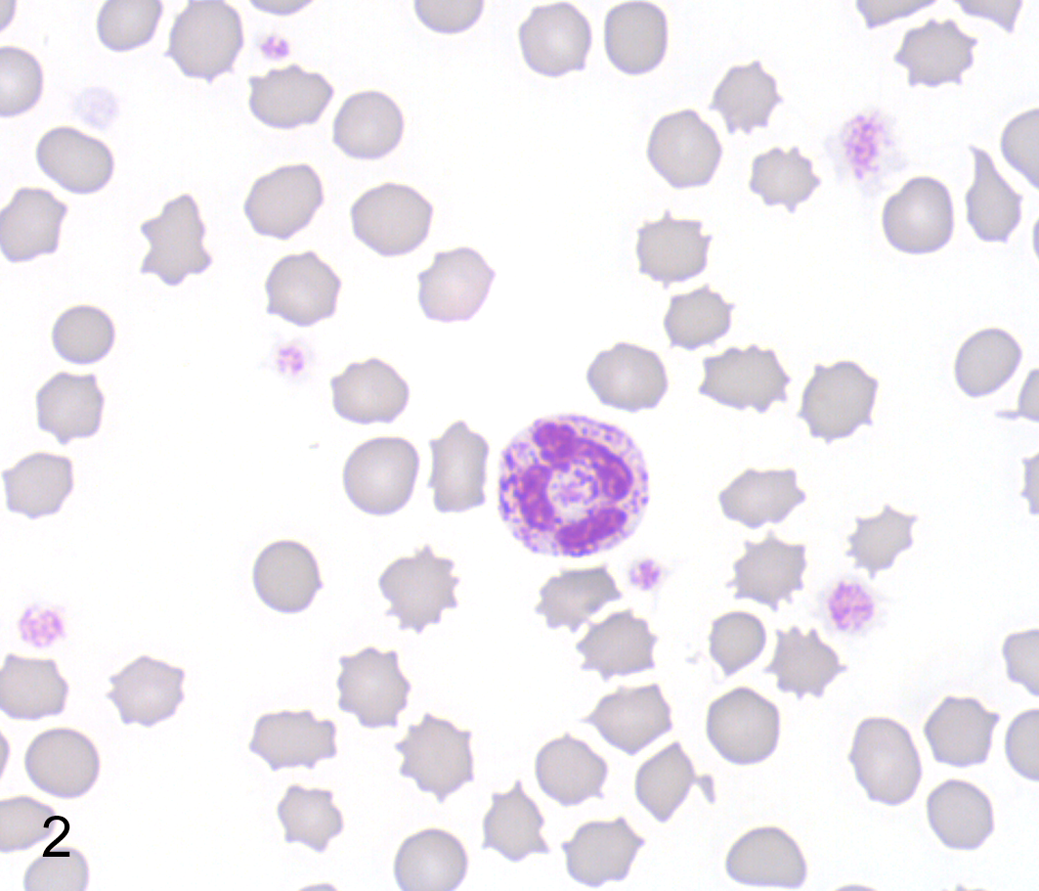
Mucopolysaccharidosis type VI, peripheral blood, Great Dane puppy. A neutrophil with many coarse metachromatic granules. Wright-Giemsa.
Histopathologic Findings
As previously described in cases of canine MPS VI, cytoplasmic vacuoles (ie, storage material) were observed within macrophages, fibroblasts, and chondrocytes throughout the body. 18,22,23 The cytoplasmic vacuolation was most notable within the corneal and uveal stroma, heart valves, Kupffer cells of liver, macrophages of spleen, laryngeal cartilage, tracheal cartilage, and cartilage of the costochondral junction of the rib. The vacuoles stained moderately to strongly positive with a periodic acid–Schiff stain and metachromatically with the toluidine blue stain. Along with the smooth muscle and fibroblast storage material, the aorta also exhibited intimal thickening due to proliferating fibroblasts and chondroid metaplasia (Fig. 3). The chondrocytes within this metaplastic focus contained similar cytoplasmic vacuoles (Fig. 4). The trachea (Fig. 5) and ribs contained severe cytoplasmic vacuolation of chondrocytes and fibroblasts with secondary chondroid degeneration and malformation.
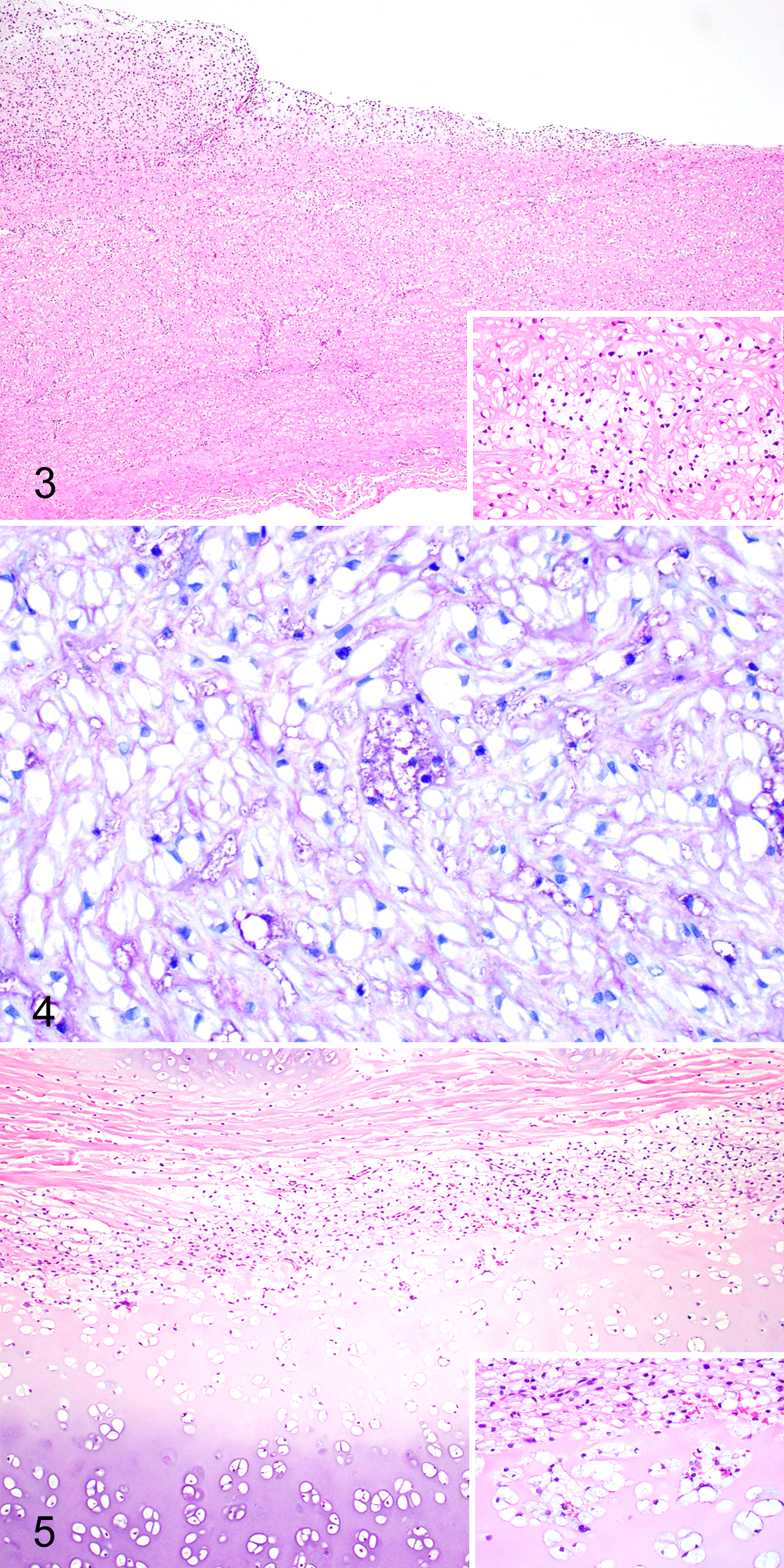
Biochemical Analyses
Urine GAG analysis
The mucopolysaccharide spot test on urine revealed a strongly positive result (Fig. 6), and chromatographic GAG separation indicated a large amount of dermatan sulfate (data not shown).
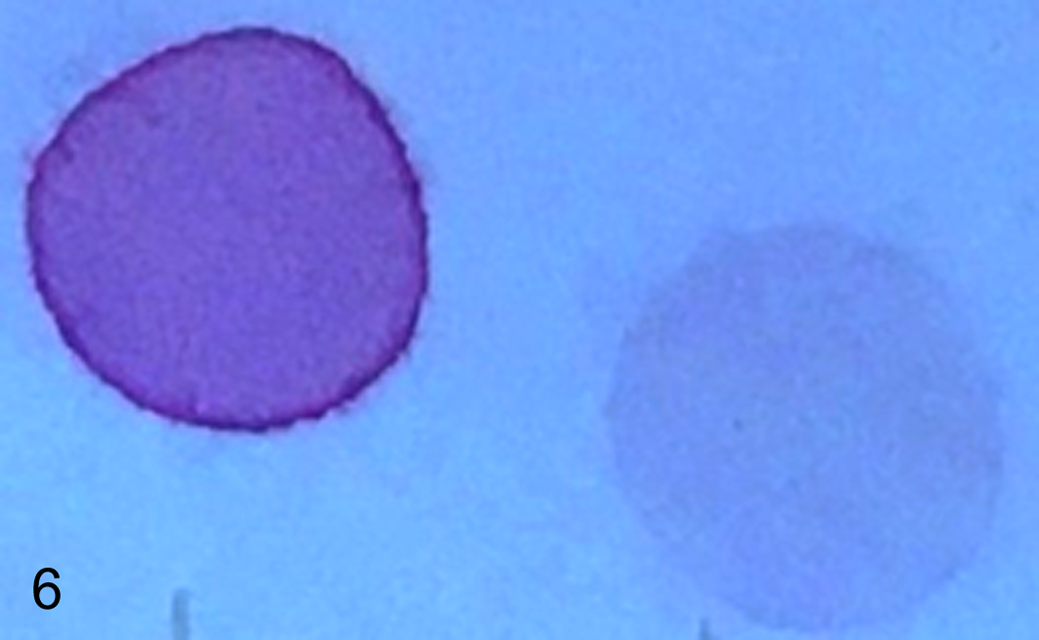
Strongly positive urinary mucopolysaccharide spot test of the suspected mucopolysaccharidosis affected puppy (left) was compared to a negative result from a normal juvenile control dog (right).
Lysosomal enzyme assays
The proband’s ARSB activities in leukocytes, liver, and spleen were severely reduced compared to the control (< 5% of control). In contrast, the enzymatic activities of α-L-iduronidase and β-D-glucuronidase were in the normal ranges (Table 1).
Gene Sequence Analysis
The canine ARSB is located on canine chromosome 3 and consists of 8 exons with the transcript length of 5785 bp coding for 535 amino acids. The genomic DNA of the proband was examined for the mutations documented in 3 other dog breeds 1,18 but was found to be homozygous normal for each of them. Exonic sequencing of the proband’s DNA revealed a single homozygous nonsense mutation at position 295 in exon 1 of ARSB, where a cytosine was exchanged by a thymine (c.295C>T), resulting in the replacement of glutamine (NCBI RefSeq #NP_001041598.1) with a premature stop codon at amino acid position 99 (p.Gln99*). Sequence analyses of DNA showed that the clinically healthy sire was heterozygous for this variant, whereas an unrelated dog was homozygous normal (Fig. 7).
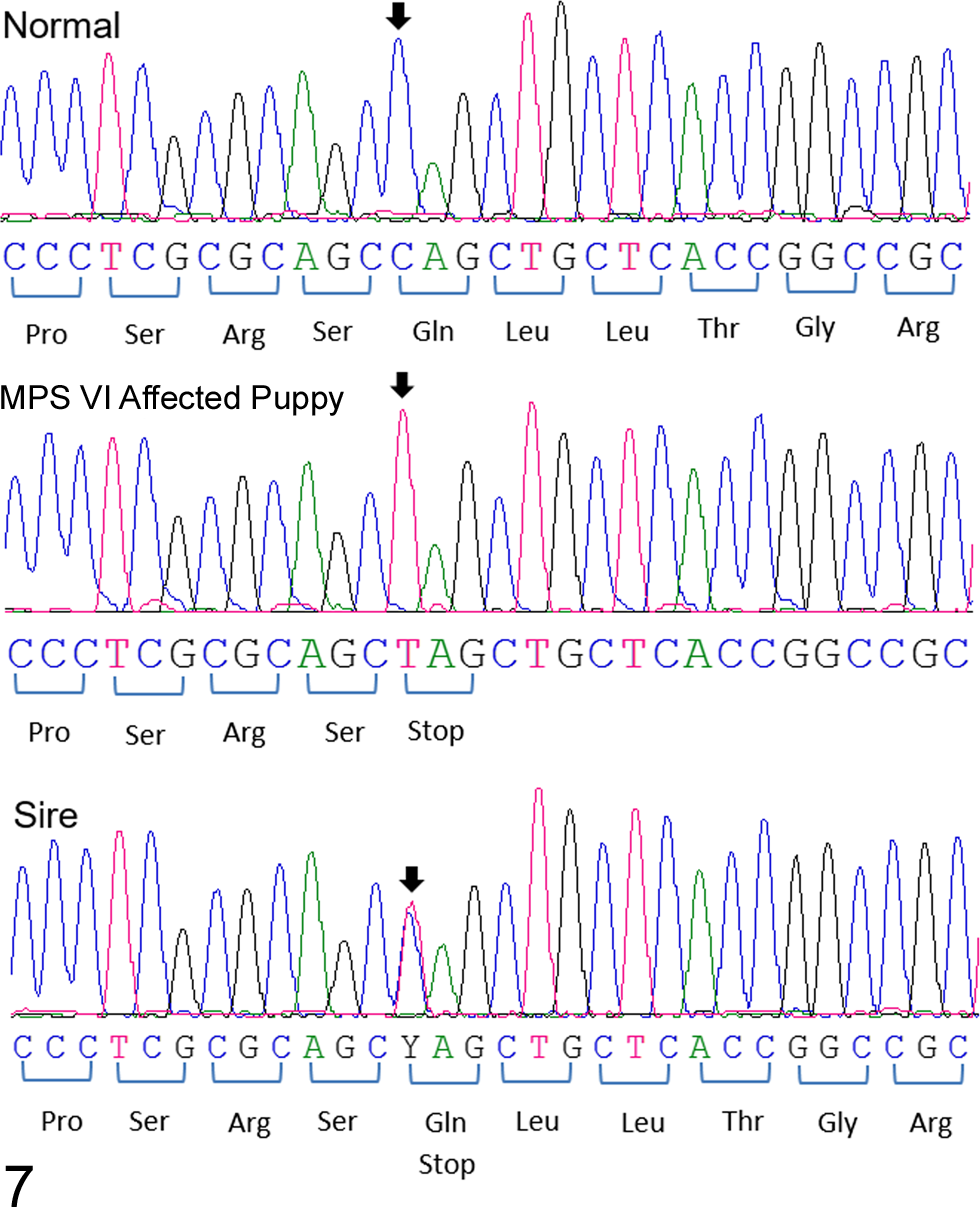
DNA sequences of a region of exon 1 of ARSB from a normal dog, the Great Dane puppy with mucopolysaccharidosis VI, and its sire.
Comparison of the sequencing results also revealed 2 synonymous silent variants in ARSB. One is an exchange of a thymine for a cytosine (c.771T>C) in exon 4. This variant was detected in the sequence (both heterozygous) of the sire and the clinically healthy dog but not the proband. Another variant was the replacement of an adenine with a guanine in exon 8 (c.1599A>G), which was observed in the proband, its sire, and the control dog. Both of the variants had been previously documented (variants rs23569849 and rs852721558, respectively, Ensembl CanFam3.1 canine reference genome assembly, Gene ID: ENSCAFG00000009168, http://www.ensembl.org). No other DNA sequence differences were observed in the coding regions between proband and the published ARSB genomic sequence and sequences obtained from the sire and normal control.
Genotyping Test Results of the Great Dane Family and Other Dogs
The PCR-RFLP–based genotyping test, which utilizes the BmtI restriction enzyme, cuts the mutant but not normal/wild-type allele, thereby clearly differentiating the 2 alleles by size on a gel. The PCR-RFLP results showed that the proband was homozygous for the C>T mutation, whereas its clinically healthy sire, dam, and grand-dam on the maternal side were heterozygous for the normal/wild-type and mutant alleles. In addition, 1 newborn puppy from a litter of 9 puppies, bred by the proband’s dam with a different sire, was also heterozygous whereas the others were homozygous normal. All PCR-RFLP results of heterozygous individuals by PCR-RFLP were confirmed by DNA sequencing showing a double peak of C and T on visual examination at position 295. All others, including 15 unrelated healthy Great Danes, were homozygous for the wild-type allele. The gel and corresponding family pedigree illustrate that the mode of inheritance of MPS VI in Great Danes follows an autosomal recessive pattern (Fig. 8).
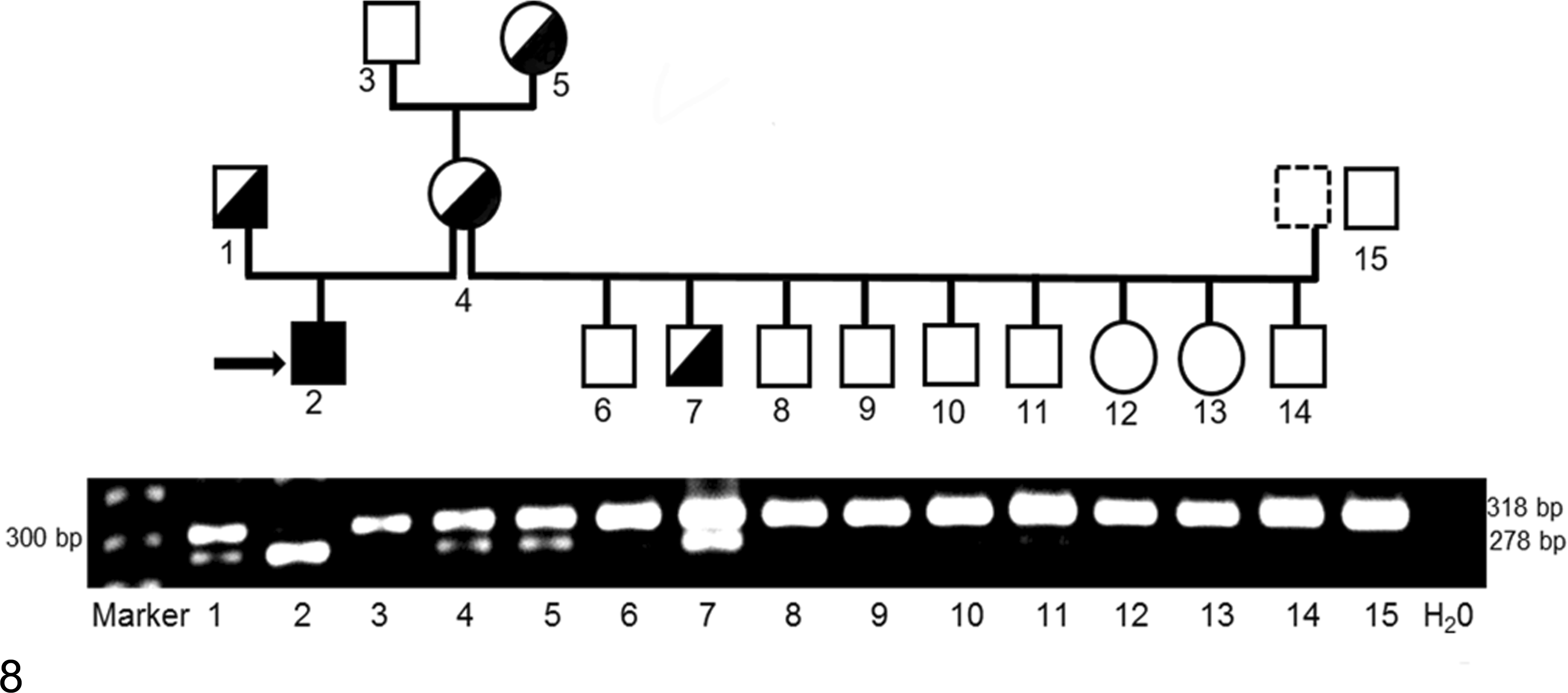
Allele-specific genotyping results of Great Dane family with mucopolysaccharidosis (MPS) VI by polymerase chain reaction (PCR)-restriction fragment length polymorphism. The intact 318 base pairs (bp) PCR fragment indicates the normal/wild-type allele. The c.295C>T nonsense mutant allele creates a BmtI restriction site, resulting in product cleavage into 278 bp and 40 bp fragments (the 40 bp fragment is too small to be visualized). A 278 bp fragment (homozygous for the C>T mutation allele) corresponds to the MPS VI affected puppy (#2, indicated by an arrow), whereas the sire (#1), dam (#4), and grand-dam (#5) on the maternal side and another puppy (#7) show both the 318 and 278 fragments, indicative of heterozygosity. Others, including an unrelated normal Great Dane (#15), display only the 318 bp fragment (homozygous normal). The numbers labeled on the gel correspond to those in the pedigree. The family pedigree with associated genotypes indicates an autosomal recessive mode of inheritance for MPS VI in Great Dane dogs. Squares and circles represent males and females, respectively. Open (white) and filled (black) symbols designate homozygous normal and mutant, respectively. Half-filled symbols represent heterozygosity.
Discussion
The storage disorder MPS VI has been described in both humans and animals including rats, 32 cats (Online Mendelian Inheritance in Animals #000666-9685), 15,17 and dogs (#000666-9615). 1,14,18,22,23 Canine MPS VI was reported in the Miniature Pinscher, 1,22 Miniature Schnauzer, 1,23 Toy Poodle, 18 Welsh Corgi, 14 and Chesapeake Bay Retriever. 14 Molecular studies of canine MPS VI have, thus far, revealed 2 deletions and 1 missense mutation in ARSB. 1,18 Furthermore, dogs and cats with MPS VI have been used as large animal models to study the pathophysiology and therapeutic safety and efficacy of novel therapies that include enzyme replacement therapy, bone marrow transplantation, and gene therapy. 5,8,24,29
In this study, the Great Dane puppy had early onset of stunted growth, skeletal deformities, corneal opacities, and abdominal respiratory effort, along with cytoplasmic granules within blood leukocytes. Histologically, cytoplasmic vacuoles consistent with storage material were observed in the fibroblasts, chondrocytes, and macrophages throughout the body. These findings, and a strongly positive urine mucopolysaccharide spot test, led to the strong suspicion of MPS.
The extreme GAG excretion shown by the simple mucopolysaccharide spot test of the proband’s urine was found to be due to excessive dermatan sulfate accumulation. The predominant storage material dermatan sulfate is a polymer with a disaccharide repeating sequence of [IdoA-GalNAc-4S]n, which contains a sulfate group that is cleaved by ARSB during the degradation process. Although chondroitin sulfate, another major GAG, is also a natural substrate for ARSB, chondroitin sulfate accumulation is not commonly observed in MPS VI patients due to its catabolism through an alternate pathway. During chondroitin sulfate degradation, the polymer chain is first clipped by the enzymes hyaluronidase and β-glucuronidase, resulting in the production of oligosaccharide fragments that are not recognized as chondroitin sulfate storage and, therefore, not degraded by ARSB. 27 Because of this alternate chondroitin sulfate catabolic pathway, cellular accumulation of dermatan sulfate is the most distinct biochemical marker for ARSB dysfunction and is indicative of MPS VI.
The diagnosis of MPS VI is generally accomplished by determining a lack of ARSB activity unless the molecular defect has already been determined for the breed. Body tissues and leukocytes contain other kinds of sulfatases than ARSB, such as arylsulfatase A (ARSA). With similar specificity for artificial substrates (4-MUS) and a similarly acidic optimal pH, ARSA may affect the ARSB assay. Chang et al 7 reported that ARSA but not ARSB was sensitive to cationic inhibitors such as Pb2+ and Ag+. To specifically measure ARSB activity, Pb2+ and Ag+ are added to the 4-MUS substrate buffer to favor and specifically observe ARSB activity in the presence of other sulfatases. The reaction-stop buffer must contain EDTA to prevent the precipitation of Pb2+ carbonate (PbCO3), which may disrupt the fluorometric readings. Clinical signs due to enzyme deficiencies are generally observed when there is severe to complete deficiency and residual activities of less than 10% are thought to be cross-reactivities by related enzymes. 3 The proband’s ARSB activity in leukocyte, liver, and spleen was less than 5% of the normal control activity. This residual activity was likely nonspecific ARSB independent as the molecular studies predict a complete lack of ARSB protein (see below). Furthermore, the activity measured in vitro may not reflect the in vivo situation, as the conditions may not be optimal regarding substrate and acidic pH. A specific ARSB deficiency should also be differentiated from the multiple sulfatase deficiency, which is a progressive neurodegenerative lysosomal storage disorder that mainly affects the nervous system. 6 Since no neurologic abnormalities were noted in the proband, this disorder could be dismissed based upon clinical signs.
It is well established that many mutations in dogs are very breed specific. Indeed, this Great Dane puppy with MPS VI did not have 1 of the known mutations in the ARSB. 1,18 However, exonic sequencing of ARSB revealed a homozygous nonsense point mutation (c.295C>T, p.Gln99*). This premature stop codon in exon 1 results in severe truncation of the protein at the equivalent of 98 amino acids instead of the wild-type length of 535 amino acids. However, degradation by nonsense-mediated mRNA decay predicts no ARSB protein synthesis. The nonsense-mediated mRNA decay, a translation-coupled pathway employed by mammalian cells, allows the body to degrade truncated mRNAs before they are translated into nonfunctional polypeptides. 25 The null mutation explains the severe phenotype of MPS VI in this Great Dane puppy. An identical mutation was previously found in human patients with severe MPS VI phenotype (c.289C>T, p.Gln97*) 19 but not in any animal models of MPS VI. However, Toy Poodles 18 and Miniature Schnauzers 1,23 also have disease-causing mutations in exon 1 of ARSB and have a similar phenotype, whereas Miniature Pinschers 1,22 with a missense mutation in exon 5 seem to have a milder phenotype.
Whereas MPS VI in Miniature Schnauzers and Miniature Pinschers has been encountered in several dogs over the past decade, the Great Dane puppy of this report and the Toy Poodle from New Zealand appear to be isolated cases. The breeder has stopped breeding with the sire and has tested all offspring from an outcross of the dam with neutering of the single carrier produced. In a very limited survey, no other Great Danes were found to be carriers. Therefore, screening of Great Danes is not generally recommended unless other affected dogs are recognized.
Siamese cats were first recognized to have MPS VI. 15,17 It is interesting that 1 of the mutations in Siamese cats (c.1427T>C, p.L476P) 31 shows a clinically severe phenotype, whereas the other (c.1558G>A, p.D520N) 30 results in a mild phenotype. Cats heterozygous for both mutations also show a mild phenotype (LA76P/D520N compound heterozygotes). 9
Currently, there is no effective treatment or cure once clinical signs of this MPS VI disorder have developed in humans and animals, and the disease is progressive and fatal. Because MPS VI affects multiple body systems, management of a diverse spectrum of disease manifestations is a common and important part of providing supportive care to patients. Clinical management may include use of adaptive or supportive devices, physical and occupational therapy, symptom-based medications, surgical interventions, and therapies to provide the deficient enzyme. 27 A more complete description of the affected systems with additional disease management information for humans can be found in the Management Guidelines for Mucopolysaccharidosis VI by Giugliani et al. 13
Experimentally, bone marrow transplantation, enzyme replacement, and gene therapy in cats 5,8,24,29 and humans 4,20 are promising as long as the treatment is instituted neonatally prior to onset of severe clinical signs. However, these therapies have been less effective than predicted for murine models. 10 Dogs and cats with naturally occurring MPS VI can be used as animal models in order to further understand the pathogenesis of MPS VI and for safety and efficacy assessment of novel therapeutic approaches in humans.
Footnotes
Supplementary material for this article is available online.
Acknowledgements
We would like to acknowledge Dr Justin Nowowiejski at VCA Animal Specialty & Emergency Center, Wappingers Falls, New York, for referring the case, collecting the specimens, and providing the radiographs and initial bloodwork, and Dr Lisa Olver at Justus Veterinary Clinic, Scott, Pennsylvania, for arranging the patient’s family members for genotyping tests.
Declaration of Conflicting Interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: Authors at the University of Pennsylvania are members of the not-for-profit PennGen Laboratories, which offer metabolic and DNA testing for mucopolysaccharidosis and other hereditary storage and other disorders.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported in part by a grant from the National Institutes of Health (NIH OD 010939).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.