
Abstract
Reported pregnancies in women with repaired truncus arteriosus are rare. As a result of improved surgical techniques, more women with conotruncal defects are now reaching reproductive age. Multifactorial inheritance rather than single gene mutation is the causative agent of most congenital heart disease. The environment can play an important and varied role in direct transmission, particularly in incidences where genes predispose a defect. This may be one of the first reported cases discussing a pregnant mother with repaired truncus arteriosus and her fetus, prenatally diagnosed with a concordant truncal defect.
Truncus arteriosus is the persistence of the fetal common arterial trunk without differentiation of the great vessels. It is reported to occur in about 1.07 of every 10,000 births. 1 Prior to substantial surgical evolution, infants with truncus arteriosus could only be cared for palliatively and would not survive beyond infancy. Few reports describe pregnancy in women with repaired truncus arteriosus because of its rarity. The following case describes a pregnant woman and her fetus, both with congenital truncus arteriosus. Although 22q11.2 deletion syndrome and pregestational diabetes are known to cause truncus arteriosus, determining the mode of transmission with familial recurrence is complex. Whole exome sequencing can test for nucleotide variants in genes reported to be associated with congenital heart disease (CHD). Further gene discovery may be merited in families with multiple affected individuals when a causative mechanism cannot be determined.
Case Report
A 21-year-old gravida 1, para 0 mother presented at 20 weeks’ gestational age for fetal echocardiography. Her history was significant for a repaired type 1 truncus arteriosus with interrupted aortic arch. Surgical correction with aortic arch repair was performed at 11 months of age. Four years later, a 16-mm right ventricular–pulmonary artery (RV-PA) homograft was placed via the LeCompte maneuver. As an adolescent, she required a bovine mitral valve replacement, and a subsequent 23-mm RV-PA conduit. Fluorescence in situ hybridization (FISH) performed in 1997 was negative for 22q11.2 deletion.
Formal fetal echocardiography was indicated due to the family history of CHD. In concordance with the mother’s cardiac defect, truncus arteriosus was evident on fetal examination with a single, large overriding great vessel, which gave rise to the branch pulmonary arteries and aortic arch. The main pulmonary artery (MPA) segment originated from the posterolateral aspect of the common trunk, and an anterior malalignment ventricular septal defect (VSD) permitted communication between the right and left ventricles. The aortic arch was found to be intact without interruption, and the truncal valve appeared to have normal integrity without stenosis or regurgitation. Initial sonographic delineation of the pulmonary artery origin classified the defect as truncus arteriosus type 1.
Amniocentesis was performed and sent for FISH (including 22q11.2), five-cell karyotype, and chromosomal microarray analysis. All cytogenetic testing confirmed a normal male, ruling out 22q11.2 deletion syndrome and other chromosomal abnormalities. Further genetic counseling prompted maternal gene sequencing with deletion and duplication analysis. A 62-gene congenital structural heart disease panel was ordered to assess genes found in association with truncus arteriosus. The panel did not detect any known disease-causing mutations in the genes sequenced, including GATA6, CHD7, and NKX2-6, which are associated with nonsyndromic truncus arteriosus.2,3
High-risk indications including family history, maternal habitus, and a fetal cardiac defect were noted at the 26-week glucose tolerance test. The patient was diagnosed with gestational diabetes mellitus (GDM) and required insulin therapy for control. Antepartum surveillance included blood glucose monitoring and twice weekly nonstress tests.
At 33 weeks’ gestational age, the patient presented for follow-up fetal echocardiography. A GE Voluson E10 ultrasound system with a C1-5-D curved-array transducer was utilized for transabdominal sonography. A cine clip documented the outflow tract, three-vessel view, and three-vessel trachea view (see online video supplement). Preceding the aortic arch at the level of the ascending aorta, the pulmonary arteries were shown branching from the common arterial trunk.
At 36 weeks’ gestational age, the patient presented to the Maternal-Fetal Medicine Division for evaluation of growth. A Philips Epiq 7W ultrasound system with a C5-1 curved-array transducer was utilized. The four-chamber fetal heart appeared normal (Figure 1); however, the five-chamber view showed a large common trunk giving rise to the aorta and pulmonary artery (Figure 2).
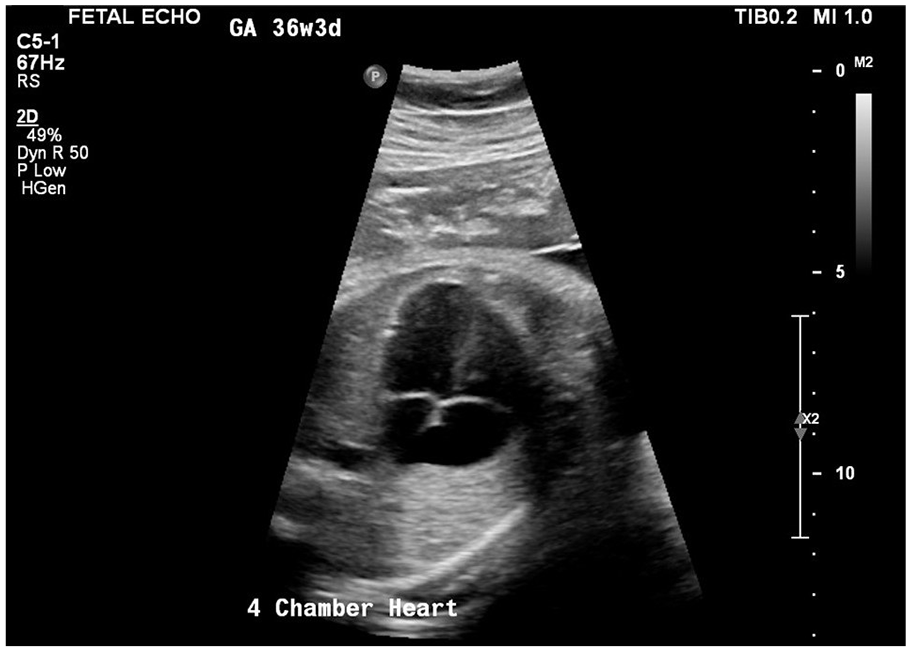
Fetal echocardiogram of a normal-appearing four-chamber heart in an apical view.
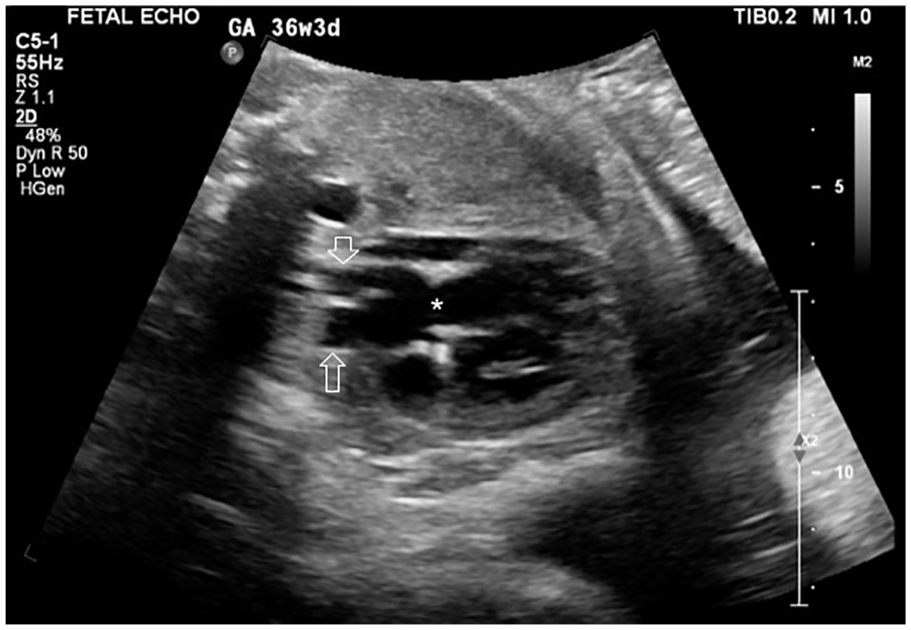
Fetal echocardiogram of a five-chamber view showing a large common arterial trunk (*) giving rise to the aorta (long arrow) and pulmonary artery (short arrow).
The fetus remained hemodynamically stable throughout the pregnancy. However, maternal cardiac status worsened slightly with an increase in aortic root dilation from 43-mm to 45-mm, and from mild pulmonic stenosis without insufficiency to significant insufficiency. Induction of labor was performed at 39 weeks with careful consideration for the maternal CHD. Precautions included an early epidural, endocarditis prophylaxis, and telemetry during labor and 24 hours postpartum. A viable male infant was delivered via forceps-assisted vaginal delivery without adverse cardiac events in either patient. On day 3 of life the newborn was transferred to a partnering institution for surgical repair and continued management.
The neonate’s first echocardiogram after birth revealed that the truncus arteriosus classification was not type 1. However, the lesion did not meet criteria for a type 2 truncus arteriosus. Instead, it fell between the two classifications and was termed type 1.5. A GE Vivid E95 ultrasound system with a 6S-D and 12S-D phased-array transducer was utilized for neonatal echocardiography. The apical four-chamber view with an anterior angle showed the overriding truncus arteriosus with a large malalignment VSD and branch pulmonary arteries arising from the truncus/ascending aorta (Figure 3). The pulmonary arteries demonstrated immediate bifurcation from their common origin on the underside of the ascending aorta, with no MPA segment. The suprasternal short-axis view showed confluent branch pulmonary arteries with normal dimensions (Figure 4). On reconstructed 3D computed tomography angiography (CTA), the unobstructed branch pulmonary arteries were seen arising directly from the truncal root (Figure 5). The aortic arch was left-sided and the common truncal valve was identified as trileaflet. In addition to the prenatally diagnosed VSD, a moderate secundum atrial septal defect (ASD) was noted. At 14 days old, the patient was taken to the operating room for truncus arteriosus repair with a 9-mm pulmonary homograft RV-PA conduit, ASD, and VSD closure. He tolerated the procedure well, despite episodes of supraventricular tachycardia (SVT) in the operating room and intermittently throughout the postoperative period. His SVT was controlled with medication and he was discharged from the hospital without incident.
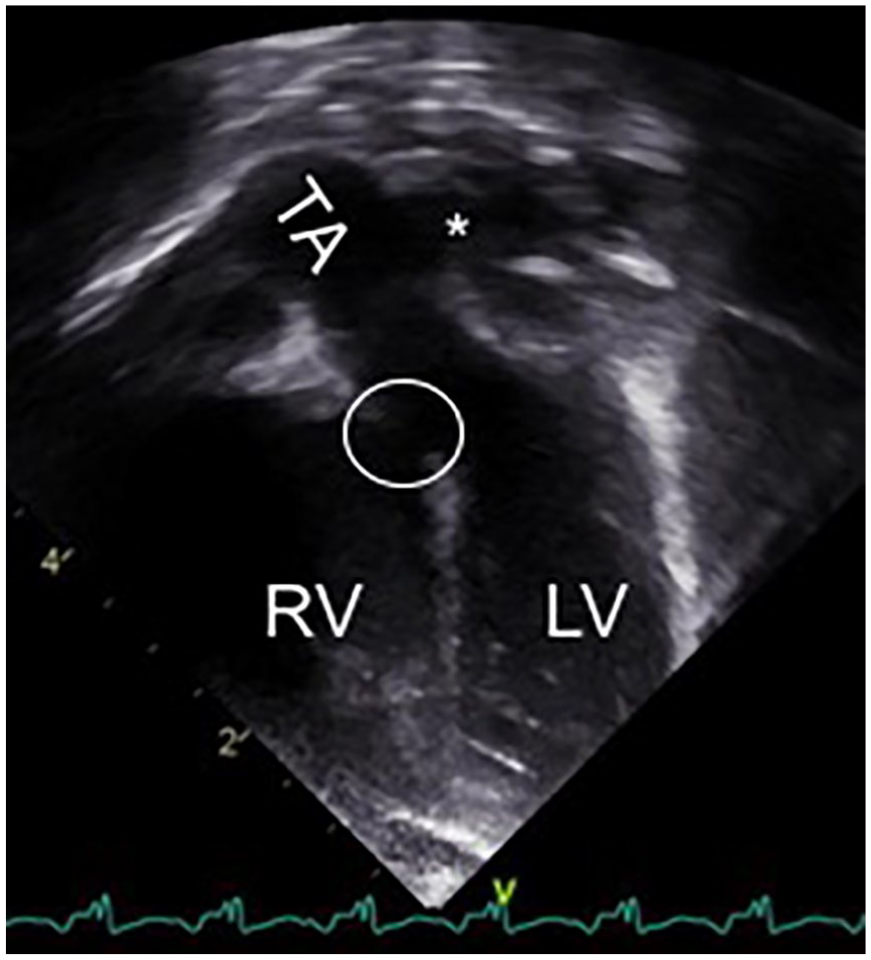
Neonatal echocardiogram of an apical four-chamber view angled anteriorly showing overriding truncus arteriosus (TA) with large ventricular septal defect (circle), and branch pulmonary arteries arising from the TA/ascending aorta (*). LV, left ventricle; RV, right ventricle.
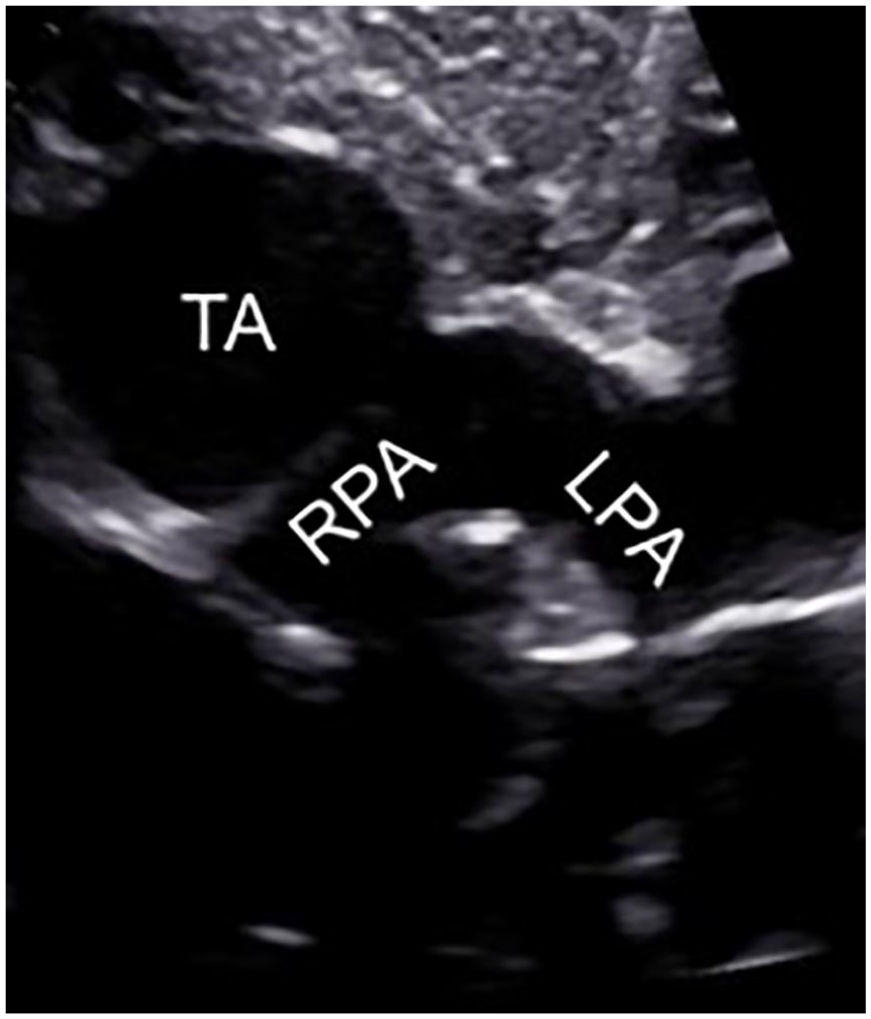
Neonatal echocardiogram using a suprasternal short-axis approach showing confluent branch pulmonary arteries. LPA, left pulmonary artery; RPA, right pulmonary artery; TA, truncus arteriosus.
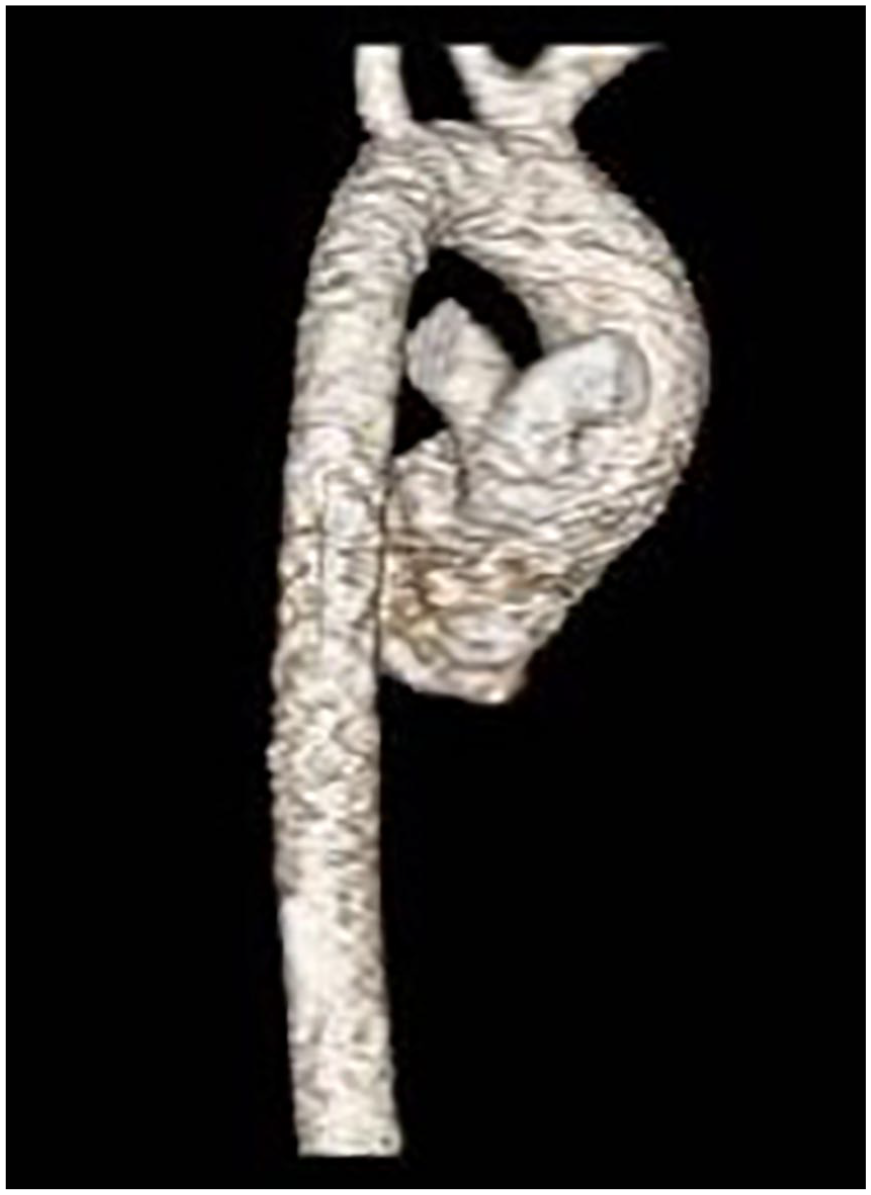
Computed tomography angiography 3D reconstruction showing branch pulmonary arteries arising directly from the truncus root.
Discussion
Truncus arteriosus is a rare and complex CHD accounting for approximately 3% of all cardiac defects among live births.4–6 Initially, common truncus arteriosus is a physiologic and transient embryologic step. 3 However, persistence throughout cardiac development without truncoconal septum formation is abnormal.6,7 During embryogenesis, the common arterial trunk is the main outlet from the heart. At approximately 27 days’ gestation, spiraled truncoconal ridges begin to fuse and partition the single truncus into two separate vessels.6,7 Complete or partial absence of the truncoconal ridges results in failure of division of the single truncus arteriosus into the pulmonary trunk and ascending aorta. 5 The subsequent congenital anomaly may be referred to as persistent truncus arteriosus, common arterial trunk, truncus arteriosus communis, or aorticopulmonary trunk.1,6,7
Truncus arteriosus is almost always associated with a VSD. 8 Truncoconal ridges that normally divide the truncus are also necessary to complete the formation of the uppermost portion of the septum. 8 Malalignment VSDs occur in the perimembranous or muscular portion of the ventricular septum, and with an anterior or posterior shift. As a result, both ventricles empty into the vessel overriding the VSD. 9 In rare occurrences, the single truncal artery may arise entirely from the right or left ventricle. 1 The number of valve leaflets forming the semilunar, truncal valve varies from two to six. 7 A tricuspid (three leaflet) truncal valve is morphologically aortic and the most common. A quadricuspid (four leaflet) truncal valve is primarily aortic with a pulmonary leaflet remnant.7,8
Collett and Edwards anatomically classified four types of persistent truncus arteriosus according to the origin of the pulmonary arteries, and the various stages of arrested septum development. The various types are further subdivided by the integrity and orientation of the aortic arch, and the presence or absence of the ductus arteriosus. 7 In type 1, a short main pulmonary trunk arises from the truncus and divides into right and left pulmonary arteries.1,7 This type represents the latest stage of arrested development as compared with type 4, which signifies the earliest. 7 In types 2 and 3, the pulmonary arteries arise as individual branches off the truncus.1,7 A type 2 defect has closely approximated PA origins, whereas a type 3 has independently arising pulmonary arteries at some distance from one another.1,7 Collett and Edwards’ original type 4 classification (absent pulmonary arteries with bronchial arteries arising from the aortic arch or descending aorta) is no longer considered a common aorticopulmonary trunk, but more accurately describes pulmonary atresia with a VSD.1,6–8
Van Praagh and Van Praagh modified the widely used Collett and Edwards classification describing two basic types of common aorticopulmonay trunk. Those with a VSD are the most common and considered type A. 8 Those without a VSD are rare and considered type B. 8 Each type is further classified in terms of the great arteries. In type A1, the aorticopulmonary septum is partly formed, resulting in an MPA. 8 In type A2, complete atresia of the aorticopulmonary septum prohibits the formation of a MPA. 8 The right and left pulmonary arteries instead branch directly from the common trunk. 8 One of the pulmonary artery branches is absent in type A3, and the other originates from the truncus.6,8 Collateral vessels maintain blood supply to the affected lung. 8 Type A4 is truncus arteriosus with an interrupted (absent) aortic arch replaced by a large patent ductus arteriosus (Figure 6).6,8 The two most commonly diagnosed types in the fetus are type 1/type A1, and type A4. 1
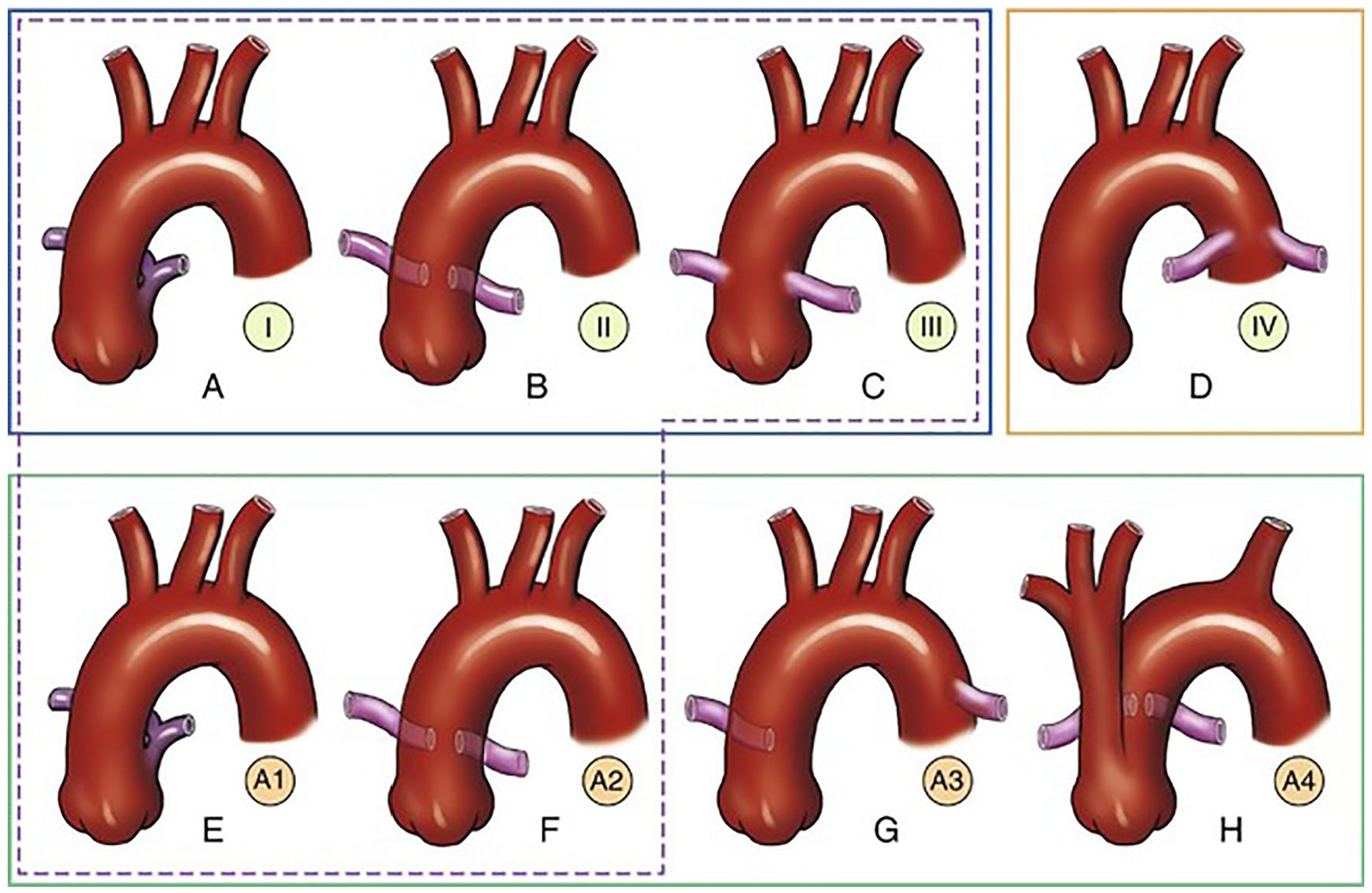
(A–D) Collett-Edwards type 1–4 classifications. (D) Type 4 is now considered pulmonary atresia with a ventricular septal defect. (E–H) Van Praagh modifications of the Collett-Edwards classifications. (A–C) Collett-Edwards types 1–3 and (E and F) Van Praagh types A1 and A2 are similar, differing only in the number and location of the pulmonary arteries originating from the truncus. (G) Van Praagh type A3 has a single pulmonary artery arising from the truncus and the second pulmonary artery from the descending aorta. (H) In Van Praagh type A4, a hypoplastic aortic arch and a patent ductus arteriosus arise from the truncus arteriosus. Pulmonary arteries originate from the posterior aspect of the truncus arteriosus. 6
This case was an example of a defect that did not meet criteria for either type 1 or type 2 of Collett and Edwards’ classifications. The fetal truncus arteriosus undoubtedly was not type 1, because there was no MPA. It did not correspond to a type 2 defect because the right and left branch pulmonary arteries originated from a common site, as opposed to separate closely spaced sites. Van Praagh’s type A2 lesion lacks an MPA with right and left pulmonary arteries branching directly from the common trunk, but the anatomic classification does not fully delineate a shared origin.
Type 1.5 best defined this defect. Despite seldom descriptions in the literature, type 1.5 is a designation commonly referenced at the institution where the patient sought care. Type 1.5 is truncus arteriosus with branch pulmonary arteries immediately bifurcating from a common origin off the truncus, without an MPA segment. This colloquial term is utilized when neither the type 1 nor type 2 classification is appropriate. Clinically, a distinction between types 1, 1.5, and 2 is not usually significant. The surgical intervention and long-term results are analogous. In 2000, the Congenital Heart Surgery Nomenclature and Database Project proposed a modified Van Praagh classification, and grouping based on surgical technique. 10 Type I, with confluent or near-confluent pulmonary arteries, incorporates Collet and Edwards’ types 1–3 and Van Praagh’s types A1 and A2. 10 Type 1.5 also fits within this designation. Surgically, use of the modified Van Praagh classification attempts to simplify the description and avoid confusion. 10
Differentially, truncus arteriosus can closely resemble tetralogy of Fallot or pulmonary artesia with VSD, and misdiagnosis is not uncommon. 1 Sonographic delineation should first identify the presence of pulmonary arteries and then distinguish the site of origin from a large truncal vessel rather than the ventricle.1,6 The diagnosis of common arterial trunk is best achieved in the five-chamber view with visualization of the malaligned VSD and overriding large vessel. 1 The heart will appear normal in the four-chamber view alone, unless the VSD is large and readily visible.1,12 Critical CHD, specifically outflow tract anomalies like truncus arteriosus, can be missed by imaging only the four-chamber view. 11 In June 2013, the American Institute of Ultrasound in Medicine (in conjunction with the American College of Radiology, the American College of Obstetricians and Gynecologists, and the Society of Radiologists in Ultrasound) updated its obstetrical ultrasound guideline to include evaluation of the ventricular outflow tracts. 11 Despite these revised guidelines stressing the importance of outflow tract imaging, prenatal diagnosis of outflow tract anomalies remains low and suboptimal. 11 Many cases of CHD occur in otherwise low-risk pregnancies, relying primarily on the screening anatomic ultrasound to detect abnormalities. 11
Without surgical correction, survival beyond the first year of life is uncommon. 1 As children with truncus arteriosus get older, worsening pulmonary vascular disease makes them unsuitable surgical candidates. Initial repair in the neonatal period results in the best long-term survival. 4 The Rastelli procedure for surgical repair consists of three parts: closure of the VSD, excision of the pulmonary arteries from the truncal root, and reattachment to the right ventricle with a conduit.1,6,13 Reoperation for RV-PA conduit replacement is inevitable as the patient grows. 1 Operative morbidity and mortality is low; however, truncal valve insufficiency is a significant source of long-term morbidity. Stenosis and insufficiency can present in utero with cardiac failure and hydrops, or later in life. 4 Deficits in exercise tolerance, functional status, and health-related quality of life are present throughout childhood. 14
Reported pregnancies in women with repaired truncus arteriosus are rare. Maternal risks are related to substantial hemodynamic changes including variations in systemic vascular resistance, increased cardiac output, blood volume, and heart rate.15,16 Sympathetic activity is heightened during labor, with pronounced anxiety, exertion, and pain placing further demand on the heart. 16 A vaginal delivery with adequate pain control is preferred over Cesarean section owing to differences in hemodynamic shifts and blood loss.15,16 The mother should be encouraged to labor in a right or left lateral position to reduce compression of the inferior vena cava by the gravid uterus and to maintain cardiac preload. 15
The modified World Health Organization (WHO) classification for maternal cardiovascular risk categorizes women into four classes according to heart condition, and the relative morbidity and mortality expected during pregnancy. 15 The patient presented in this case falls into class III because of her complex CHD and 45-mm dilated aortic root. Those in class III have a significant increase in maternal mortality or severe morbidity and should be monitored by cardiac and obstetric specialists throughout pregnancy, childbirth, and the puerperium. 15 The potential for lesion progression during pregnancy and postpartum is also a concern. 13 Severe truncal valve regurgitation has been reported in the third trimester, prompting early delivery. 16 Risks to the fetus and neonate include a higher preterm birth rate, especially to mothers with complex CHD, and a higher perinatal mortality. 15 Preconception counseling is imperative and should establish a clear understanding of the potential risk of pregnancy for the mother and her offspring. 15
The aforementioned case was unique because the fetus presented with a rare CHD, identical to the mother, and not related to a 22q11.2 deletion. An autosomal dominant mode of transmission was suspected initially. However, transmission of a pathogenic variant in genes currently known to be associated with truncus arteriosus was excluded through panel genetic testing. The suspicion of the healthcare team shifted to an unknown gene, yet to be identified as the causative agent of the concordant truncal defect. As in this case, recurrence risk for nonsyndromic, nonchromosomal CHD is substantially higher if the affected parent is the mother, rather than the father or sibling.17,18
Variations in incidence are further dependent on the specific type of CHD in the affected relative.17,18 When focusing on conotruncal cardiac defects, the risk of CHD is actually higher among siblings (4.4%) than parents (1.5%). 19 The risk of CHD in a sibling of an individual specifically affected with truncus is 9.4%, compared with 3.9% in a parent. 19 Among affected parents of probands with conotruncal defects, 39% have a concordant truncal lesion, whereas 69% of affected siblings have a similar anomaly. 19 Discordance between parents and siblings may reflect survival bias and reproductive success. 19 The rarity of passing on an identical truncal defect is exemplified by this unique case. The offspring of a mother with truncus arteriosus is certainly at increased risk for CHD, but not typically the same defect.
Single gene mutations are the least common cause of cardiac malformations. 20 Multifactorial inheritance explains most CHD, and the risk to future offspring increases with the number of affected individuals in the family. 20 In this mode of transmission, a multitude of genes interact with a variety of environmental influences at a vulnerable period of cardiac development. 20 Isolating the exact environmental factors acting on an individual predisposed to malformation may be difficult. However, even in multifactorial inheritance, genetic factors may be entirely responsible for a cardiac anomaly. 20
Diabetes is a well-documented maternal factor influencing CHD.2,21 The most susceptible time for development of structural CHD is before the seventh week of gestation. 2 A fivefold (3%–5%) increase in CHD compared with the general population is associated with pregestational diabetes mellitus. A higher relative risk is noted for specific defects including 4.72% for truncus arteriosus. GDM and insulin resistance diagnosed later in pregnancy are not linked to an increased risk of CHD in the fetus. 18 Therefore, GDM was not likely responsible for the established structural fetal heart defect in this case. Even if undiagnosed pregestational diabetes mellitus had influenced this pregnancy early on, two generations with the same heart condition is rare, and likely attributed to more than diabetes alone.
Conclusion
As more women with repaired conotruncal defects reach reproductive age, it is important to discuss preconceptually the increased risk for CHD in the fetus. Although the hemodynamic stress of pregnancy can pose a risk for women with repaired truncus arteriosus, maternal cardiac complications are not common. 13 Sonography plays a key role in the prenatal diagnosis of truncus arteriosus, and the lifetime surveillance of sequelae with growth and reoperation. When a single gene mutation cannot be identified, a model of multifactorial inheritance should be considered. As this case highlights, families demonstrating direct transmission of an unknown etiology are perceivable candidates for potential gene discovery.
Footnotes
Acknowledgements
Lisa Howley, MD, and Bettina Cuneo, MD, for anatomic clarification, and ascertainment of images. Kestutis Micke, MS, CGC, for genetic discernment. Julia Drose, BA, RDMS, RDCS, RVT, and Teresa Bieker, MBA, RDMS, RDCS, RVT, RT, FAIUM, for guidance and direction.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.