
Abstract
Ewing’s sarcoma is a primary malignant bone tumor affecting individuals in the second decade of life. Primary sarcomas of the spine are rare and the occurrence of primary Ewing’s sarcoma in the spine is very rare. There are 2 types of Ewing’s sarcoma of the spine, Ewing’s sarcoma of the sacral spine which is very aggressive with a poor prognosis and Ewing’s sarcoma of the non-sacral spine which is an extremely rare occurrence. The patient may have a neurological deficit when the tumor extends into the spinal canal, causing compression of the spinal cord. Magnetic resonance imaging (MRI) is very sensitive in diagnosing the tumor and Evaluation of the extent of the tumor. Here, we report the case of a 12-year-old boy who presented with low back pain, weakness of both lower limbs and bilateral spastic paraplegia progressively evolving since 1 month. The CT and MRI scans showed the presence of a tissue lesion process centered on the vertebral body of L1, heterogeneously enhanced after injection of Gadolinium respecting the posterior arch, without bulging of the posterior wall with epiduritis, endocanal extension and spinal cord compression. The patient underwent decompression with surgical biopsy and posterior stabilization of the spine. Histopathology and immunohistochemistry studies confirmed the diagnosis of Ewing’s sarcoma and the patient was referred to an oncopediatric center for combined chemotherapy and radiotherapy, but died at home a few days later before the start of treatment.
Introduction
Ewing’s sarcoma, first described by James Ewing in 1921 as diffuse malignant endothelioma of bone, is a highly malignant primary bone tumor, preferentially affecting children and adolescents, and more rarely adults. 1 It accounts for only 3.5% to 14.9% of all primary bone sarcomas. The age of presentation ranges from 12 to 24 years (average : 21 years).2,3 It can affect any part of the skeleton, but the most affected parts are the ilium and the diaphyses of the femur and tibia. 2 Primary involvement of the non-sacral spine is extremely rare and accounts for approximately 0.9% of all cases.2,4 We report a case of primary spinal Ewing’s sarcoma in a 12-year-old child.
Case report
A 12 year old child with no notable history, who presented for 1 month with lumbar pain, of slight intensity progressively worsening, for which he was treated with analgesics without improvement, the evolution was marked by an accentuation of the pain, a weakness of both lower limbs. There was no history of trauma. Clinical examination found tenderness at the spinous processes of the dorsolumbar hinge, bilateral spastic paraplegia, and mild hypoesthesia of all sensations below the level of D11. A CT scan of the spine was ordered and showed a lesional process in the epidural space extending from D11 to L2, centered at D12-L1, infiltrating the vertebral bodies and the holes of conjugation (Figure 1). The MRI complement showed the presence of a lesion process centered on the vertebral body of L1 in T1 hyposignal, T2 hyposignal, heterogeneously enhanced after injection of gadolinium respecting the posterior arch, without bulging of the posterior wall with epiduritis, endocanal extension and spinal cord compression (Figure 2). A biological assessment was performed and showed an elevated level of LDH (373 IU / L) and the rest of the assessment was without particularities. The child underwent decompressive laminectomy with surgical biopsy as the first line of treatment with analgesics, there was no use of corticosteroids. The diagnosis of Ewing’s sarcoma was confirmed by the anatomopathological study of the surgical specimen. Indeed, the histopathology showed a malignant tumor proliferation made of round cells of more or less lobulated architecture with pseudorosettes in some places (Figure 3a). The tumor cells were monomorphic, with a rounded or oval nucleus, regular outlines, homogeneous finely granulated vesicular chromatin containing a small nucleolus. Immunohistochemistry showed diffuse positive membrane staining of tumor cells with the anti-CD99 antibody (Figure 3b) with an estimated high proliferation index of 40%. Staining with anti CD45; Desmin; Actin smooth muscle; EMA; PS100; and synaptophysin antibodies remained negative. A thoracic-abdominal-pelvic CT scan was performed as part of the extension workup and showed multiple diffuse bilateral pulmonary nodules involving both lung fields with a random distribution realizing a “Release of balloons” sign indicating secondary locations (metastases) (Figure 4), the evolution was marked by a good initial improvement of motor weakness. The patient was referred to a specialized oncotherapy center for combined chemotherapy and radiotherapy but died at home a few days later before the start of treatment.

Lumbar CT scan in parenchymal window (a) and bone window (b and c) showing a lesional process centered on the epidural space extended from D11 to L2, centered at the level of D11-D12 slightly enhanced after injection of contrast medium, infiltrating the vertebral bodies and the conjugation holes opposite and the adjacent soft parts.

Sagittal and axial sections of a dorsolumbar MRI showing a lesional process centered on the vertebral body of L1 in T1 hyposignal, heterogeneous T2 signal, discretely and heterogeneously enhanced after injection of Gadolinium, without bulging of the posterior wall or involvement of the posterior arch, associated with epiduritis, endocanal extension and spinal cord compression.
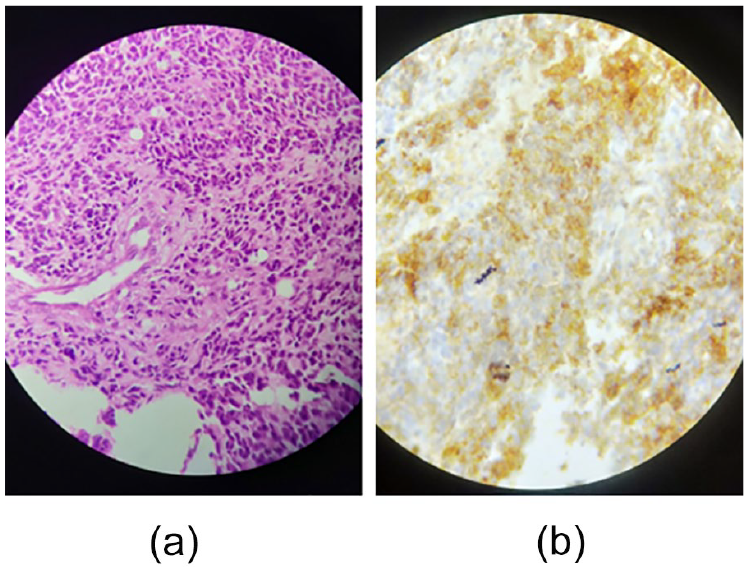
(a): Round cell tumor proliferation with pseudorosette images; HE staining at ×40 magnification. (b): Immunostaining with anti CD99 antibody: diffuse membrane staining.

Axial (a and b) and coronal (c) sections of a chest CT scan in the parenchymal window showing multiple diffuse bilateral pulmonary nodules with random distribution realizing the balloon-release appearance related to pulmonary metastases.
Discussion
Ewing’s sarcoma is a small round cell tumor. 1 Its origin has been the subject of much discussion. An endothelial origin was initially proposed by James Ewing, later a neural origin was widely suspected based on the observation of the t(11; 22) translocation, and an epithelial origin was evoked due to the expression, albeit inconsistent, of cytokeratin and tight junction proteins.1,5,6 It is the second most common primary malignancy in bone after osteosarcoma. 1 Its peak incidence is in the second decade of life and it is very rare after the age of 30. It occurs mainly during childhood, when the bones are growing. It usually affects the metaphyseal plates of the long bones and rarely affects the spine, the most common location being the sacrum. Involvement of the non-sacral spine has been reported, but is rare.7,8
Due to the expansive nature of the lesion, local swelling and pain are the most common manifestations in cases of long bone involvement. Systemic symptoms may also be present. Involvement of the non-sacral spine usually manifests as spinal cord compression, often late in the course of the disease. The presentation of acute paraplegia without significant localized pain and swelling has not been reported in the literature. 9
The radiological signs are not pathognomonic and the visible lesions are patches of osteolysis with destruction of the cortex. There is corticoperiosteal proliferation, lamellar, or in the form of spicules when the cortex is destroyed associated with significant infiltration of the adjacent soft tissues. 6 Magnetic resonance imaging represents the gold standard for early diagnosis, assessment of the extent of the tumor in the soft tissues and determination of the therapeutic strategy. 10
In this location, neurogenic tumors, malignant lymphoma, rhabdomyosarcoma, synoviolosarcoma, and Histiocytosis are the representative differential diagnoses; it is difficult to make a definitive diagnosis of Ewing sarcoma. 10
The diagnosis of Ewing’s sarcoma is based on histology, immunophenotyping and the demonstration of one of the specific translocations. The tumor expresses CD99 in 90% of cases. This marking is non-specific and can be found in some non-Hodgkin’s malignant lymphomas, alveolar rhabdomyosarcoma and undifferentiated carcinoma. These diagnoses were ruled out by the negativity of the respective markers (CD45; demsin and EMA), as well as neuroblastoma (negative synaptophysin). Cytogenetic analysis, by Fluorescence in situ hybridization (FISH) technique, allows to confirm the diagnosis. 11 This technique allows to find the reciprocal translocation t(11;22)(q24;q12) involving chromosome 22 located on EWS-FLI 1 in more or less 90% of cases. 2 This technique is not available in our training.
The extension workup includes magnetic resonance imaging to determine local extension, a standard chest X-ray, a chest CT scan to look for lung metastases, and a technetium bone scan, which may reveal bone locations other than the primary tumor. 1
When deciding on the treatment of Ewing’s sarcoma of the mobile spine, the determining factor is the presence of neurological deficits which, once present, are often rapidly progressive. In such circumstances, only rapid surgical decompression can offer a maximum chance of recovery. 9 The approach is defined by the type of injury. Anterior decompression is warranted in cases where cord compression is due to body extension. Ewing’s sarcoma often tends to invade the spinal canal from the paravertebral soft tissue component through the intervertebral foramen, compressing the cord circumferentially. This makes laminectomy an effective approach for cord decompression. 7 In either case, postoperative chemotherapy for micrometastasis control and local control with radiation therapy are warranted. In cases where the diagnosis is anticipated prior to neurological involvement, confirmation by needle biopsy is advised, and once made, the patient should undergo a 3- or 4-drug neoadjuvant chemotherapy regimen. 9 This not only reduces the primary tumor, thus increasing the chance of complete excision, but also manages micrometastases and gives an idea of the tumor’s responsiveness to adjuvant therapy. 7 The classical chemotherapy protocol of ES is VACA (vincristine, dactinomycin, cyclophosphamide and doxorubicin), many other drugs have been added like isofosfamide and or etoposide (VAC/IE) which improved the outcome. 12 Chemotherapy as first-line treatment for Ewing’s Sarcoma of the spine can achieve similar results to primary surgery in preserving neurological function, even in cases of major neurological deficits. Neoadjuvant chemotherapy is associated with improved overall survival compared with primary surgery. In addition, induction chemotherapy contributes to a higher rate of en bloc resection with an R0 margin, which is an independent prognostic factor for improved overall survival. 13 This is followed by surgery or radiotherapy or both. Primary radiotherapy is not recommended in these cases because post-treatment edema will lead to the development or progression of neural compression. 9
In a study by Marco et al 14 to evaluate the oncologic outcome of patients treated with chemotherapy and radiotherapy for Ewing’s spine sarcoma, the disease-free survival rate was 49% at 5 years and 36% at 10 years.
Conclusion
Primary Ewing’s sarcoma of the non-sacral spine is an extremely rare primary malignant bone tumor, which should be suspected in young patients with rapidly worsening low back pain. The diagnosis is guided by imaging and confirmed by histology and molecular biology data. Treatment with multimodal chemotherapy, radiotherapy and surgery at the earliest possible stage improves survival, but the prognosis is poor when the disease is diagnosed at the metastatic stage or when the serum lactate dehydrogenase level is elevated before treatment is started.
Footnotes
Author Contributions
All authors contributed equally to this work.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.