
Abstract
Canine influenza virus (CIV) is a recently emergent pathogen of dogs that has caused highly contagious respiratory disease in racing Greyhounds, pet dogs, and shelter animals. Initial characterizations of CIV-induced respiratory disease suggested alveolar macrophages may be susceptible to virus infection. To investigate the role of the alveolar macrophage in the pathogenesis of CIV infection, primary alveolar macrophages were inoculated with CIV and studied from 0 to 48 hours later. Virus titers in alveolar macrophage culture supernatants increased significantly (P < .05, n = 7) from 3 to 24 hours following virus inoculation. Virus matrix gene expression was significantly increased (P < .05, n = 14) at 3, 6, and 12 hours after inoculation, peaking at 6,445-fold the level of RNA detectable immediately following inoculation. Virus-inoculated macrophages demonstrated significantly (P < .05, n = 5) decreased viability (30% trypan blue positive) by 12 hours after inoculation compared with mock-inoculated cells (5% trypan blue positive). By 12 hours after inoculation, tumor necrosis factor-alpha (TNF-α) and interleukin-10 (IL-10) mRNA levels were significantly (P < .05, n = 11) increased over those immediately following inoculation. Only TNF-α protein levels were significantly increased (P < .05, n = 11) at 12 hours after inoculation. In conclusion, the results indicate that CIV replicates in canine alveolar macrophages and induces TNF-α expression and cell death.
Keywords
Introduction
Influenza A viruses are enveloped, segmented, single-stranded, negative-sense RNA viruses belonging to the family Orthomyxoviridae.20 Canine influenza virus (CIV) was first isolated in 2004 during the investigation of an outbreak of respiratory disease in Greyhounds in Florida.10 CIV is an influenza A virus closely related genetically to contemporary H3N8 equine influenza viruses.10 Serologic evidence suggests the virus was present in the racing Greyhound population as early as 2000.11 Since the discovery of CIV, serologic and virologic evidence indicates the virus has spread within the racing Greyhound population, as well as in other nonracing dogs throughout the United States.11, 28
Clinical disease associated with CIV typically is observed within 5 days of infection and is characterized by anorexia, lethargy, fever, and serous to purulent nasal discharge as well as by nonproductive cough that may persist for several weeks.11 A subset of infected dogs develop pneumonia, which is complicated by secondary bacterial infection, and it is in these animals that mortality associated with CIV is most commonly seen.11 A more severe form of disease has been seen in a minority of infected racing Greyhounds that die peracutely with severe hemorrhagic pneumonia characterized by extensive pulmonary, mediastinal, and pleural hemorrhage.10 The disease is highly transmissible, with seroconversion rates of up to 95% in groups of exposed animals.10
Histologic evaluation of naturally and experimentally infected animals revealed neutrophilic to lymphohistiocytic tracheitis and bronchitis, with necrosis and hyperplasia of surface and glandular epithelium. Bronchopneumonia, when it occurs, is typically neutrophilic to histiocytic, with interstitial edema and minimal to extensive hemorrhage.10, 11, 28 Immunohistochemical studies on infected animals revealed influenza antigen (H3) in the cytoplasm of bronchial surface and glandular epithelium, bronchiolar epithelium, and macrophages in airway lumens and alveolar spaces.10
Alveolar macrophages play an important role in the pathogenesis of influenza virus infection. Cytokines produced by macrophages in the lung are important in establishing an innate immune response, as well as in determining the magnitude of the inflammatory response to influenza infection. Tumor necrosis factor-alpha (TNF-α), an important pro-inflammatory cytokine produced by macrophages, is pivotal in the establishment of an acute inflammatory response through its actions of activating endothelium and leukocytes and induction of increased vascular permeability, which in the lung may lead to increased pulmonary recruitment of inflammatory cells as well as the development of pulmonary edema and hemorrhage. Various influenza viruses have been shown to differentially induce the expression of TNF-α in macrophages.7, 36 The degree to which specific influenza viruses induce TNF-α expression has been shown to be strain dependent and influenced by viral genes such as HA and NA.7, 36
Interleukin-10 (IL-10) is a functionally complex cytokine with an important role in increasing susceptibility to bacterial pneumonia following influenza infection.42 IL-10 has been shown to impair host response to Streptococcus pneumoniae pneumonia in mice,41 as well as to have a role in the development of bacterial pneumonia concomitant with sepsis.39 The source of IL-10 in the pulmonary environment during and after influenza infection is currently poorly defined but may be from both T-cells and/or macrophages. It is important to know if CIV induces IL-10 in macrophages since IL-10 may predispose virus-infected dogs to develop secondary bacterial pneumonia.
Influenza viruses induce cell death in macrophages through either apoptosis or necrosis, in a strain-dependent manner.24, 36 Several studies have shown that alveolar macrophages play a role in limiting disease severity following influenza virus infection.19, 40 Furthermore, loss of alveolar macrophage viability or function after influenza infection could potentially diminish innate immunity to bacterial challenge and thus contribute to the development of secondary bacterial pneumonia.2, 27, 34
The objectives of this study were to determine if canine alveolar macrophages support replication of canine influenza virus and, if so, to determine if virus replication induces high levels of pro-inflammatory cytokines such as TNF-α as well as macrophage death.
Materials and Methods
Alveolar macrophage isolation
In preliminary studies, we assessed the suitability of dog macrophages from 3 sources for studies on CIV replication and cytokine expression: 1) macrophages lavaged from lungs of laboratory-maintained beagle dogs; 2) macrophages lavaged from euthanatized (cadaver) dogs at a local community animal shelter; and 3) macrophages derived in vitro from peripheral blood monocytes. Initial viral replication and TNF-α induction studies on lavaged macrophages from beagle dogs and cadaver dogs yielded comparable results. However, only small numbers of macrophages were recoverable by pulmonary lavage from normal beagle dogs. Macrophages derived from peripheral blood monocytes had poor survival in culture using several culture methods,4, 16, 30 and only minimal TNF-α production in response to virus inoculation was found in blood-derived macrophages compared with primary alveolar macrophages. All subsequent studies were performed on macrophages lavaged from mixed-breed male and female dogs from a local shelter, within 30 to 90 minutes after euthanasia with sodium pentobarbital. Dogs were excluded from the study if they had any of the following abnormalities: 1) gross evidence of pneumonia or pulmonary hemorrhage; 2) greater than 10% neutrophils in their pulmonary lavage fluid; or 3) positive culture of lavage fluid for mycoplasma (on SP-4 agar) or greater than 5 bacterial colonies on sheep blood agar (SBA). Approximately two thirds of the total dogs studied were positive for Dirofilaria immitis infestation based upon antemortem serologic testing and upon identification of adult nematodes in the heart at necropsy.
Alveolar macrophage isolation methods via pulmonary lavage were adapted from previously published reports.16, 37, 38 The trachea was dissected and isolated in the mid-cervical region, and an incision was made through which lavage tubing was passed to the level of the thoracic inlet. String was used to create a seal between the trachea and tubing, and 700 to 1,000 ml of cold Dulbecco's phosphate buffered saline (D-PBS) was introduced through the tubing and into the lungs under approximately 25 cm H2O pressure. Fluid was drained from the lung into a sterile flask, and a second similar wash was performed. Lavage fluid was cultured for bacteria and mycoplasma on SBA and SP-4 agar, respectively, at 37°C in 5% CO2. Tryptic soy broth was also inoculated with 0.1 to 0.2 ml lavage fluid and similarly cultured to screen for trace bacterial contamination. SBA plates and tryptic soy broth were read at 24–48 hours after inoculation. SP-4 plates were read at 5 days after inoculation. Washings were filtered through a single layer of sterile gauze and centrifuged for 10 minutes at 4°C, 500 × g. Supernatants were removed, and the cell pellets were resuspended in a small amount of D-PBS and transferred to 50-ml conical centrifuge tubes. Tubes were filled to the 50-ml mark with D-PBS, and then centrifuged for 5 minutes at 4°C, 500 × g. Cell pellets were again resuspended in 50-ml sterile D-PBS and centrifuged. The resulting cell pellet was resuspended in 10 ml of cold MEMα (Gibco, Grand Island, NY) supplemented with 10% fetal bovine serum (Sigma-Aldrich, St. Louis, MO), 100 IU/ml penicillin, 100 μg/ml streptomycin and 0.25 μg/ml actinomycin (Sigma-Aldrich). A 0.1-ml quantity of cell suspension was diluted 1:100 and 1:1,000 for nucleated cell counts (using a hemacytometer) and for manual cell differential counts (cytospin preparations, Wright-Giemsa staining), respectively.
Macrophages were purified by adhesion. The cell suspension was divided between two 150-cm2 tissue culture flasks (Corning, Lowell, MA) and diluted to 25 ml (per flask) with supplemented MEMα medium. Cells were incubated for 2–4 hours at 37°C in 5% CO2. The flasks were gently rocked at hourly intervals during incubation. Nonadherent cells were then gently resuspended by rocking the flasks, and the suspension was removed leaving adherent cells. A 15-ml quantity of warm supplemented MEMα medium was added, and the cells were incubated overnight at 37°C in 5% CO2. The following day, the cells were washed 3 to 4 times with 5–10 ml warm MEMα to remove any loosely adherent or nonadherent cells. Adherent cells were eluted from the tissue culture flasks by first washing once with warm D-PBS, followed by two 1–3 minute incubations with 37°C 10× Trypsin-EDTA solution (Sigma-Aldrich) diluted 1:10 in D-PBS, with gentle agitation of the flasks. The trypsinized cells were collected into equal volumes of supplemented MEMα medium. The resulting cell suspension was centrifuged for 5 minutes at 4°C, 500 × g, and the cell pellet was resuspended in cold medium and placed on ice until further processing. A 0.1-ml amount of the cell suspension was diluted 1:10 and 1:100 for trypan blue exclusion assay (for cell viability), cell counting, and differential counts. The cell suspension was then seeded into 24-well plates at a concentration of 5 × 105 viable macrophages per well, and incubated overnight at 37°C in 5% CO2, prior to viral inoculation studies.
Macrophage purity was assessed by cytology after Wright-Giemsa staining in each experiment and agreed well with immunocytochemical staining for CD68 (Biocare, Concord, CA). Macrophage purity was typically >85%, with remaining cells consisting almost entirely of small mononuclear cells consistent with either immature macrophages/monocytes or lymphocytes.
Virus and virus titers
Virus used in all studies was a second or third passage influenza A/Canine/FL/04 virus, propagated in MDCK cells from a stock kindly provided by Dr. Ed Dubovi (Cornell University, Ithaca, NY). Infectivity of stock and supernatant in macrophage cultures was assessed by plaque assay, adapted from previous reports14, 35 as cytopathic effect on confluent cultures of MDCK cells with a 0.5% agarose overlay.
Macrophage Inoculation
Alveolar macrophages were inoculated with virus diluted in 1 ml cold MEMα at a multiplicity of infection (MOI) of 2 unless otherwise noted. Cells were incubated with virus for 1 hour at 37°C in 5% CO2. Mock-inoculated cells were incubated with MDCK cell lysate prepared as per virus stock without seed virus added. The MDCK lysate was diluted identically to the virus inoculum in cold MEMα. At the end of the 1-hour inoculation period, the inoculum was removed and replaced with 1 ml warm supplemented MEMα. The time point referred to as 0 hour in all graphs is thus 1 hour after virus or mock inoculation.
In a second set of experiments to determine whether there was persistence of virus inoculum that was being detected at the 3-hour time point, macrophages were inoculated at MOI of 0.1 and 0.01. For the MOI of 0.1 experiment, 2 wells, in separate 24-well plates, were seeded with 2 × 106 macrophages. Prior to inoculation, culture medium was removed from one of the wells, and the cells were frozen at −80°C for 30 minutes. Each of the wells was inoculated with 2 × 105 plaque-forming units (PFU) of third passage Influenza A/Canine/FL/04 (MOI = 0.1) suspended in 0.5 ml MEMα, and incubated at 37°C for 1 hour. The inoculum was removed and placed in a centrifuge tube. The wells were washed once with 2 ml of warm MEMα. The wash was collected and added to the centrifuge tube containing the inoculum. Half a milliliter of warm fresh supplemented medium was added to the wells. The combined wash and inoculum was centrifuged for 5 minutes at 500 × g to collect any suspended macrophages. The cell pellet was resuspended in 5 ml of warm MEMα and centrifuged for 5 minutes. The resulting cell pellet was then resuspended in 0.5 ml warm supplemented medium and returned to the well from which it originated. At 3, 12, and 24 hours after the inoculation period, 0.5 ml of culture medium was collected and stored at −70°C, and 0.5 ml of warm fresh supplemented medium was replaced into wells. Titers were measured by standard infectious plaque assay. A third well was tested at MOI of 0.01 in a similar manner except the well with viable macrophages was inoculated with 2 × 104 PFU of canine influenza virus.
Real-time reverse transcriptase–polymerase chain reaction (RT-PCR)
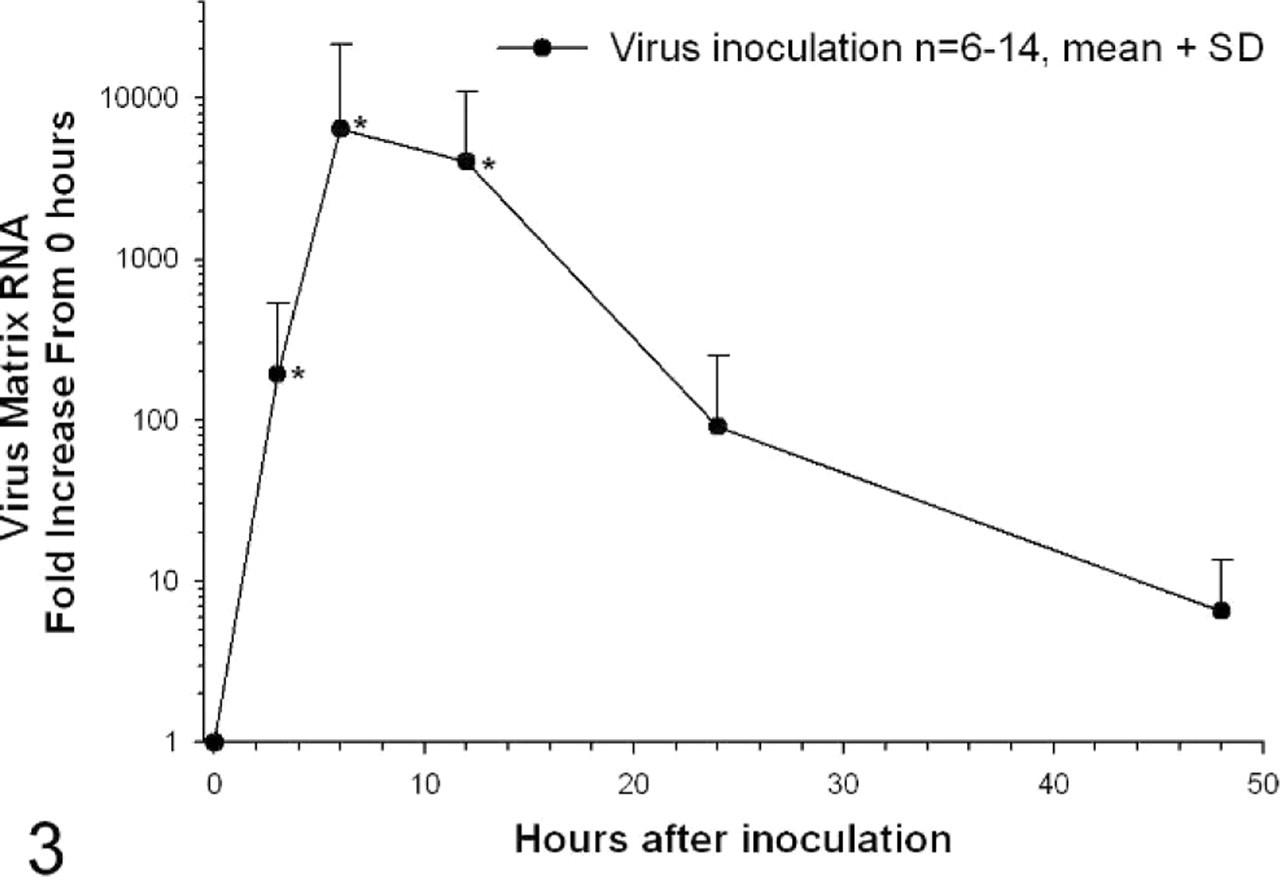
Total RNA was isolated from inoculated and mock-inoculated macrophages at the specified time points using the RNeasy Mini Kit (Qiagen, Valencia, CA). Supernatants were removed from macrophage cultures, centrifuged at 10,000 rpm in a microfuge for 5 minutes, and cell pellet collected. Lysate buffer was added to adherent cells in wells, and then transferred to cell pellet from the supernatant. Lysates were stored at −70°C until processed further as per manufacturer's instructions. Total RNA was measured by optical density at 260 nm. cDNA was synthesized using the Advantage RT for PCR kit (Clontech, Mountain View, CA). Viral gene, G3PDH and cytokine mRNA, was quantified by real-time PCR using the DNA engine Opticon II system (MJ Research/Bio-Rad Laboratories, Hercules, CA) at conditions of 95°C for 10 minutes, 95°C for 15 seconds, and 60°C for 1 minute (40 cycles). Primer and probe sequences for canine G3PDH, TNF-α, and IL-10 were obtained from the literature, and are listed in Table 1. Virus matrix gene primer sequences were kindly provided by R. Donis (Centers for Disease Control, Atlanta, GA). Virus probe sequences were generated using Primer Express Software for Real Time PCR, version 3.0 (Applied Biosystems, Foster City, CA) from the NCBI influenza virus resource. Viral matrix gene and cytokine mRNA levels were normalized to G3PDH mRNA expression. As a positive control for cytokine expression, alveolar macrophages were incubated with lipopolysaccharide (LPS) at concentrations ranging from 1 to 1,000 ng/ml culture supernatant (n = 7). Maximal TNF-α mRNA expression occurred at 6 hours after inoculation, averaging 32-fold the level of expression in mock-inoculated macrophages. Maximal IL-10 mRNA expression also occurred at 6 hours after inoculation, averaging fivefold the level of expression in mock-inoculated cells. As a negative control, alveolar macrophages were incubated with ultraviolet (UV)-inactivated virus (n = 3). At 12 hours after inoculation, macrophages incubated with inactivated virus averaged 1.2-fold the level of TNF-α expression, and 0.9-fold the level of IL-10 expression in mock-inoculated cells.
Primer and probe sequences.
Cytokine protein quantification
Canine TNF-α and IL-10 were quantified by enzyme-linked immunosorbent assay (ELISA) (R&D Systems, Minneapolis, MN). Supernatants were collected from macrophage cultures at the specified times after the inoculation period and stored at −70°C until further processing as per the manufacturer' instructions. As a positive control, alveolar macrophages were incubated with LPS at concentrations ranging from 1 to 1,000 ng/ml (n = 7). Average TNF-α protein levels peaked at 368 pg/ml supernatant by 6 hours after LPS exposure. Average IL-10 protein levels peaked at 226 pg/ml supernatant by 12 hours after LPS exposure.
Macrophage viability
To assess virus effects on viability, alveolar macrophages were inoculated as described above and cultured in 0.5 ml supplemented MEMα medium. At the specified time, the culture medium was collected. To remove adherent macrophages, 0.5 ml of 1× trypsin/EDTA was added to the well and repeatedly resuspended over the cells. The resulting cell suspension was added to the medium, and cell counts and trypan blue exclusion assay were performed using a hemacytometer.
H3 antigen staining in alveolar macrophages
Alveolar macrophages were inoculated with virus or mock inoculated at an MOI of 4 in order to assure that a large number of macrophages being sampled would be successfully infected. We were uncertain of the efficiency of infection. Cells were incubated with inoculum for 1 hour. The inoculum was removed and replaced with fresh supplemented MEMα, and further incubated at 37°C in 5% CO2 for 12 hours. The medium was collected, and adherent cells were trypsinized and added to the collected medium. Cytospin preparations were then made on glass slides, and the slides were fixed in 10% neutral buffered formalin for 1 hour. Air dried slides were stained for H3 influenza A antigen using an H3 mouse anti-influenza A monoclonal antibody (Chemicon-Millipore, Temecula, CA). Bound antibody was detected using a STAT-Q 3-step peroxidase staining system (Innovex Biosciences, Richmond, CA), and slides were counterstained with hematoxylin.
Results
Virus replication
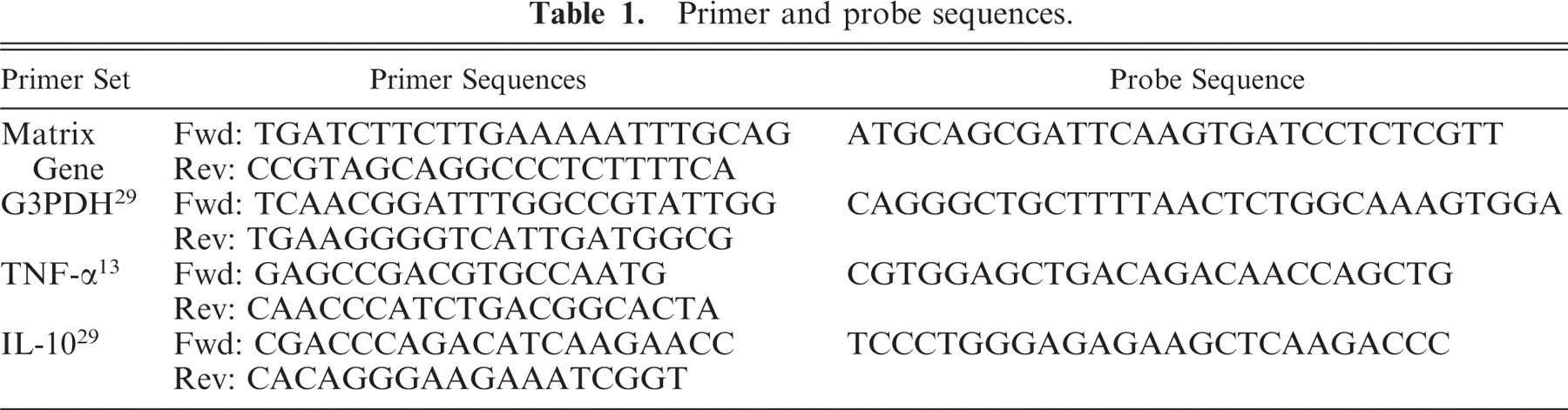
Culture supernatants from 7 experiments were titrated by plaque assay for infectious virus. Average virus titer in culture supernatant from 7 experiments increased 15-fold by 24 hours after the 1-hour inoculation period (time 0 on all graphs), and thereafter declined (Fig. 1). The 3-, 6-, 12-, and 24-hour titers were significantly (P < .05) increased over the average titer immediately after inoculation. By comparison, titers from virus incubated with freeze-thaw killed macrophages rapidly declined to undetectable levels by 6 hours.
Infectious virus titers in alveolar macrophage culture supernatant. Supernatant from alveolar macrophage cultures inoculated with canine influenza virus was titrated by plaque assay at the times indicated after the inoculation period. Virus titer following inoculation of freeze-thaw killed macrophages was undetectable at 6 hours after inoculation. PFU = plaque-forming units; n = 7 for 0 to 12 hours after inoculation, n = 5 for 24 and 48 hours after inoculation; ∗ = significantly different to 0 hour titer, P < .05, analysis of variance (ANOVA), Student-Newman-Keuls method of pairwise multiple comparisons.
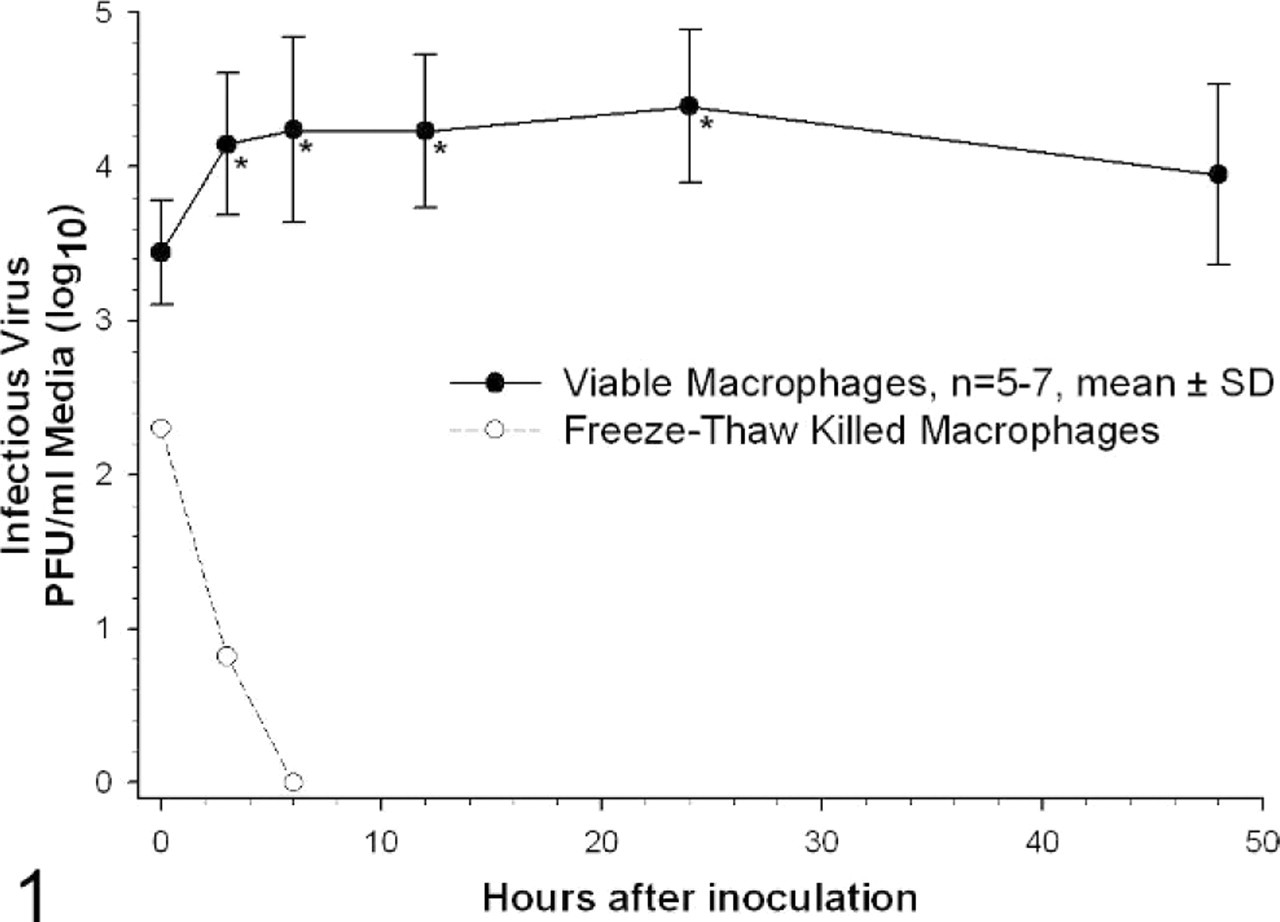
Because increased titers of virus were noted in wells with virus-inoculated viable cells as early as 3 hours, it was possible that the virus initially bound to viable cells and then was subsequently released into culture medium at early time points. We did additional studies at multiplicities of infection of 0.1 and 0.01 (Fig. 2). Productive virus replication was not detectable at MOI of 0.01. Data at MOI of 0.1 are consistent with there being initial binding of inoculum virus to viable cells and subsequent release of virus into medium at 3 hours. Free virus appears to degrade rapidly under the culture conditions without viable cells. Inoculated virus binding and then release into medium cannot account for the steady-state level of virus assayed at 12 and 24 hours after inoculation.
Infectious virus titers in viable and freeze-thaw killed alveolar macrophage culture supernatant. Supernatant from 2 × 106 alveolar macrophage cultures inoculated with canine influenza virus at a multiplicity of infection of 0.1 and 0.01 was titrated by plaque assay at the times indicated after the inoculation period. PFU = plaque forming units.
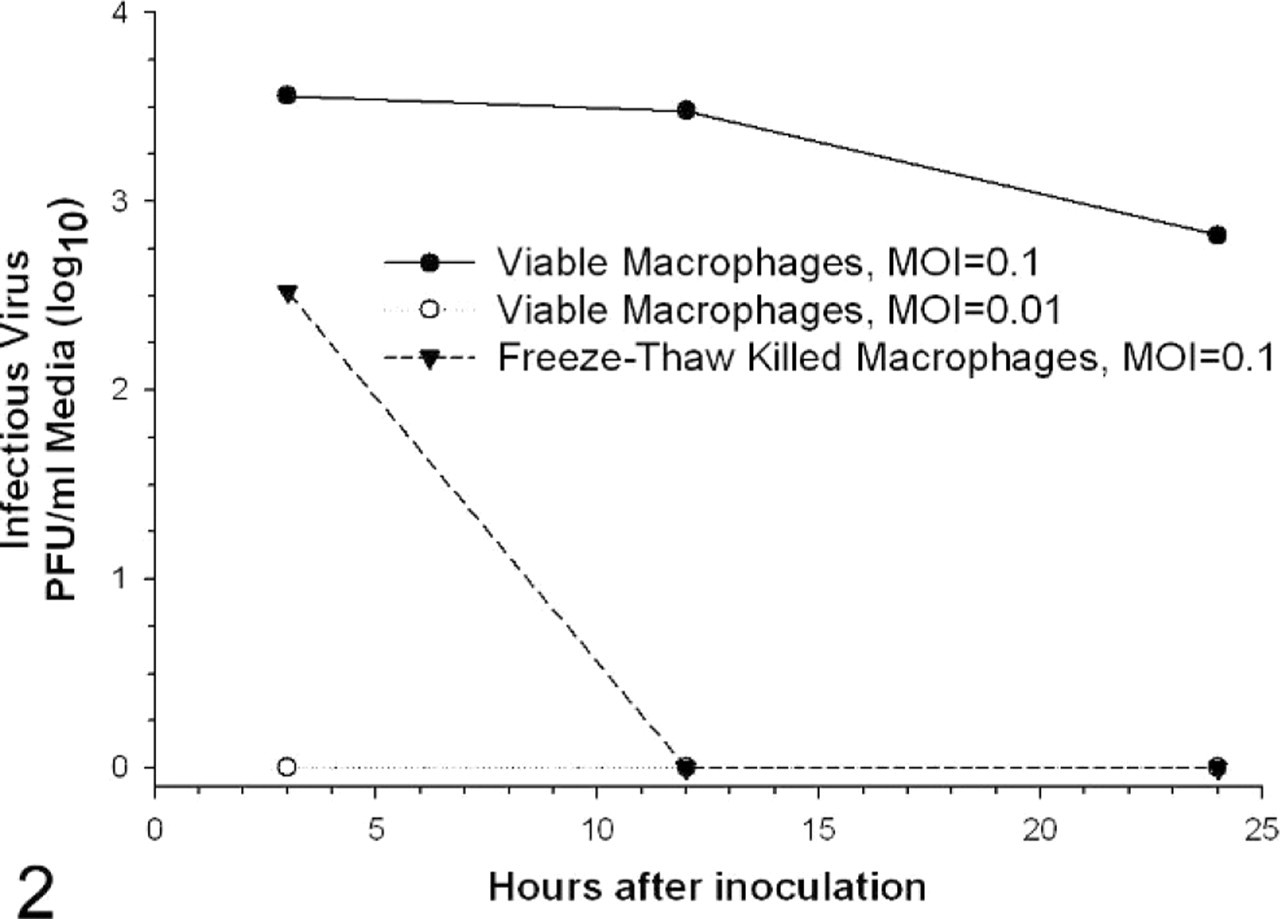
Virus matrix gene expression detected in cell lysates significantly increased beyond that immediately after inoculation at the 3-, 6-, and 12-hour time points (Fig. 3, P < .05). Matrix gene expression was maximal at 6 hours after inoculation, averaging 6,445-fold the matrix gene RNA level immediately after the inoculation period. Matrix gene expression was not detectable in mock-inoculated cells.
Virus matrix gene expression in alveolar macrophages. Viral matrix RNA levels as determined by real-time RT-PCR are expressed as fold-increase over the levels present immediately after the inoculation period. n = 14 for 0 and 12 hours, n = 12 for 6 hours, n = 11 for 3 hours, n = 8 for 24 hours, n = 6 for 48 hours; ∗ = significantly different from 0-hour expression, P < .05, Kruskal-Wallis one-way ANOVA, Dunn's Method of pairwise multiple comparisons.
Influenza A H3 antigen was evident using immunocytochemistry in virus-inoculated cells. Approximately 70% of macrophages inoculated with virus (MOI = 4) showed diffuse or peripheral cytoplasmic staining for H3 antigen (Fig. 4) at 12 hours after inoculation. Mock-inoculated cells uniformly stained negatively for H3 antigen at 12 hours.
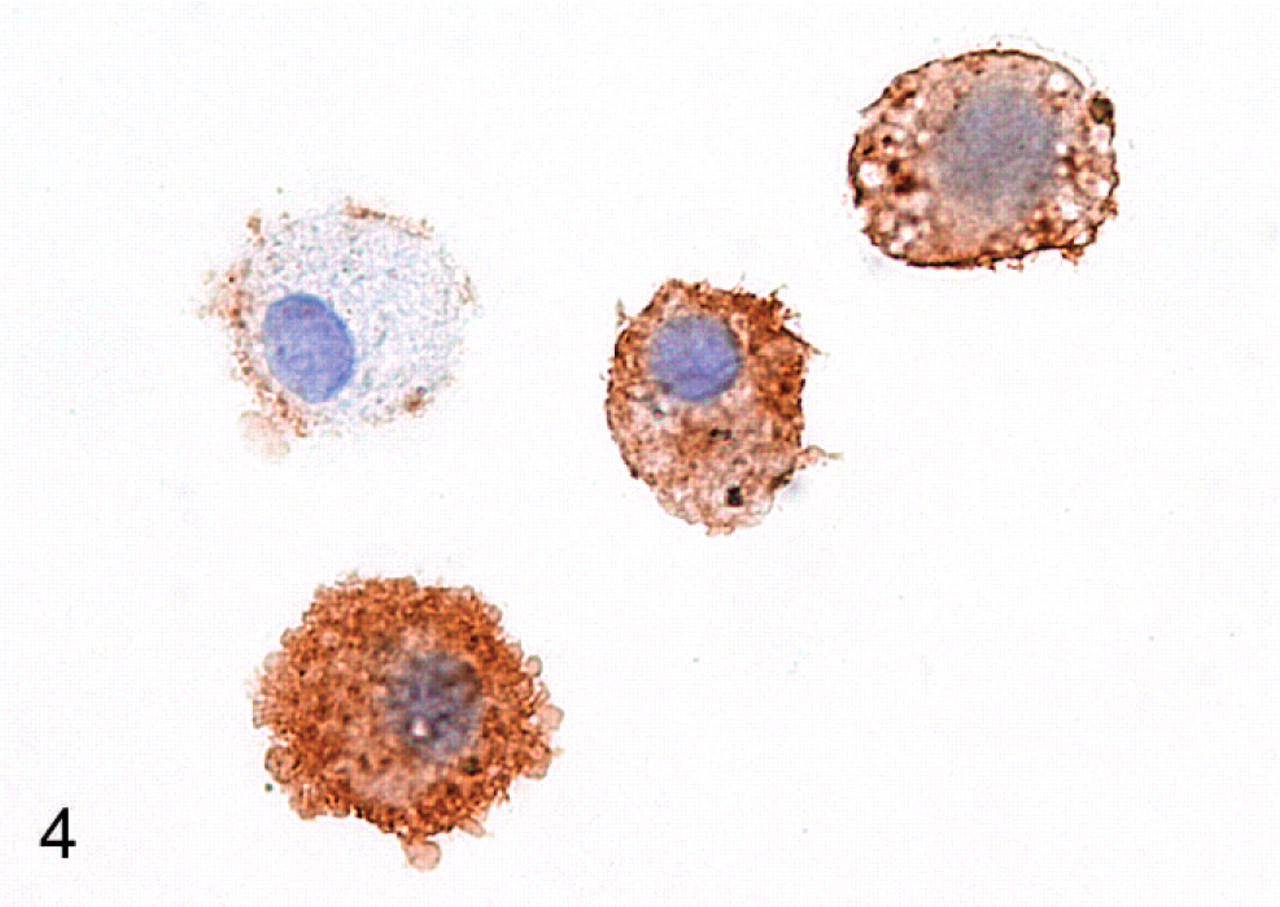
Canine alveolar macrophages. Cultured alveolar macrophages are immunocytochemically positive (brown) for H3 hemagglutinin protein. Immunoperoxidase with hematoxylin counterstain.
Loss of macrophage viability
Virus inoculation resulted in decreased macrophage viability by 12 hours after inoculation as measured by trypan blue exclusion assay (Fig. 5, P < .05). Mock-inoculated cells averaged greater than 95% trypan blue negative at all time points, whereas virus-inoculated cells averaged less than 70% trypan blue negative from 12 hours after inoculation onwards. Cell counts of trypan blue negative cells were significantly less (P < .05) in virus-inoculated cells compared with mock-inoculated cells at 24 hours after inoculation. Viable cell counts from virus-inoculated wells averaged 135,475 cells (27% of the original macrophages seeded into wells) compared with 295,708 cells (59%) in mock-inoculated wells.
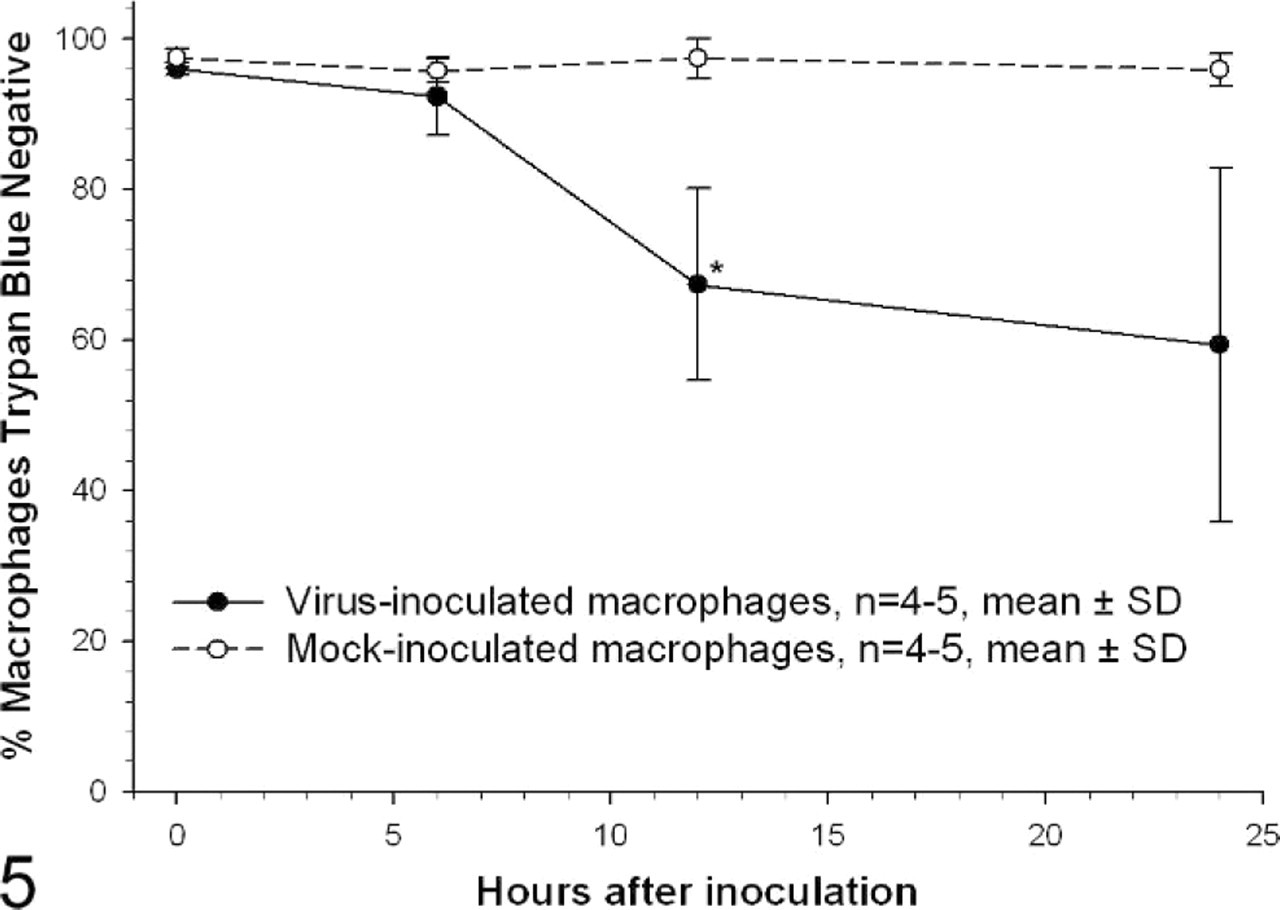
Trypan blue exclusion assay on virus- and mock-inoculated alveolar macrophages. Results are expressed as percentage trypan blue negative, an indicator of cell viability. Cells were inoculated at a multiplicity of 2. n = 4 for 6-hour time point, n = 5 for 0-, 12-, and 24-hour time points; ∗ = significantly different from corresponding mock-inoculated time point, P < .05, Kruskal-Wallis one-way ANOVA on ranks, Dunn's method of pairwise multiple comparisons, arcsine square root transformed data.
Cytokine production
TNF-α mRNA levels in virus-inoculated cells were significantly increased at 12 hours after inoculation, averaging 28-fold higher than mock-inoculated cells (Fig. 6, P < .05). TNF-α protein concentrations in supernatants from virus-inoculated cells differed significantly from mock-inoculated cells at 12 hours after inoculation, averaging 154 pg/ml supernatant compared with 4 pg/ml supernatant for mock-inoculated cells at the same time point (Fig. 7, P < .05). By comparison, IL-10 mRNA levels were marginally but significantly increased at both 6 and 12 hours after inoculation (P < .05) peaking at 1.7-fold the level in mock-inoculated cells by 6 hours after inoculation (Fig. 8). IL-10 protein levels in virus-inoculated supernatants did not increase significantly above mock-inoculated levels at any time point (Fig. 9).
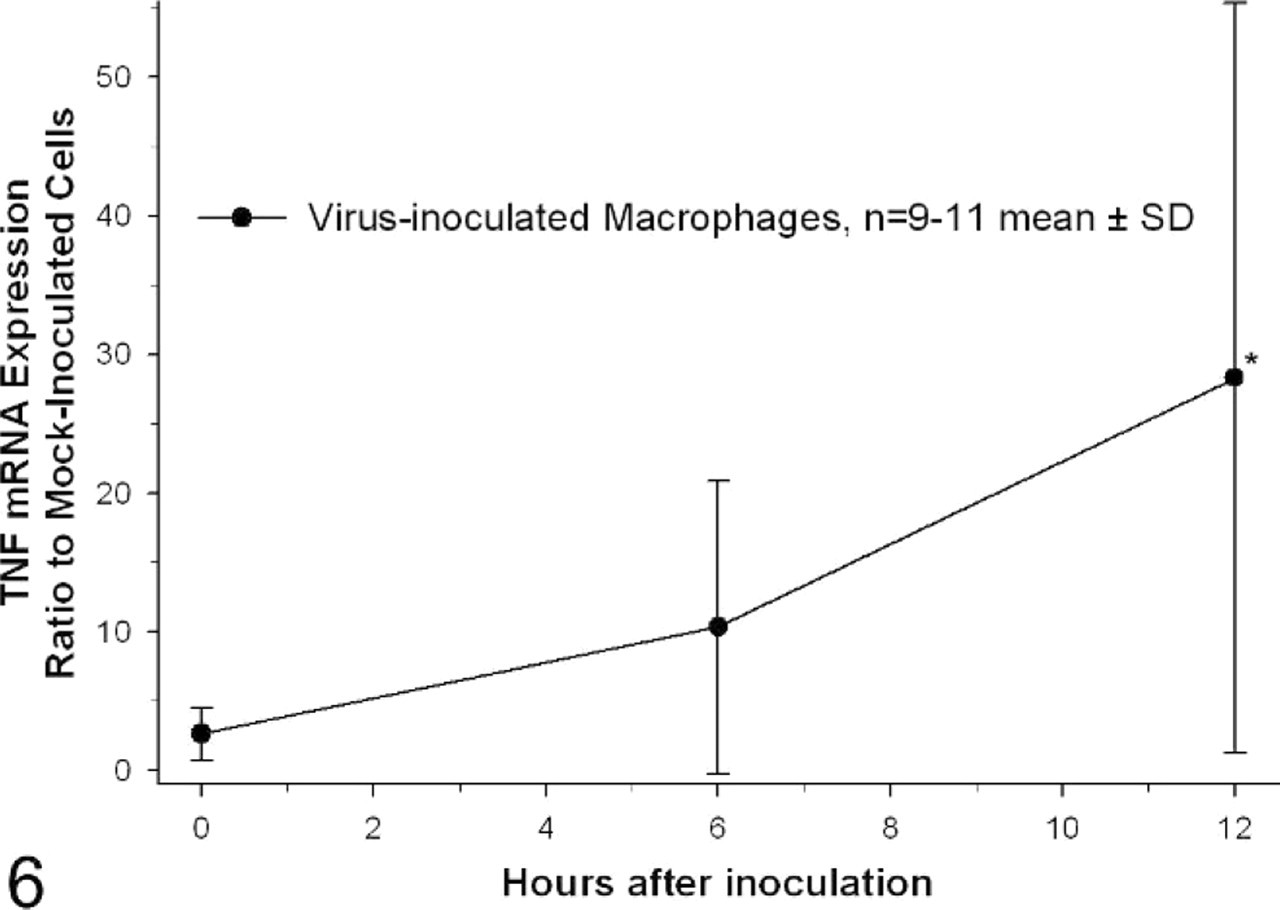
TNF-α mRNA in virus-inoculated alveolar macrophages. Results are expressed as a ratio to TNF-α mRNA levels in mock-inoculated cells at each time point. TNF-α mRNA levels were normalized to G3PDH mRNA expression. n = 11 for 0- and 12-hour time points, n = 9 for 6-hour time point; ∗ = significantly different from ratio immediately after the inoculation period, P < .05, Kruskal-Wallis one-way ANOVA on ranks, Dunn's method of pairwise multiple comparisons.
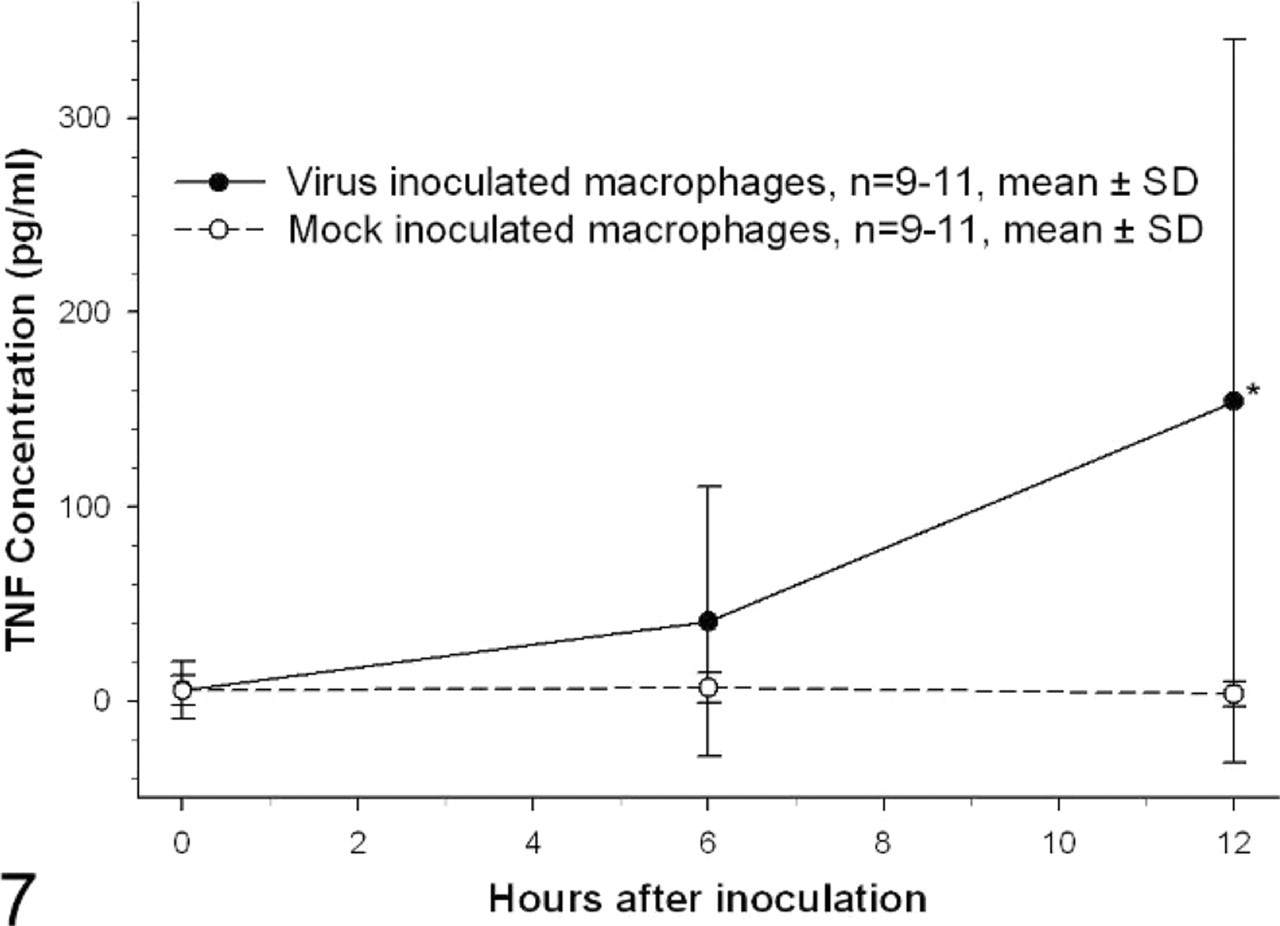
TNF-α protein concentration in alveolar macrophage culture supernatant. Concentration of TNF-α protein, as determined by ELISA, is expressed as pg/ml of culture supernatant. n = 11 for 0- and 12-hour time points, n = 9 for 6-hour time point; ∗ = significantly different from mock-inoculated cells at the same time point, P < .05, Kruskal-Wallis One-way ANOVA on ranks, Dunn's method of pairwise multiple comparisons.
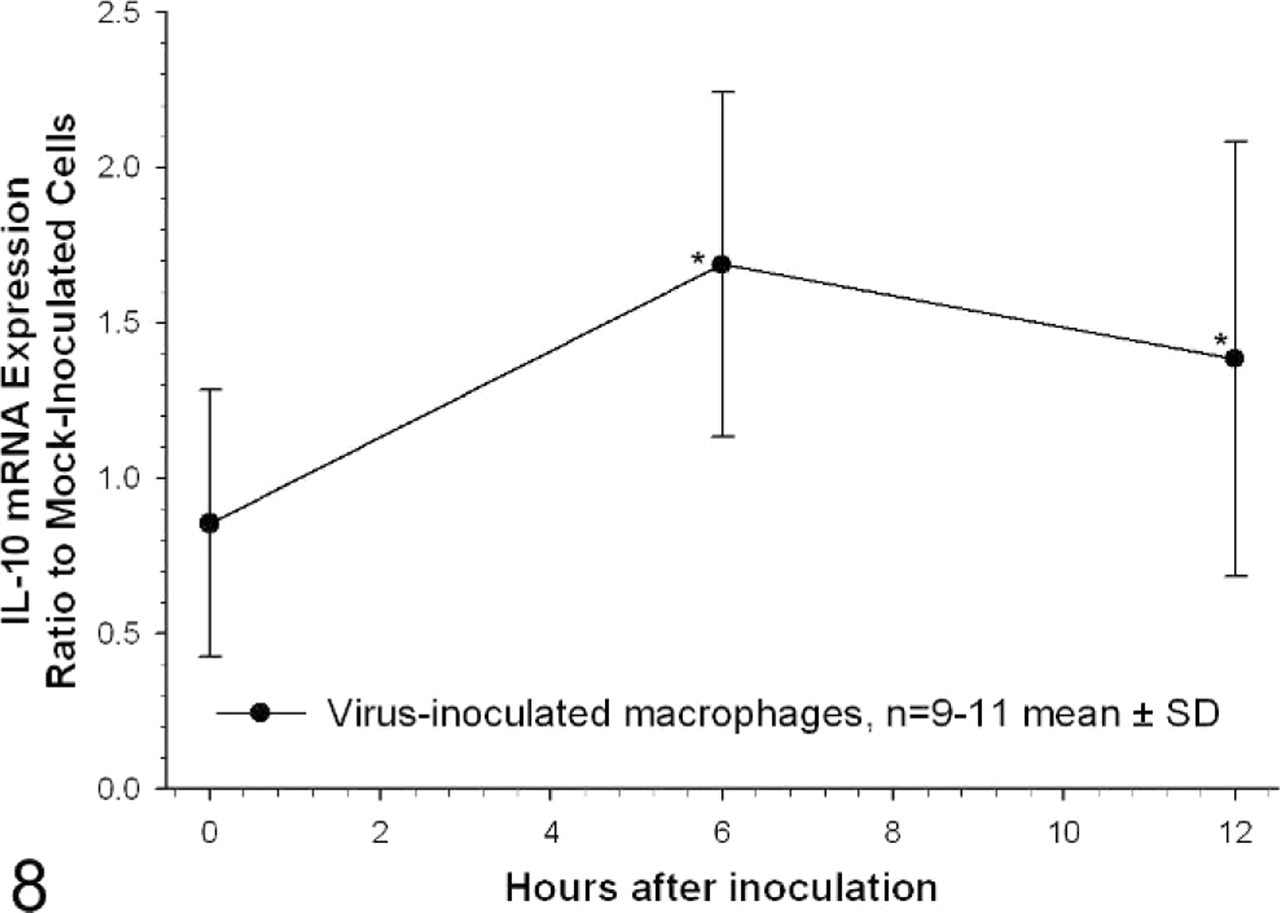
IL-10 mRNA in virus-inoculated alveolar macrophages. Results are expressed as a ratio to IL-10 mRNA levels in mock-inoculated cells at each time point. IL-10 mRNA levels were normalized to G3PDH mRNA expression. n = 11 for 0- and 12-hour time points, n = 9 for 6-hour time point; ∗ = significantly different from ratio immediately after the inoculation period, P < .05, one-way ANOVA, Dunn's Student-Newman-Keuls method of pairwise multiple comparisons.
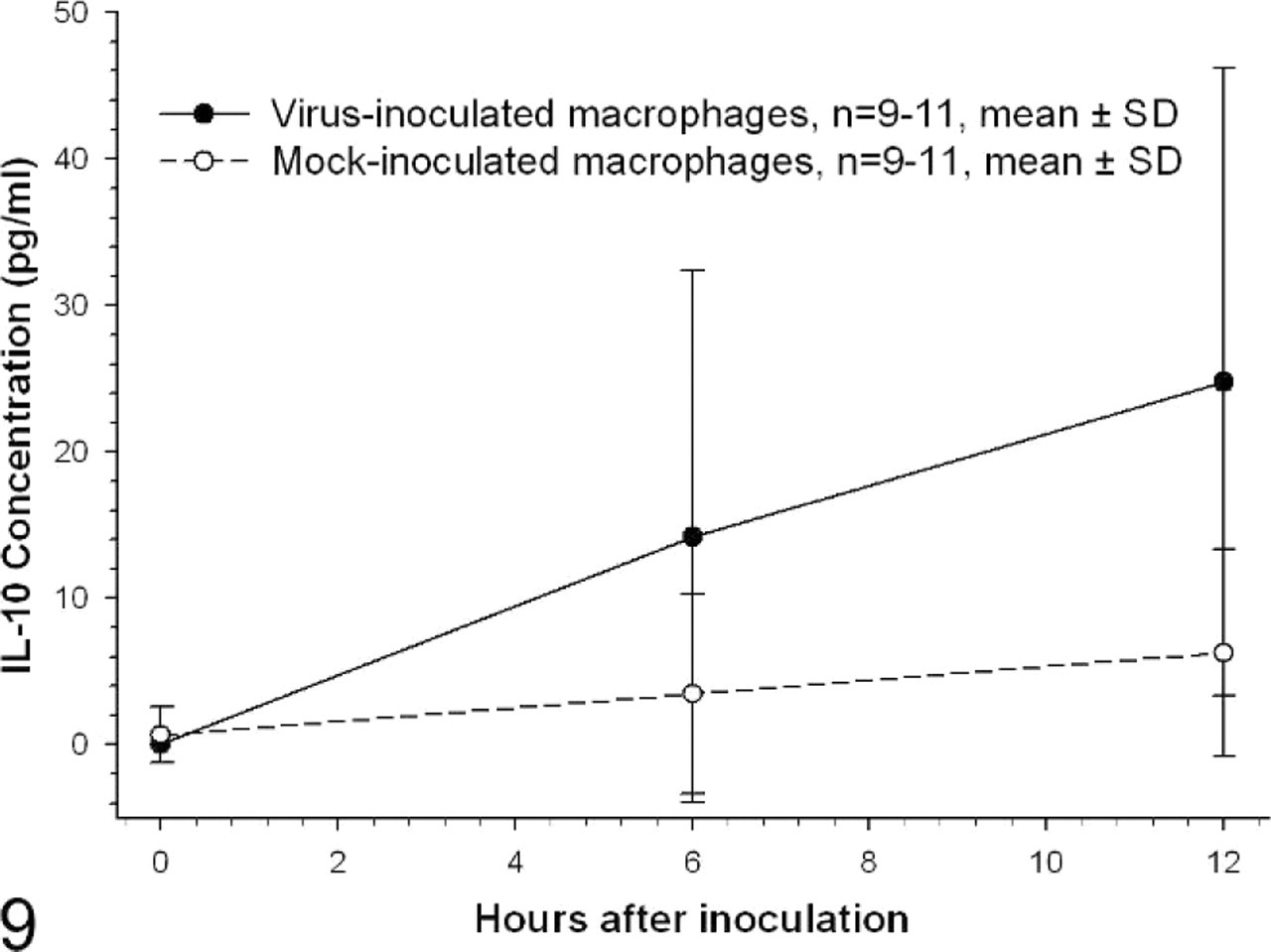
IL-10 protein concentration in alveolar macrophage culture supernatant. Concentration of IL-10 protein, as determined by ELISA, is expressed as pg/ml of culture supernatant. There are no significant differences among data from virus- and mock-inoculated macrophages at each of the time points. The 12-hour value for virus inoculated cells differed significantly from the virus- and mock-inoculated 0-hour values. n = 11 for 0- and 12-hour time points, n = 9 for 6-hour time point; P < .05, Kruskal-Wallis one-way ANOVA on ranks, Dunn's method of multiple comparisons.
Discussion
CIV is an important emerging pathogen of dogs that causes highly transmissible respiratory disease. Investigations into the initial outbreaks of disease associated with CIV showed that alveolar macrophages were positive for influenza virus antigen by immunohistochemistry. However, it was unclear whether the cytoplasmic antigen was secondary to phagocytosis of debris from other infected cells or associated with active replication of the virus in macrophages.10 Studies using other influenza viruses, including H5N1 viruses, have shown that infection of macrophages with influenza viruses may lead to excessive cytokine production.7 It has been postulated that this cytokine induction, particularly of TNF-α, may play an important role in the severity of disease in human H5N1 influenza virus infection.7, 21 Results from this study are consistent with the conclusion that canine alveolar macrophages support both virus RNA synthesis and production of viral protein and infectious viral particles at a low level. Macrophages also respond to virus infection with TNF-α production.
The level of productive viral replication by CIV in macrophages appears to be very low since the viral growth curve shown in Fig. 1 remains flat out to 12 or 24 hours after inoculation. The virus rapidly degrades in media without viable cells present. If there were not synthesis and release of at least small numbers of new infectious virus particles into the medium, we would have expected the slope of the virus curve in Fig. 1 to point downward from 6 to 24 hours after inoculation. While the combination of data of diffuse viral hemagglutinin protein staining in macrophage cytoplasm, matrix gene expression in macrophages, and prolonged release of infectious particles in macrophage culture medium support the conclusion that CIV replicates in canine alveolar macrophages, the level of productive virus replication remains undefined.
Influenza virus has been shown to infect monocytes or macrophages in a variety of species, including mice, swine, and humans.17, 23, 31–33, 36 Viral infection of macrophages in vitro may be abortive or productive and, when productive, usually only results in small numbers of infectious virions.3, 26, 32, 36 CIV replication kinetics were comparable with those from experiments in mice macrophage cell lines,3, 26 where maximal virus production occurred at approximately 24 hours after inoculation. In dog macrophages, production of virus RNA was intercurrent with cytokine mRNA and protein production.
CIV infection resulted in increased death of isolated pulmonary macrophages over 12 to 24 hours after inoculation as indicted by the trypan blue staining and viable cell counts. The studies in this report provide no insight into the question of whether virus-induced cell death resulted from apoptosis or necrosis. However, several studies indicate that influenza A virus induces apoptosis in monocytes or macrophages from humans and mice.12, 22 A study in swine could not demonstrate apoptotic processes in influenza-associated cell death of macrophages.36 Recently, a novel viral protein, PB1-F2, has been shown to be instrumental in the induction of apoptosis in cells of the monocytic lineage, including human monocytes.6 Not all influenza viruses harbor intact PB1-F2 proteins, and the presence of the protein is now considered to be a virulence determinant of some strains of influenza viruses.8 CIV is thought to contain an intact PB1-F2 protein. However the PB1 gene belongs to a novel phylogenetic clade, which may influence the function of PB1-F2 protein.43 Further investigations into the role of CIV PB1-F2 protein in alveolar macrophage apoptosis are required. Macrophage death induced by CIV could contribute to depression of pulmonary bacterial defense mechanisms and increase susceptibility to virus infection. However, the level of virus-induced macrophage death could be offset by increased recruitment of monocytes into the lung with differentiation to functional macrophages during the pulmonary inflammatory response to virus.
CIV induced differential expression of TNF-α and IL-10 in alveolar macrophages. TNF-α was strongly induced with 28-fold increases in mRNA associated with 39-fold increases in released protein. In contrast, there was only a 1.7-fold increase in IL-10 mRNA following virus inoculation, and protein levels increased fourfold from control levels.
Influenza virus can induce TNF-α expression through several transcription factor-mediated mechanisms18 including activation of nuclear factor kappa B (NF-κB), activating protein (AP)-1, and signal transducers and activators of transcription (STATs), although NF-κB is recognized as one of the most important mechanisms in macrophages. Influenza A viruses differ in their capacity to induce TNF-α in macrophages. Hyperinduction of TNF expression in human macrophages following H5N1 influenza virus infection has been shown to be p38 mitogen-activated protein (MAP) kinase dependent.21 Hyperinduction of pulmonary TNF-α can contribute to disease pathogenesis through at least several mechanisms7, 15, 25 acting in the local tissue environment on endothelial cells, epithelial cells, and leukocytes to induce increased vascular permeability, edema, and hemorrhage as well as to induce increased leukocyte-mediated tissue injury and secondary waves of cytokine release that can have massive systemic effects.
There was considerable variation in influenza virus-induced TNF-α expression in alveolar macrophages that were recovered from cadaver dogs. Mean levels of TNF-α mRNA expressed by CIV-infected macrophages were in a similar range to those seen with macrophages exposed to 1 to 1,000 ng/ml lipopolysaccharide used as a positive control (see Materials and Methods section). Both antemortem disease and husbandry events as well as postmortem handling of the dogs used in this study undoubtedly induced variability into macrophage function assays. Nevertheless, dog-to-dog variability in the assays for TNF-α probably reflect, at least in part, genetically controlled variations in TNF-α response among dogs. Humans have considerable variation in TNF-α transcriptional response that is under control of TNF-α promoter polymorphisms, and variations in TNF-α expression have been linked to disease susceptibility and severity.1 Although it was highly desirable to get a uniform population of primary dog alveolar macrophages for these studies, logistical problems in obtaining large enough numbers of cells for the studies from laboratory-housed dogs required the use of macrophages recovered from cadavers. Studies focused on differences in macrophage TNF-α production among dog breeds could provide important information relevant to genetically determined disease susceptibility. These studies could be particularly insightful if focused on Greyhound dogs that are highly susceptible to fatal disease associated with CIV infection.10
IL-10 responses to CIV infection were considerably lower than those found for TNF-α. IL-10 is produced by regulatory T cells, macrophages, dendritic cells, and other myeloid and lymphoid cells, and it plays an important immunoregulatory role in host response to infectious agents.9 Stimulators of IL-10 in macrophages include LPS and CpG from bacteria, many protozoa, and fungi. These stimuli act through activation of toll-like receptor (TLR)-2 and TLR-4 mechanisms, Fc receptor ligation by immune complexes, and CD40 ligation.5, 9 Increased IL-10 expression in the lung following influenza infection may be more the result of production by T cells than by macrophages.9, 42 The small increase in IL-10 produced by dog macrophages inoculated with CIV could be a secondary effect of virus-induced type I interferon, rather than a direct effect of virus.5 CIV infection studies in immunocompetent dogs would be required to critically assess the roll of IL-10 from both T cells and macrophages in pulmonary pathogenesis and impaired host response to bacteria.41
In conclusion, the data from this study indicate that CIV can replicate in canine alveolar macrophages and induce TNF-α production that may be important in respiratory disease pathogenesis. IL-10 is stimulated to a lesser extent, and virus inoculation results in a decrease in macrophage viability.