
Abstract
Primary renal Ewing sarcoma (ES) is an extremely rare and aggressive malignancy, with fewer than 100 cases reported worldwide. Diagnosis is difficult because its clinical and radiologic features mimic more common renal tumors, particularly in children, and the challenge increases in resource-limited settings where molecular testing may not be available. We report a 12-year-old Palestinian boy presenting with flank pain, weight loss, and night sweats. Contrast-enhanced CT revealed a large left renal mass, and radical nephrectomy was performed. Histopathology demonstrated small round blue cells with strong CD99 and NKX2.2 expression and negative WT1 staining, supporting the diagnosis of primary renal Ewing sarcoma. Confirmatory EWSR1 rearrangement testing was unavailable. The patient-initiated adjuvant chemotherapy and showed no radiologic recurrence at 3 months. This case highlights the important diagnostic role of immunohistochemistry when molecular testing is limited.
Keywords
Introduction
Ewing sarcoma (ES) was first reported as a primary bone tumor in 1921, but later recognized to arise in extra skeletal sites, including the kidney. 1 Less than 100 cases have been recorded globally since the first report of renal ES in 1975, 1 representing 1% of all renal malignancies and 5% of extra skeletal Ewing sarcoma cases.2 -4
Molecularly, ES is characterized in approximately 85% of cases by translocations involving the EWSR1 gene, most commonly t(11;22; q24;q12), resulting in fusion with FLI1. 1 While identification of these genetic alterations provides definitive molecular confirmation, access to such testing may be limited in resource-constrained settings. In these contexts, immunohistochemistry (IHC) plays a critical supportive role in establishing the diagnosis.
Previous studies have demonstrated the utility of markers such as CD99 and NKX2.2 in supporting the diagnosis of Ewing sarcoma when interpreted in conjunction with morphologic features. 5 However, these markers are highly sensitive but not entirely specific, and molecular confirmation remains the gold standard where available. Clinically, renal Ewing sarcoma often presents with flank pain, weight loss, hematuria, or a palpable mass.3,6 Notably, up to 66% of patients may have metastatic disease at diagnosis, 7 underscoring the importance of early recognition and prompt multimodal management. This report describes a case of primary renal Ewing sarcoma in a pediatric patient from Palestine and highlights the diagnostic and therapeutic challenges encountered in a resource-limited healthcare setting.
Case Presentation
A 12-year-old male from the Hebron region of Palestine, with a medical history of type 1 diabetes mellitus managed with insulin glargine (12 IU daily) and rapid-acting insulin as required was referred to our tertiary oncology center with a 5-day history of severe, unremitting left flank pain. The patient’s parents reported significant weight loss (9 kg over 2 months) and drenching night sweats, though the boy denied fever, hematuria, or urinary symptoms. Physical examination revealed a cachectic adolescent with left costovertebral tenderness and a palpable, firm left upper quadrant mass measuring approximately 10 cm in greatest dimension
Initial laboratory investigations showed normocytic anemia (hemoglobin 9 g/dl, MCV 82 fl) with normal renal function (creatinine 0.6 mg/dl). Urinalysis demonstrated microscopic hematuria (20 RBCs/hpf) without proteinuria. Serum tumor markers revealed isolated elevation of CA-125 (144 U/ml), with normal AFP (8 ng/ml) and CEA (1.2 ng/ml).
Contrast-enhanced abdominal CT imaging disclosed a 10 × 11 × 8 cm heterogeneously enhancing left renal mass (non-contrast HU 40, arterial phase HU 110) with central necrosis but no calcifications or fat density (Figure 1). The mass distorted the renal architecture without evident vascular invasion. Metastatic workup, including chest radiograph and bone scintigraphy, showed no evidence of distant disease (Figure 2).
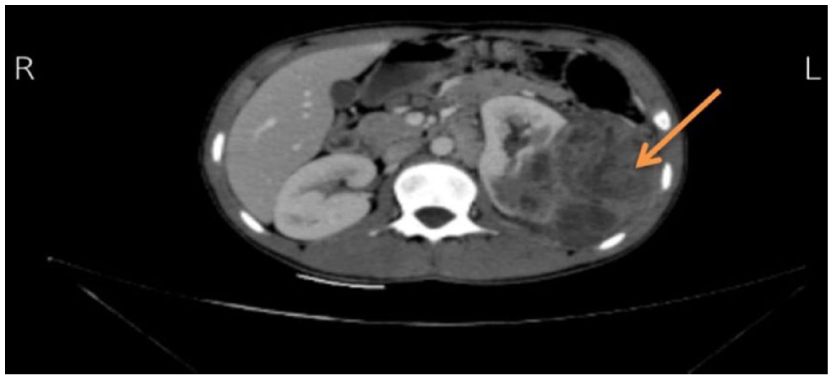
Axial contrast-enhanced CT image of the abdomen in a pediatric patient showing a large heterogeneous renal mass arising from the left kidney.
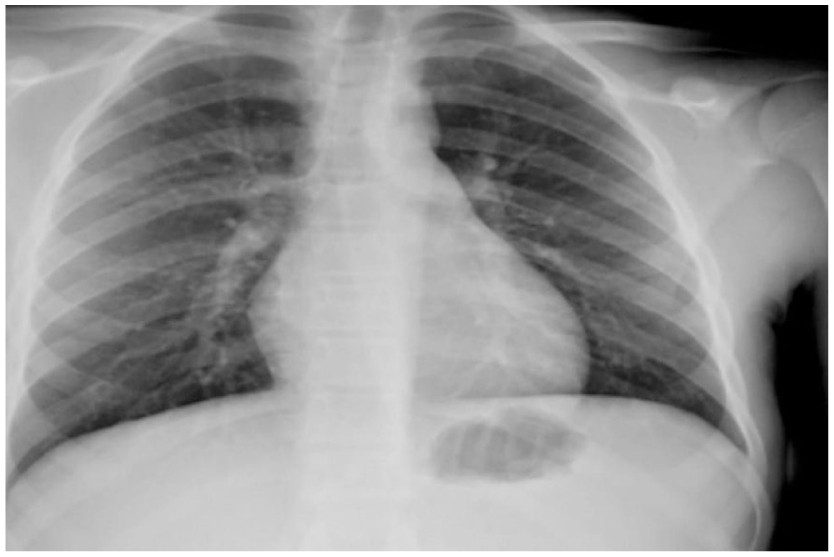
Chest X-ray (posterior–anterior view) performed during initial staging work-up, showing clear lung fields with no evidence of pulmonary metastases or pleural effusion.
The patient underwent urgent open radical nephrectomy. Intraoperative findings included complete replacement of the left kidney by a friable, tan-gray tumor with focal hemorrhage and adherence to perirenal fat. Frozen section analysis confirmed malignant small round blue cells infiltrating the renal capsule.
Histopathologic evaluation of the nephrectomy specimen revealed near-complete replacement of the renal parenchyma by a highly cellular small round blue cell tumor exhibiting extensive geographic necrosis. The neoplastic cells were arranged in solid sheets and displayed scant clear to eosinophilic cytoplasm, finely stippled chromatin, and brisk mitotic activity (15 mitoses per 10 high-power fields; Figure 3A). The vascular, ureteric, and perirenal resection margins were uninvolved by tumor. Immunohistochemical staining showed strong, diffuse membranous CD99 expression and nuclear positivity for NKX2.2 (Figure 3B and C). The tumor cells were negative for LCA, TdT, cytokeratin, WT1, desmin, and chromogranin, thereby excluding lymphoma, leukemia, nephroblastoma (Wilms tumor), rhabdomyosarcoma, and neuroblastoma. Targeted molecular panel testing identified pathogenic variants in TP53 (p.R248Q, variant allele frequency 31.62%) and STAG2 (p.Q609*, variant allele frequency 3.17%). However, confirmatory EWSR1 fusion testing was not available.
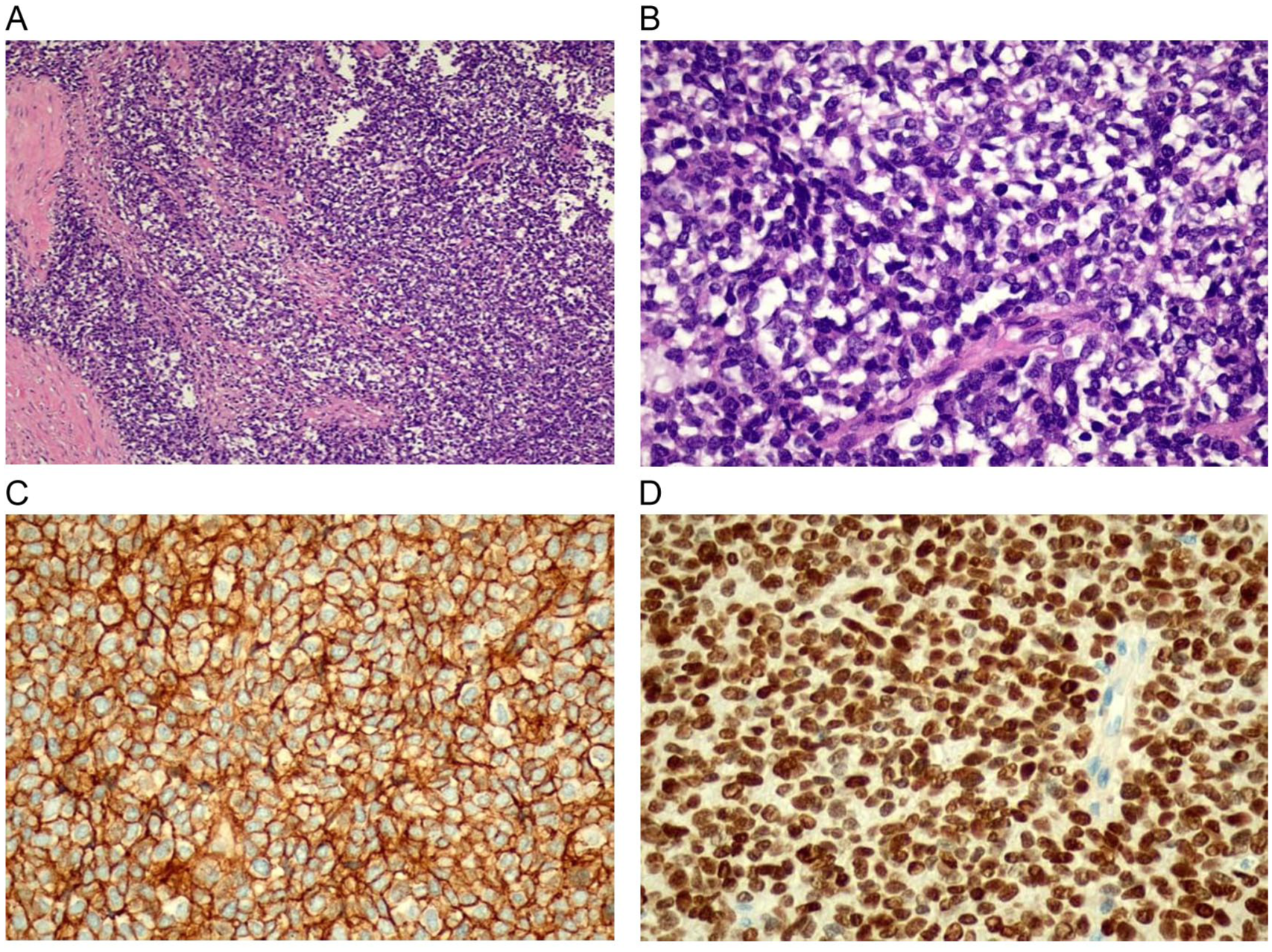
(A) This tumor is composed of solid sheets and islands of undifferentiated small round blue cells separated by fibrous bands; this tumor infiltrates the kidney and extends into the perinephric fat. The tumor exhibits areas of necrosis (not included in this image); 10× H&E, (B) high power shows sheets of tumor cells exhibiting small hyperchromatic nuclei and inconspicuous nucleoli. The cytoplasm is clear. This tumor is mitotically active; 40× H&E, (C) tumor cells show diffuse membranous positivity for CD99 immunostain; 40× CD99, and (D) tumor cells show diffuse nuclear positivity for NKX2.2 immunostain; 40× NKX2.2.
Postoperatively, the patient received adjuvant chemotherapy per the Euro-E.W.I.N.G. 99 protocol: 3 cycles of vincristine (2 mg/m2), doxorubicin (75 mg/m2), and cyclophosphamide (1.2 g/m2) alternating with ifosfamide (1.8 g/m2/day × 5) and etoposide (100 mg/m2/day × 5). Treatment was complicated by grade 3 neutropenia requiring two dose reductions. At the 3-month follow-up, contrast-enhanced MRI showed no evidence of local recurrence or metastatic disease, with the patient reporting complete resolution of flank pain and gradual weight recovery.
Given the patient’s age, Wilms tumor was initially considered in the differential diagnosis. However, several features argued against this diagnosis. Wilms tumor typically presents in younger children, with a peak incidence between 3 and 4 years of age and is uncommon after 10 years. Radiologically, Wilms tumors often demonstrate a more heterogeneous pattern with possible pseudo capsule formation, whereas the present tumor exhibited extensive necrosis without classic nephroblastoma features. Histopathologically, the absence of triphasic components (blastemal, epithelial, and stromal elements) and the presence of uniform small round blue cells further reduced the likelihood of Wilms tumor. Immunohistochemistry was decisive: strong membranous CD99 expression and nuclear NKX2.2 positivity, combined with negative WT1 staining, strongly favored primary renal Ewing sarcoma over nephroblastoma. In this case, the diagnostic approach progressed from radiologic suspicion of a malignant renal tumor to histopathologic confirmation of a small round blue cell neoplasm, with immunohistochemical profiling ultimately establishing the diagnosis of primary renal Ewing sarcoma.
Discussion
Primary Renal Ewing Sarcoma (RES) is a rare neuroectodermal tumor in the kidney that primarily affects children and adolescents, with a reported male predominance and a male-to-female ratio ranging from 2:1 to 3:1, with an average age of 29 years. 8 First reported in 1975, it represents less than 1% of renal tumors 3 and is considered part of the Ewing sarcoma family of tumors (ESFT), characterized histologically by small round blue cells. 9 Over 85% of RES cases are characterized by the translocation t(11;22; q24;q12) resulting in the fusion of the EWSR1 gene on chromosome 22 with the FLI1 gene on chromosome 11. 10
The nonspecific clinical presentation and imaging features make RES a diagnostic challenge and may lead to initial misdiagnosis as renal cell carcinoma (RCC) or, in pediatric patients, Wilms tumor. Distinguishing it from other similar entities is vital as the management differs, such as Wilms tumor, rhabdoid tumor, neuroblastoma, lymphoma, clear cell sarcoma, small cell carcinoma, synovial sarcoma and desmoplastic small round cell tumor. In our case, the patient’s age initially supported consideration of Wilms tumor; however, several findings argued against this diagnosis. Wilms tumor most commonly presents in children aged 3 to 4 years and is less frequent after the first decade of life. Radiologically, the lesion lacked classic nephroblastoma features. Histopathologic evaluation did not demonstrate the typical triphasic pattern of Wilms tumor but instead revealed uniform small round blue cells with high mitotic activity. Furthermore, immunohistochemistry showed strong CD99 and NKX2.2 positivity with negative WT1 staining, supporting the diagnosis of primary renal Ewing sarcoma over nephroblastoma.
Common symptoms include flank pain and hematuria. Ultrasound, CT and MRI are used to characterize the mass, though they may not definitively distinguish RES from other renal malignancies. 7 Investigations may include mercaptoacetyltriglycine renal scan and histologic evaluation requiring expert personnel; with immunostaining for CD99 and Synaptophysin, in addition to FISH studies. 11 In resource-rich settings, such comprehensive work-up is standard. In contrast, in resource-limited settings, each diagnostic investigation must be carefully weighed against availability, cost, and accessibility constraints.
In 2022, Li et al. analyzed imaging modalities for diagnosing RES and proposed contrast-enhanced ultrasound as a useful initial modality due to its cost-effectiveness and lack of radiation exposure, while emphasizing that definitive diagnosis still relies on histopathology and ancillary studies. 7 Although advanced imaging techniques may not always be readily accessible in low-resource settings, their judicious use remains important in facilitating timely diagnosis.
Definitive diagnosis relies on histopathology, immunohistochemistry (IHC), and cytogenetic studies with CD99 being a commonly used IHC marker. Among small round cell tumors, the nuclear protein WT1 has been expressed in the majority of Wilms tumors and desmoplastic small round cell tumors, but not typically in primary renal Ewing sarcoma (RES), neuroblastomas, or rhabdomyosarcomas. 10 Jimenez et al. found that all their examined RES cases in their series were strongly CD99-positive. They also demonstrated FLI-1 expression was observed 63% of RES cases while none of the Wilms tumors expressed FLI-1, and WT1 expression was observed in 78% of Wilms tumors but in none of the RES cases. 10 These findings highlight the value of immunohistochemistry—particularly FLI-1 and CD99—in supporting the diagnosis of RES; however, such markers should be interpreted in conjunction with morphologic features and, where available, molecular studies.
Compared with previously published cases of primary renal Ewing sarcoma, our patient shares several common features, including male sex, large tumor size at presentation (>10 cm), and symptomatic flank pain. However, unlike many reported cases in which metastases are present at diagnosis—reported in up to 60% to 65% of patients—our patient had localized disease at presentation without radiological evidence of distant spread.
Treatment strategies for RES typically involve neoadjuvant chemotherapy, surgical resection and adjuvant chemotherapy. Standard Ewing sarcoma protocols commonly describe induction chemotherapy followed by local control and completion of systemic therapy, often totaling approximately 14 to 15 cycles.12,13 However, management of primary renal Ewing sarcoma may vary based on disease presentation and institutional context. In this case, upfront radical nephrectomy was performed because the tumor was localized and deemed surgically resectable at presentation. The patient subsequently initiated adjuvant multiagent chemotherapy in accordance with Ewing sarcoma treatment principles. 14 Completion of standard systemic therapy was planned; however, documentation beyond 3 cycles was limited due to transfer barriers and follow-up constraints related to healthcare access challenges.
Prognosis for RES is guarded, particularly in metastatic cases at diagnosis. Early recognition and appropriate multimodal management are crucial for improving outcomes. This case highlights the challenges of managing rare pediatric malignancies in resource-limited settings. Beyond limitations in molecular diagnostics, barriers in healthcare access and treatment continuity may influence long-term outcomes.
A limitation of this report is the relatively short follow-up duration of 3 months. Given the aggressive nature of primary renal Ewing sarcoma and its potential for recurrence, longer-term surveillance is necessary to determine durable oncologic outcomes.
Conclusion
Our report emphasizes that early recognition, careful integration of histopathologic and immunohistochemical findings, and coordinated multidisciplinary care are essential for optimizing outcomes in primary renal Ewing sarcoma, particularly in under-resourced environments. While molecular confirmation may not always be accessible, accurate diagnosis and adherence to multimodal treatment principles remain critical. This case illustrates how healthcare system constraints can influence diagnostic pathways, treatment continuity, and follow-up in rare pediatric malignancies, underscoring the importance of strengthening oncologic care capacity in under-resourced regions.
Footnotes
Abbreviations
ES: Ewing sarcoma
IHC: immunohistochemistry
VDC/IE: vincristine/doxorubicin/cyclophosphamide + ifosfamide/etoposide
Ethical Considerations
According to institutional policy at Palestinian Ministry of Health, formal ethical approval was not required for publication of a single anonymized case report. The study was conducted in accordance with local legislation and institutional requirements.
Consent for Publication
Written informed consent for publication of the clinical details and accompanying images was obtained from the patient’s legal guardian.
Author Contributions
Dr. Abd Alfattah Shalalfa conceptualized the case report. Dr. Abd Alfattah Shalalfa and Dr. Jakub Khzouz managed the patient and collected clinical data. Dr. Neveen Shalalfa, Dr. Mays Najjar, Dr. Amenah Shahin, and Dr. Ghadeer Eideh conducted the literature review and interpreted findings. Dr. Amenah Shahin, Dr. Mays Najjar, and Dr. Ghadeer Eideh drafted the manuscript. Dr. Mays Najjar, Dr. Jakub Khzouz, and Dr. Neveen Shalalfa critically revised it. All authors approved the final manuscript and are accountable for all aspects of the work.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.