
Abstract
Abstract
Purpose
Congenital clubfoot is a serious birth defect that affects nearly 0.1% of all births. Though there is strong evidence for a genetic basis of isolated clubfoot, aside from a handful of associations, much of the heritability remains unexplained.
Methods
By systematically examining the genes involved in syndromic clubfoot, we may find new candidate genes and pathways to investigate in isolated clubfoot.
Results
In addition to the expected enrichment of extracellular matrix and transforming growth factor beta (TGF-β) signalling genes, we find many genes involved in syndromic clubfoot encode peroxisomal matrix proteins, as well as enzymes necessary for sulfation of proteoglycans, an important part of connective tissue. Further, the association of Filamin B with isolated clubfoot as well as syndromic clubfoot is an encouraging finding.
Conclusion
We should examine these categories for enrichment in isolated clubfoot patients to increase our understanding of the underlying biology and pathophysiology of this deformity. Understanding the spectrum of syndromes that have clubfoot as a feature enables a better understanding of the underlying pathophysiology of the disorder and directs future genetic screening efforts toward certain genes and genetic pathways.
Level of evidence
V
Introduction
Congenital clubfoot, also called talipes equinovarus, is a common and serious birth defect, affecting an estimated one of every 1000 live births. 1 Clubfoot is characterized by structural defects of several tissues of the foot and lower leg, which leads to abnormal positioning of foot and ankle joints. 2 If left untreated, it can become a severe disability and deformity. 3 Approximately 80% of clubfoot cases are isolated birth defects, having an idiopathic aetiology. 4 The remaining 20% of cases are due to associated malformations, chromosomal abnormalities and known genetic syndromes, such as distal arthrogryposis (DA) and myleomeningocele. 5
However, there is strong evidence for a genetic basis for isolated clubfoot. Approximately 25% of all isolated cases report a family history of clubfoot. 6 Data from twin studies shows a higher concordance in monozygotic (33%) than dizygotic (3%) twins, 4 and more recent data estimates heritability of isolated clubfoot at around 30%. 7 Monochorionic triplets all affected with bilateral isolated clubfoot have been observed, 8 further supporting a genetic aetiology for isolated clubfoot. In addition, prevalence of clubfoot varies across ethnic populations, from 0.39 cases per 1000 births in Chinese populations to seven cases per 1000 births in Hawaiians and Maoris.9–11 Further, the ratio of isolated clubfoot among males and females is 2:1, and this ratio is consistent across ethnic groups.11,12 Taken together, this data points to an obvious role of genetics in isolated clubfoot. As the clinical presentation between the isolated and syndromic forms can be similar, it is possible that by examining the genes involved in syndromic clubfoot, i.e. those disorders that often have clubfoot as one symptom of many, we may find further clues to the underlying mechanisms of isolated clubfoot.
Genetics of isolated clubfoot
PITX1-TBX4 pathway
Although few causative genes are known, progress has been made on the genetics of isolated clubfoot. The strongest genetic evidence is the PITX1-TBX4 pathway, the proper function of which is required for normal hindlimb development.13,14 Variation in this pathway has been linked to isolated clubfoot phenotypes through a segregating dominant mutation in PITX1, 15 inherited TBX4 microduplications,12,16,17 Pitx1 mouse knockout studies 18 and copy-number variants. 19
Homeobox (HOX) genes
In addition to the PITX1-TBX4 pathway, HOX genes also contain some of the more convincing genetic associations with clubfoot phenotypes. HOX genes comprise four gene clusters (HOXA-D) and these clusters are known to coordinate and mediate limb development. In fact, these genes play critical roles in skeletal patterning throughout the axial and appendicular skeleton. 20 Single nucleotide polymorphism (SNPs) in HOXD12 and HOXD13 were found to be associated with idiopathic clubfoot. 21 A HOXD10 missense mutation segregated with a related disorder, called congenital vertical talus, in a British family, 22 and that same mutation was also described as segregating in a family of Italian descent with both clubfoot and Charcot-Marie-Tooth Disease. 23 Four SNPs in the HOXA cluster showed altered transmission in a case-control study, but gene-gene interactions were identified between HOXA and HOXD variants and previously associate SNPs in mitochondrial-mediated apoptotic genes. 24 However, a functional analysis of a SNP in HOXA9 created allele-specific nuclear protein interactions and caused higher promoter activity, suggesting that HOXA9 promoter variants alter expression, thus playing a functional role in the underlying mechanisms of isolated clubfoot. 25 Most recently, HOXC microdeletions were shown to overlap a noncoding region upstream of HOXC13. The authors found a missense SNP in HOXC11 to segregate in a family with isolated clubfoot, and a missense SNP in HOXC12 was enriched in clubfoot patients. 26
Muscle contractile genes
There is conflicting evidence of the role of muscle contractile genes in isolated clubfoot. While they are good candidate genes due to their expression either embryonically or perinatally, which is the period during which isolated clubfoot develops, and are part of Type II muscle, which is known to be decreased in clubfoot patients, 27 no groups have found any evidence of contractile gene association with isolated clubfoot.27,28 This suggests a different pathophysiology than the syndromic form of clubfoot so often seen in DA syndromes, for which muscle contractile genes have proved of importance. However, a study performed two years later found an association with two SNPs in TNNC2 and isolated clubfoot, as well as SNPs in TPM1 and TPM2. 29 Functional analyses of SNPs in TPM1 and TPM2 have been shown to cause allele-specific higher promoter activity, suggesting a functional role for these gene promoters in isolated clubfoot. 25
Environmental in utero causes
Smoking during pregnancy has been associated with birth defects including clubfoot.2,30 N-acetylation genes NAT1 and NAT2 modulate the biotransformation of exogenous substances such as tobacco smoke, and one study found that there were significantly more slow NAT2 acetylators among clubfoot cases. 31 A SNP in CYP1A1, a nicotine metabolism gene, was also weakly associated with isolated clubfoot. 30 Similarly, low folate levels during pregnancy can lead to congenital malformations. An interaction between genotype at a missense SNP in the methylenetetrahydrofolate reductase gene and maternal folic acid usage was found, leading to decreasing relative risk for isolated clubfoot in an allele dosage type manner. 32
Apoptotic genes
Apoptotic genes involved in the cell death cascade that aid in shaping the developing limb (CASP8, CASP10 and CFLAR) had been previously associated with microsatellite markers spanning a deletion of chromosomal region 2q31-33 linked with isolated clubfoot. 33 However, after further genotyping of 40 SNPs in seven genes involved in apoptosis was performed no significant associations were found. 34
Other genes
Filamin B (FLNB) is an actin-binding protein that crosslinks actin cytoskeleton filaments into a dynamic structure. 35 Three novel missense mutations in FLNB have been associated with isolated clubfoot. 36 In the only genome-wide association study for clubfoot to date, a SNP on chromosome 12q24.31 between NCOR2 and ZNF664 was associated with clubfoot in both the initial and replication datasets. Suggestive SNPs were identified near FOXN3, SORCS1 and MMP7, suggesting a role for common variants in several non-candidate genes. 37
Genetics of syndromic clubfoot
Distal Arthrogryposis
One of the most common syndromic causes of clubfoot is arthrogryposis. It occurs in one of 3000 to one in 5000 live births. 38 However, given how many specific subtypes there are, each is relatively rare. This term is used to describe multiple congenital contractures. Arthrogryposis is not a diagnosis in itself, but rather a symptom, and implies contractures in multiple regions of the body. It is present in over 400 specific conditions. 39 In utero, arthrogryposis is often associated with decreased foetal movement, which results in connective tissue abnormalities and muscle atrophy, among other features. DA is a group of syndromes with predominantly distal contractures of the hand and foot. DA in many cases has an underlying genetic cause (Table 1 40 ). However, unlike isolated clubfoot, DA has most consistently been shown to be caused by variants in sarcomeric muscle proteins responsible for muscle contraction, many of which are only expressed early in development.41–44
Distal arthrogryposes (DA) and associated genes adapted from Hall et al (2017)(40)

While distal arthrogryposis type 3 (DA3) and distal arthrogryposis type 5 (DA5) as well as the phenotypically similar Marden-Walker Syndrome are caused by autosomal dominant mutations in PIEZO2,45,46 a separate phenotype in individuals lacking PIEZO2 causes muscular atrophy with spinal deformities and DA as a symptom. 47 This gene encodes an ion channel critical for proprioception. FBN2 is a component of connective tissue and elastic fibre assembly, mutations in which cause distal arthrogryposis type 9 (DA9).
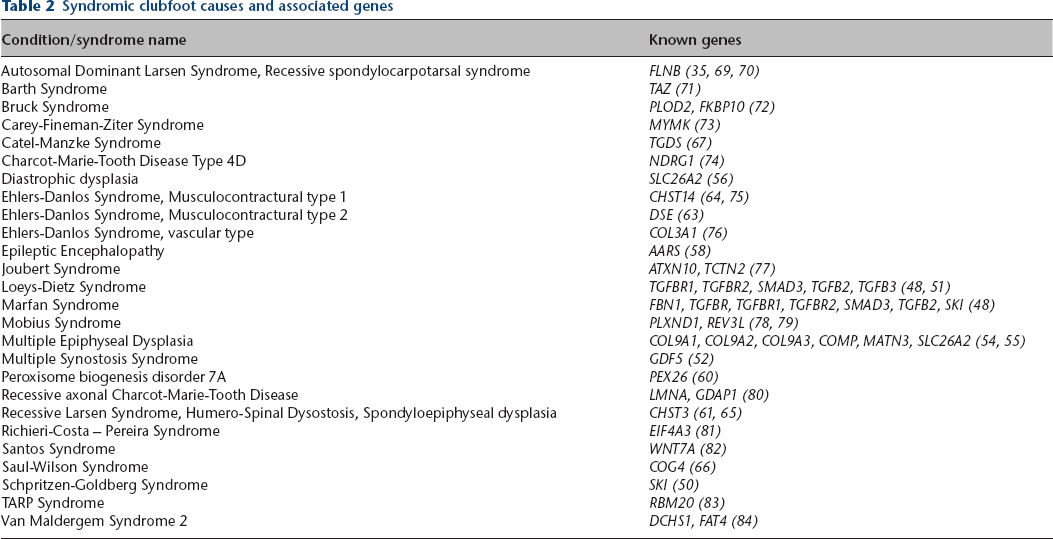
Many syndromes besides the distal arthrogryposes also have clubfoot as a symptom and are known to have a genetic basis (Table 2). Some, though not all of these genes, fall into known categories that can easily be understood in the context of the pathogenesis of clubfoot.
Syndromic clubfoot causes and associated genes
Transforming growth factor beta (TGF-β) signalling
TGF-β signalling regulates cellular processes including proliferation, apoptosis, differentiation and extracellular matrix formation and remodelling. It is also involved in skeletal, vascular and hematopoietic homeostasis.48,49 Genes in this pathway including TGFBR1, TGFBR2, SMAD3, TGFB2, TGFB3 and SKI have been implicated in hereditary connective tissue disorders including Marfan Syndrome, Loeys-Dietz Syndrome and Schprintzen-Goldberg Syndrome.48,50,51 GDF5, a bone morphogenetic protein and part of the TFG-β family that was previously referred to as cartilage derived morphogenetic protein 1 (CDMP-1), is a growth factor that is expressed during several critical periods of skeletal development. Mutations in this gene are associated with multiple syndromes including synostosis syndrome and brachydactyly type C. 52
Extracellular matrix (ECM)
The ECM provides structural support for organs, tissues and cell membranes. They also play a role in cell differentiation, proliferation survival and migration. Extracellular matrix binding helps to regulate TGF-β signalling. 53 Mutations in genes encoding ECM proteins COL9A1, COL9A2, COL9A3, COMP and MATN3 as well as the transmembrane glycoprotein involved in matrix organization, SLC26A2, have been associated with multiple epiphyseal dysplasia.54,55 SLC26A2 mutations have also been associated with distrophic dysplasia, a non-lethal form of de la Chapelle dysplasia. 56 Mutations in the ECM protein encoded by FBN1 are known to cause Marfan Syndrome.48,57 COL3A1 mutations cause a vascular type of Ehlers-Danlos syndrome. 58
Peroxisomal defects
Peroxisomes are organelles that play an essential role in several cellular and metabolic pathways. GDAP1 is involved in the fission of peroxisomes, and patients with GDAP1 mutations display a Charcot-Marie-Tooth phenotype where mitochondria and peroxisomes are elongated in cells. Mutations in peroxisomal biogenesis factors (PEX) genes including PEX26 can disrupt import of peroxisomal matrix proteins. 59 Mutations in PEX26 are a known cause of peroxisome biogenesis disorder. 60 Both of these disease phenotype sequelae include clubfoot.
Proteoglycans
Proteoglycans are a component of connective tissues that consist of glycosaminoglycan (GAG) polymer chains attached to core proteins. GAG sugar composition (dermatan, chondroitin, heparin) helps determine the biological roles and tissue distributions of the macromolecules produced. 61 Dermantan sulfate proteoglycans are components of diverse connective tissues, defects in which can result in abnormal collagen fibril assembly. It is also known to interact with heparin cofactor II and can modulate thrombus formation. 62 CHST14 and DSE encode two enzymes necessary for dermatan sulfate biosynthesis. Mutations in these genes cause the musculocontractural types of Ehlers-Danlos syndrome, both of which present with clubfoot.63,64 CHST3 encodes an enzyme that catalyzes sulfation of chondroitin containing proteoglycan, which is a necessary part of connective tissues. 65 Mutations in CHST3 have been associated with skeletal dysplasias that can present with clubfoot, including humero-spinal dysostosis and spondyloepiphyseal dysplasia, as well as recessive Larsen syndrome.61,65 Initiation and polymerization of GAG occurs in the Golgi apparatus. A mutation in COG4 has been found to disrupt this process, resulting in a rare cause of dwarfism that presents with multiple limb malformations including clubfoot, known as Saul-Wilson syndrome. 66 Lastly, TGDS, a member of the short-chain reductase family, is also suspected to be involved in proteoglycan synthesis or sulfation. Mutations in this gene cause Catel-Manzke syndrome, which can present with clubfoot.67,68
Discussion
Here we have shown that a variety of pathways and categories of genes are involved in both isolated and syndromic clubfoot. To our knowledge, this is the first time that genes involved in syndromic clubfoot have been categorized in an attempt to better understand the biology of this deformity. While some of the categories were known or expected, such as TGF-β signalling and extracellular matrix components, others including peroxisomal defects and proteoglycans, were novel.
We posit that examination of rare variants in syndromic clubfoot genes could yield associations with isolated clubfoot. It is promising that FLNB has already been associated with both isolated clubfoot and with autosomal dominant Larsen syndrome. We believe that by elucidating new genes and pathways that underlie clubfoot, we will both be able to increase the explained amount of heritability of isolated clubfoot, as well as increase our overall understanding of the disease process.