
Abstract
Mitral regurgitation (MR) is a frequent and heterogeneous disease associated with significant morbidity and mortality. Normal mitral valve closure requires a complex interplay between left ventricular shape and dynamics, papillary muscles, mitral annulus, and both mitral leaflets. Anomalies of the left ventricle or the left atrium can therefore cause functional mitral regurgitation (FMR), which can be improved by related therapeutic interventions. However, heart failure therapies (revascularization, resynchronization, and pharmacological treatment) are incomplete treatments for FMR, which remains frequent. Another key component in FMR is the valve itself, as the leaflets undergo significant changes in the process. Those changes can be beneficial and prevent FMR, or detrimental and contribute to valve dysfunction. Mitral valve remodeling starts with biological changes occurring at the cellular level and is ultimately reflected macroscopically by leaflet enlargement and/or thickening. This remodeling also modifies the biomechanical properties of the valve, critical for adequate coaptation. Some conditions result in adequate valve enlargement, which remains proportional with the annulus and ventricular sizes, preventing FMR despite significant myocardial disease. Other conditions, ischemic heart disease in particular, are associated with maladaptive valve changes with excessive thickening and fibrotic remodeling. This adverse evolution is associated with the presence of transforming growth factor beta in the leaflets, which can be influenced pharmacologically. Clinical observational studies and experimental works are suggesting potential therapeutic targets to prevent FMR by influencing the valve biology. While those interventions still require clinical validation, they could represent an entirely new strategy for this common and morbid disease.
Keywords
Key points
Mitral valve remodeling has an important role in the genesis of functional mitral regurgitation.
Compensatory valve enlargement can prevent mitral regurgitation even in the most severe left ventricle dilatation.
Adverse remodeling leading to a fibrotic valvular phenotype is associated with secondary mitral regurgitation, especially in patients after myocardial infarction.
More studies are needed to understand the transition between compensatory enlargement and adverse fibrotic remodeling, as this could lead to new therapeutic opportunities.
Introduction
Valvular heart diseases are frequent, with a heavy burden on morbidity and mortality. 1 As prevalence increases with age, it is estimated that more than 10% of those 75 years old and older have a moderate or severe valve disease. With aortic stenosis, mitral regurgitation (MR) represents the most common condition. MR can be broadly classified as organic/primary when the mitral valve is abnormal, or functional/secondary when the valve dysfunction is caused by left ventricle (LV) or left atrium (LA) anomalies. Ischemic MR (IMR) is the term used for functional mitral regurgitation (FMR) caused by myocardial infarction (MI). While FMR terminology implicitly assumes normal leaflet biology (in opposition to primary MR), the valve undergoes significant microscopic and macroscopic changes induced by the underlying cardiac disease with a crucial impact on valve function. Those changes can result in improved mitral geometry and decreased regurgitation, or evolve into a maladaptive state with fibrotic remodeling contributing to MR. A better understanding of the underlying processes could potentially lead to new therapeutic opportunities. This manuscript will review the current knowledge regarding mitral valve remodeling in FMR, integrating the respective roles of ventricular, atrial, and valvular components in the genesis of FMR.
Valve Development, Normal Structure, and Ageing
Cardiac valve development is a complex process constantly influenced by changing cardiac morphology and hemodynamics. Both mitral and tricuspid valves are derived from endocardial cushions. Detailed embryology is beyond the scope of the current review, but developmental processes involve several growth factors from the transforming growth factor beta (TGF-β) superfamily, driving the endothelial cells to acquire mesenchymal cells properties (endothelial-to-mesenchymal transformation [EMT]). 2 The valve ultimately evolves into three distinct histological layers (Figure 1). The first layer (atrialis) consists of lamellar collagen fibers and elastin. The mid-leaflet layer is the spongiosa, which consists of a loose connective tissue that is rich in proteoglycans and glycosaminoglycans. The layer situated on the ventricular side of the valve is the fibrosa, formed by dense collagen fibers. The mitral leaflets are covered by valvular endothelial cells, with quiescent interstitial cells contributing to homeostatic remodeling of the extracellular matrix. Macroscopically, the mitral leaflets are asymmetric, the anterior being longer while the posterior has a wider implantation and constituted of typically 3 scallops. The mitral annulus has a non-planar saddle shape and dynamic motion throughout the cardiac cycle. The chordae tendineae are fibrous strings connecting the leaflets to the papillary muscles (PM) and ensuring adequate leaflet coaptation. Primary chordae are attached to the free edge of the leaflets, while secondary chordae, thicker, are found in the body of the leaflets. 3 During ventricular systole, intraventricular pressure rises and is opposed to tethering forces that restrict leaflet closure. 4 Under the rising systolic pressure, leaflet extensibility results in an acute increase of leaflet area, contributing to normal valve closure. 5 Importantly, adequate leaflet coaptation requires an excess of leaflet area in relation to the closure area, allowing free edges joining to form a coaptation seal.
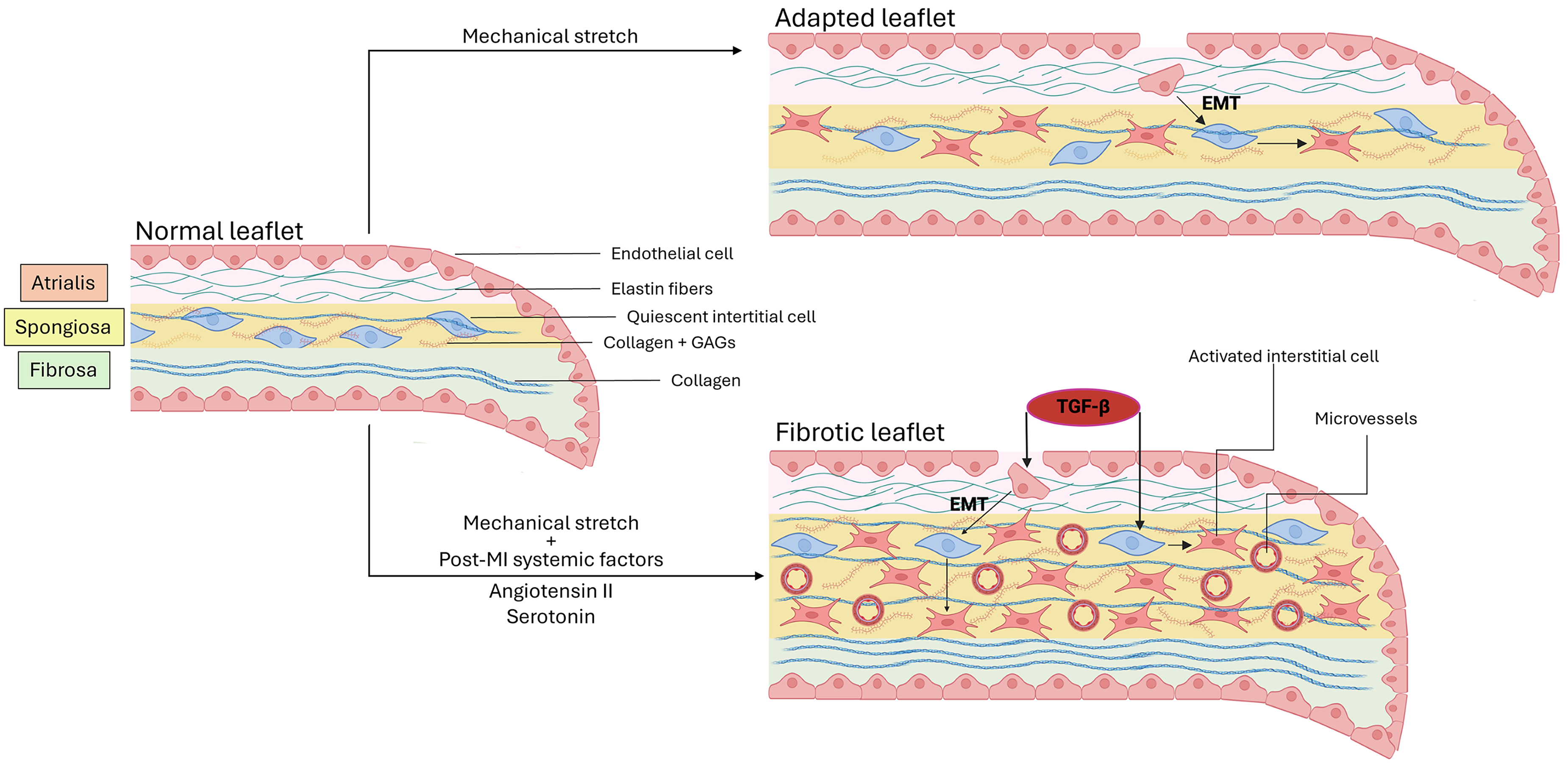
Leaflet Response to Mechanical Stress and Ischemic Environment
While the valve overall structure remains relatively stable in adult life, changes associated with ageing have been reported. Progressive and significant thickening of the valve has been described in autopsy studies of normal hearts, without clear relation with body surface area and without significant sex related differences. 6 Changes in microscopic structure are also described, including progressive collagen fibers disorientation, lipid accumulation, calcification, and decreased cellularity. 7 While these changes may not always directly affect valve function, leaflet stiffness, and thickness can ultimately impact biomechanical properties of the leaflets and their coaptation, 8 potentially explaining in part the increased prevalence of regurgitation with age. Adequate mitral function therefore depends on a very delicate balance of forces and geometry.
Valve Remodeling: Ventricular and Atrial Mechanisms
Any cardiac disease impairing the mitral valve surrounding structures can affect its function, causing secondary regurgitation. The most typical scenario are patients with primary LV disease, causing a variable combination of decreased closure forces (decreased trans-mitral gradient in systole) and PM displacement away from the annulus because of LV dilatation and dysfunction. Those geometric alterations will result in an apical shift of the valve, increasing the closure area and reducing the amount of leaflet excess to form the coaptation (Figure 2). As MR itself causes more LV dilatation and remodeling, a vicious cycle occurs and accelerates heart failure. 9 Although virtually any cause of LV dysfunction can induce FMR, ischemic heart disease is the most common and by far the most studied cause. IMR has been described to affect nearly a third of patients after MI, 10 and is associated with adverse clinical events, even when the MR is not severe. 11 Although any infarcted territory can ultimately cause IMR, inferior MI affecting the PM or the underlying myocardium is associated with higher incidence and greater severity of regurgitation despite often milder infarct size and LV dysfunction.12,13 Single blood flow in the posteromedial PM also make it more vulnerable to ischemia and dysfunction. 14 Multiple other factors, including PM angle, interpapillary muscle distance and PM symmetry are linked with FMR. 15 Papillary muscle rupture is the most extreme manifestation of MR after MI. While this complication is rare (∼1%), its consequences are disastrous and will require surgery in most cases. 16 Partial PM rupture is also possible, with sometime more subtle presentations but still associated with significant morbidity. 17 Non-ischemic heart disease can also modify LV anatomy and create FMR. 18 LV dyssynchrony is another factor that contributes to FMR. 19 In patients eligible for cardiac resynchronization therapy, reverse remodeling can occur, and sustained significant decrease in FMR can be obtained with impact on prognosis. 20 With those mechanisms in mind, the first therapeutic options for ventricular FMR are oriented on the LV: revascularization and resynchronization when indicated, as well as the pharmacological armamentarium for systolic heart failure. The use of more recently approved sodium-glucose cotransporter 2 (SGLT2) inhibitors in heart failure has been associated with favorable outcome on FMR. 21
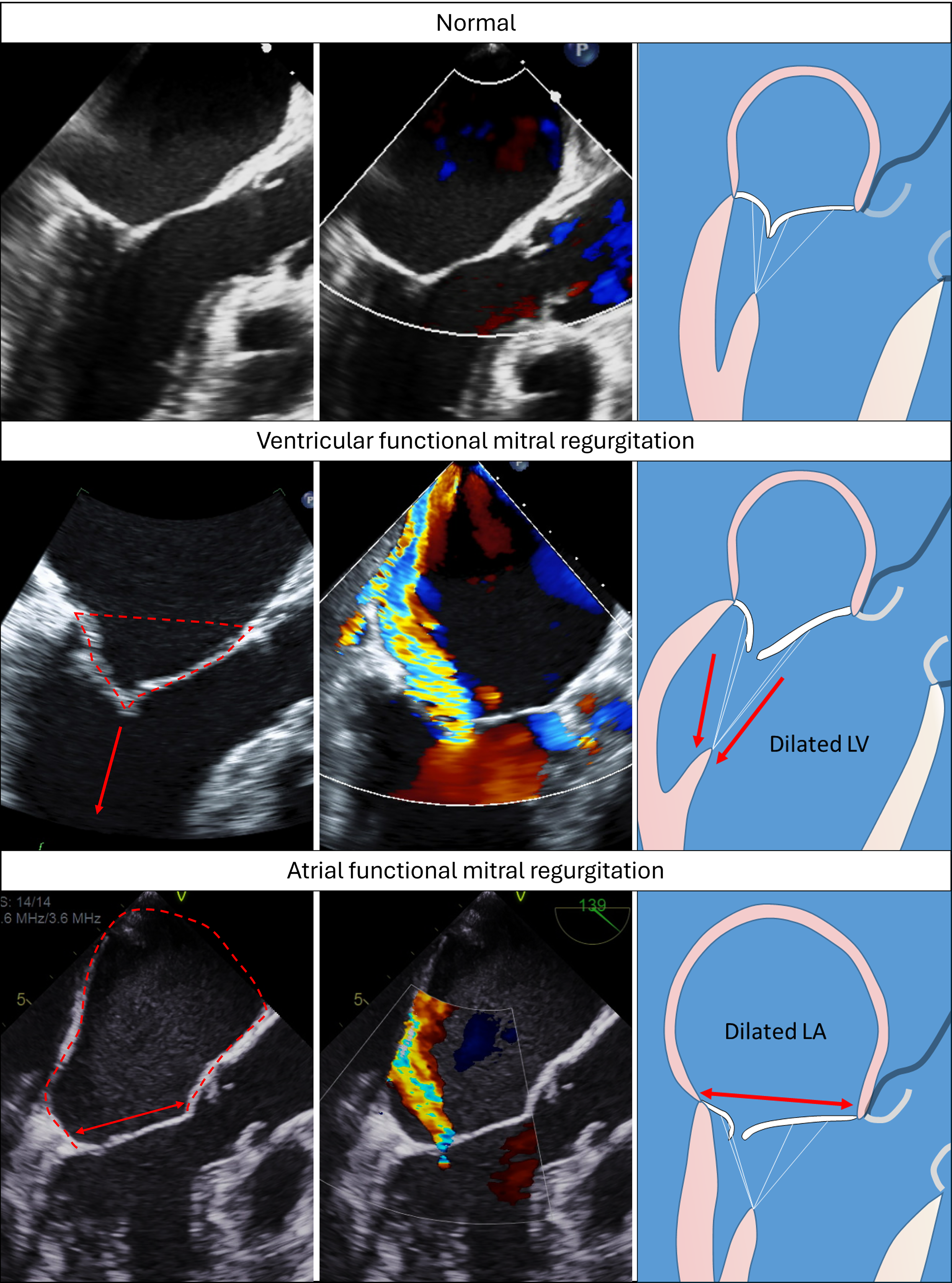
Geometrical Deformations Leading to Functional Mitral Regurgitation. Transoesophageal echocardiography images with and without color Doppler and mechanistic illustrations showing normal mitral valve (top row), ventricular functional mitral regurgitation (middle row) and atrial functional mitral regurgitation (bottom row). Ventricular functional mitral regurgitation is associated with significant leaflet tenting (dotted red line) and apical displacement of the coaptation point as the leaflets are pulled by the papillary muscles (red arrows). Atrial functional mitral regurgitation is caused by left atrial dilatation (dotted red line), annular dilatation (red arrow) and annular deformation despite a normal left ventricle. The leaflets are losing their normal concavity towards the left ventricle. In both cases, the closure area increases. Without leaflet enlargement, the excess of leaflet available for coaptation is reduced, resulting in mitral regurgitation.
Atrial FMR happens with predominantly annulus enlargement from LA dilatation, without significant LV anomaly. 22 In a normal situation, the increase in intracavity pressure during systole causes the MV leaflets to be concave toward the LV. With annular dilatation, the leaflets straighten or even become convex toward the LV (Figure 2). Mitral annulus dimension and dynamics, LA size and leaflet geometry all participate in regurgitation. 23 Epidemiologically, a community study found that nearly 40% of FMR were of atrial origin. 24 Compared with ventricular FMR, patients with atrial FMR are often older, with more female representation and high atrial fibrillation (AF) burden. While ventricular FMR has a higher rate of mortality and cardiovascular events, mortality in atrial FMR is still clearly excessive compared to the normal population.25,26 By being two syndromes strongly associated with atrial myopathy, lone AF and heart failure with preserved ejection fraction are frequent culprit of atrial FMR. In AF, the mitral annulus has reduced dynamic change in area between systole and diastole, causing higher closure area. In patients with restored sinus rhythm, gradual increase in annular dynamics happens, owed predominantly to increased presystolic contraction. The improved annular dynamics is associated with reduction in LA size and annular dimension with subsequent decreased mitral regurgitation severity.27–29 Heart failure with preserved ejection fraction is also a cause of atrial remodeling in relation with chronically elevated filling pressures and LV diastolic dysfunction, even in the absence of AF.30,31
Although ventricular and atrial changes are clearly related to FMR, regurgitation severity is not always proportional to the severity of LV or LA dilatation or dysfunction.13,32 This suggests additional factors involved in the genesis of FMR. Current treatments for FMR aim to attenuate the LV/LA disease; those approaches are often insufficient and surgical or percutaneous interventions can be performed. Recent data with transcatheter edge-to-edge repair in FMR show positive results that are however not applicable to all cases,33,34 leaving a significant proportion of patients without therapeutic option.
Leaflet Response to Mechanical Stretch
Several observations suggest that stretch imposed on mitral leaflets can induce biological changes resulting in leaflet adaptation, counterbalancing the effects of atrial and ventricular changes. One of the first and most compelling evidence of mitral valve plasticity comes from an autopsy study analyzing mitral valve size in patients with aortic valve pathologies. This study demonstrated significantly enlarged mitral leaflet size in patients with LV dilatation. 35 Later, the development of three-dimensional (3D) imaging modalities (3D echocardiography and cardiac computed tomography) allowed non-invasive evaluation of mitral valve morphology, including the precise computation of total leaflet area. 36 The measure of mitral leaflet area can be expressed as a ratio with the systolic closure area. An excess of leaflet area roughly around 1.5 times the systolic closure area allows the leaflets to fold and form a coaptation seal.36–38 Using these non-invasive imaging techniques, observational studies have shown consistently increased leaflet size in patients with heart failure from ischemic or non-ischemic causes.36–38 Echoing the autopsy results in patients with aortic disease, 3D echocardiography in patients with chronic aortic regurgitation and LV dilatation showed valve leaflet size 31% greater than control patients without heart disease. 37 Crucial data have emerged from the comparison of heart failure patients with versus without FMR. Larger valves are observed in patients without FMR, suggesting that valve adaptation may not be equal for all individuals.36,38 This suggest that even the largest LV size can be free of FMR if the leaflet area can adapt to remain proportional to the systolic closure area. Mitral leaflet compensation has similarly been shown in patients with isolated annular dilatation, 39 indicating that the atrial causes of FMR can also generate mitral leaflet adaptation. In this case, leaflet enlargement becomes insufficient with larger annulus dilatation coinciding with the appearance of MR, suggesting a potential limit to valvular adaptation. Leaflet adaptation has been also suggested in the tricuspid valve in patients with pulmonary hypertension and dilated right ventricle, 40 as well as in aortic valves of patients with aortic root dilatation. 41 In both cases, the importance of leaflet enlargement was inversely associated with valve regurgitation. Similarly to the mitral valve, aortic valve enlargement seems to have a limit in cases of more severe or asymmetric aortic dilatation.
Those clinical observations are supported by experimental prospective data from animal models with serial imaging combined with histopathologic analyses. Turbulent flow associated with MR by itself is a trigger for mitral valve remodeling, causing change in collagen synthesis, increased expression of matrix metalloproteinases and elastin. 42 However, leaflet remodeling has been shown to occur without MR in models of mechanical stretch imposed by PM retraction without heart failure and without MR. In this model, pure mechanical stretch was associated with leaflet enlargement and thickening. 43 Biological changes were seen in the leaflets and chordae, including the presence of endothelial cells expressing the endothelial-mesenchymal transdifferentiation marker alpha-smooth muscle actin (αSMA) and collagen deposition. It is noteworthy to mention that active adaptation of mitral leaflets was found in a relatively short period of time (60 days) after the application of mechanical stretch. Additional animal models of heart failure from different causes have demonstrated similar dynamic valve biology and morphological changes. Rat models of aortic regurgitation showed acute changes in mitral leaflets biology as early as 48 h after induction of aortic regurgitation, with subsequent leaflet thickening and elongation. 44 Large animal (ovine) models of aortic regurgitation have also shown increased valve area in the first months after induction of aortic regurgitation, with evidence of cell proliferation and extracellular matrix remodeling. 45 Tachycardia-induced cardiomyopathy models have also showed leaflet length augmentation and remodeling near the leaflet edge. 46 Increased cell density, loss of normal leaflet layer structure, and increased turnover of collagen and elastic fibers is reported in such models. 47 These models focus on pathological conditions, but the mitral valve is also subject to dynamic adaptation in non-pathological condition such as pregnancy, which is associated with an increase in blood volume increasing LV and LA dimensions. 48 Although mild degree of valvular regurgitation can occur during pregnancy, occurrence of pathological MR is quite uncommon. 49 Bovine models of pregnancy showing changes in heart physiology similar to humans have shown significant mitral valve remodeling, including increased leaflet area, increased number of attachments by chordae tendineae, increased leaflet thickness and total collagen concentration suggesting de novo collagen synthesis.50,51 Those morphological changes were associated with altered mechanical properties, with leaflet stiffness increasing during early pregnancy but returning to normal by late pregnancy. Whether these changes are caused only by leaflet stretch from dilated left heart or influenced by the neurohormonal changes seen in pregnancy remains to be determined. Interestingly, those changes are not limited to the mitral valve but seem to occur in all cardiac valves, including aortic and pulmonary valves that are not influenced by LV dilatation. 52
While those studies collectively suggest that adaptive valve enlargement can prevent FMR, increase in mitral leaflet area is not always associated with better valve function. For instance, hypertrophic cardiomyopathy (HCM) is associated with mitral regurgitation from systolic anterior motion of the valve, which has been attributed to different explanations, including altered subvalvular flow dynamics 53 and abnormal PM position. 54 Anatomopathological analyses of these valves showed increased mitral leaflet area, leaflet thickness and leaflet mass. 55 Imaging studies using noninvasive 3D echocardiography 56 also revealed mitral valve enlargement in patients with HCM, which was greater in patients with LV outflow tract obstruction. Additional magnetic resonance imaging studies also demonstrated mitral valve changes, which were seen even in genotype-positive patients without LV hypertrophy. The ratio between anterior leaflet length and LV outflow tract diameter was associated with LV obstruction. 57 Although the presence of mitral leaflet enlargement and its contribution to systolic anterior motion and LV outflow tract obstruction is well documented, the underlying mechanisms are less clear and possibly multiple. The presence of leaflet anomalies in positive genotype patients without hypertrophy 57 could suggest a direct mutational effect. However, a paracrine effect from the adjacent myocardium with subsequent valvular effect 58 and/or contribution of mechanical stress from stretched leaflets secondary to systolic anterior motion itself have also been suggested. Leaflet elongation is therefore well described in different physiological and pathological states, demonstrating the capacity of cardiac valves to change their surface in adult life. This phenomenon can prevent MR in some situations (dilated ventricle but increased mitral surface area and overall preserved ratio of leaflet size to closure area) but can also contribute to valve dysfunction in patients with HCM and associated regurgitation.
These findings put more depth into the mechanisms of FMR, which might not only be expressed in terms of LV/LA dilatation, but also linked to mitral leaflet size, which can change in response to stretch. The capacity of leaflets to actively enlarge and the factors limiting this adaptation are therefore critical variables to consider, keeping in mind that leaflet elongation can also be detrimental in some conditions.
Excessive Leaflet Thickening and Fibrotic Remodeling
Other works have suggested the possibility of maladaptive mitral valve remodeling. Mitral leaflets and chordae from end-stage heart failure were analyzed after heart transplantation and compared with normal control valves obtained from autopsy. Those apparently normal valves revealed significantly abnormal morphological and biomechanical properties compared with control valves. These abnormalities included different thickness, increased stiffness, decreased viscosity and reduced extensibility.59,60 Biochemical studies showed that these valves had higher DNA and collagen content, increased glycosaminoglycan concentration and less water content. These changes imply that the valve extracellular matrix was fibrotic. Computer modeling suggests that alteration in leaflet thickness and stiffness can interfere with normal coaptation, which requires leaflet extensibility and flexibility. 8 Imaging studies with echocardiography and cardiac magnetic resonance also suggest abnormal thickening and fibrotic remodeling after MI; which are also associated with the occurrence of MR.61,62 These contrasting evidences suggest that leaflet remodeling in some conditions could ultimately result in maladaptive fibrotic remodeling that could be considered a secondary organic contribution to FMR: the valves may be larger, but not normal in terms of composition and biomechanical function (Figure 1). 60
It is critical to understand whether these changes are only secondary to mechanical stretch (therapeutic target would remain the preservation of LV and LA normal geometry) or if other additional factors are contributing to this fibrotic remodeling. Ischemic MR is the most studied pathology in that setting and a growing body of data suggest that leaflet changes are not solely caused by mechanical stretch. In experiments of apical MI without visible leaflet deformation and without associated MR, changes in leaflet biology were observed, including near doubled thickness, evidence of EMT and presence of TGF-β. 61 Interestingly, similar valve remodeling after MI is also observed in aortic 63 and tricuspid valves, 45 despite preserved size of the aorta and normal right ventricular function. Taken together, those findings suggest that myocardial ischemia can alter leaflet biology without the presence of mechanical stretch. Furthermore, the implication of the other valves (not subjected to deformation or stretch) supports the idea of circulating factors influencing valve biology. The circulating factor hypothesis is also supported by in-vitro studies completely removing the effect of mechanical stress: isolated interstitial mitral valve cells were exposed to serum collected pre- and post-myocardial infarction. Post-MI serum was associated with overexpression of TGF-β, αSMA, and collagen compared to pre-MI serum. 64 These post-MI valvular changes seem to interfere with the normal process of valve enlargement in response to mechanical stress. In controlled models of stretch with and without the addition of MI, the presence of ischemic heart disease modified the valve biology, with marked increase of leaflet thickening, presence of microvessels, CD45 positive cells and αSMA positive cells.65,66 Extracellular matrix turnover was also activated with TGF-β and matrix metalloproteinases. Those changes were different and much more pronounced than leaflets exposed to similar mechanical stretch but without MI. In another mechanistic model of aortic regurgitation with versus without the presence of MI, infarction was again associated with biological changes in the leaflets. However, MI was also linked with attenuated leaflet enlargement and increased mitral valve regurgitation. 45 This experimental work is supported by clinical observation data: although the mitral valve is larger than normal in patients with ischemic heart disease, this elongation may not be equal in all individuals and not as pronounced when compared with patients with aortic regurgitation for similar LV size. 37
Specific mechanisms of leaflet remodeling after MI are still under investigation. The presence of TGF-β has been identified recurrently in post-MI leaflets and is likely involved in the process. TGF-β is a multifunctional cytokine involved in many physiological processes such as tissue embryogenesis, growth and cell differentiation, but has also a critical role in fibrosis. In heart valves, TGF-β has an important role in promoting the differentiation of valvular interstitial cells into active myofibroblasts. Once activated, these cells secrete collagen and accumulation of extracellular matrix, potentially contributing to valve fibrosis and stiffening. TGF-β can be modulated by angiotensin II, which also plays an important role in post-MI global myocardial remodeling. Angiotensin receptors are present in valvular endothelial cells, and their stimulation can induce EMT. 67 Experimental studies showed that post-MI induced mitral valve remodeling (leaflet thickening, collagen deposition, endothelial activation, TGF-β downstream signaling) can be prevented by the administration of losartan. 68 Similarly, a clinical echocardiographic study suggested attenuated leaflet thickening after MI for patients treated with maximal doses of renin-angiotensin blocking agents (angiotensin converting enzyme inhibitors or angiotensin receptor blockers). 61 This re-emphasizes the role of post-MI anti-remodeling therapies, which in addition of preserving myocardial function can also have a direct impact on valvular biology. Of note, angiotensin blockers or angiotensin-converting enzyme inhibitors have also been suggested to influence the progression of aortic stenosis by slowing the valvular fibrosis.69,70 Whether this pharmacological option can be used more broadly in the goal of treating or preventing valvular heart disease will require more data from randomized controlled studies.
Other potential causes for increased TGF-β in the mitral valve after MI have been suggested. Serotonin has been reported to be elevated in patients after myocardial infarction, 71 with potential impact on post-MI mood disorder and thrombotic events. 72 Serotonin can also regulate TGF-β and is well known to cause valvular fibrosis in patients with carcinoid syndrome 73 or in the setting of drug induced valve disease involving the serotonin type 2B receptor. Serotonin levels post-MI are neither as elevated nor sustained compared with carcinoid syndrome, but could contribute to affect valve biology in a critical moment where the leaflets are acutely adapting to post-MI ventricular remodeling. In an experimental study, the serotonin blocker cyproheptadine was able to attenuate leaflet thickening and prevent FMR after myocardial infarction. 64
Clinical Translation and Future Studies
Experimental and clinical observational studies place leaflet remodeling as a critical actor in the development of FMR. Adequate adaptation explains the absence of FMR in patients with dilated LV, while fibrotic remodeling with inadequate leaflet compensation is seen in ischemic MR (Figure 3). Excessive leaflet elongation can also contribute to MR in situations like hypertrophic cardiomyopathy. While the phenomenon of valve adaptation is mostly described in functional MR, its role in other disease, including other cardiac valves, is understudied. Those concepts are still to be translated into clinical practice. While advanced non-invasive imaging metrics (leaflet area and closure area by echocardiography or computed tomography) allowed significant progress in our understanding of valve biology, their specific value in the diagnostic clinical routine remains limited given the current absence of therapy influencing leaflet biology. Potential pharmacological targets to prevent fibrotic remodeling have been suggested, but there is no current clinical recommendation to influence leaflet remodeling in order to prevent or treat FMR. Angiotensin II is an important target for patients with heart failure and reduced ejection fraction; related drugs are therefore already widely used in patients with ventricular FMR. The specific place of those molecules for patients without LV dysfunction (atrial FMR in particular) remains to be explored. While the use of serotonin blockers after MI to prevent FMR has been suggested in experimental studies, this concept is not currently supported by clinical data. The use of cyproheptadine in that setting is currently under investigation. Exploration of more specific agents targeting the 5HT2B receptor could also deserve future studies. Interestingly, leaflet remodeling in myxomatous valve degeneration has also been linked with serotonin. 74 Whether pharmacological intervention targeting serotonin can also apply to patients with primary MR is currently unknown.
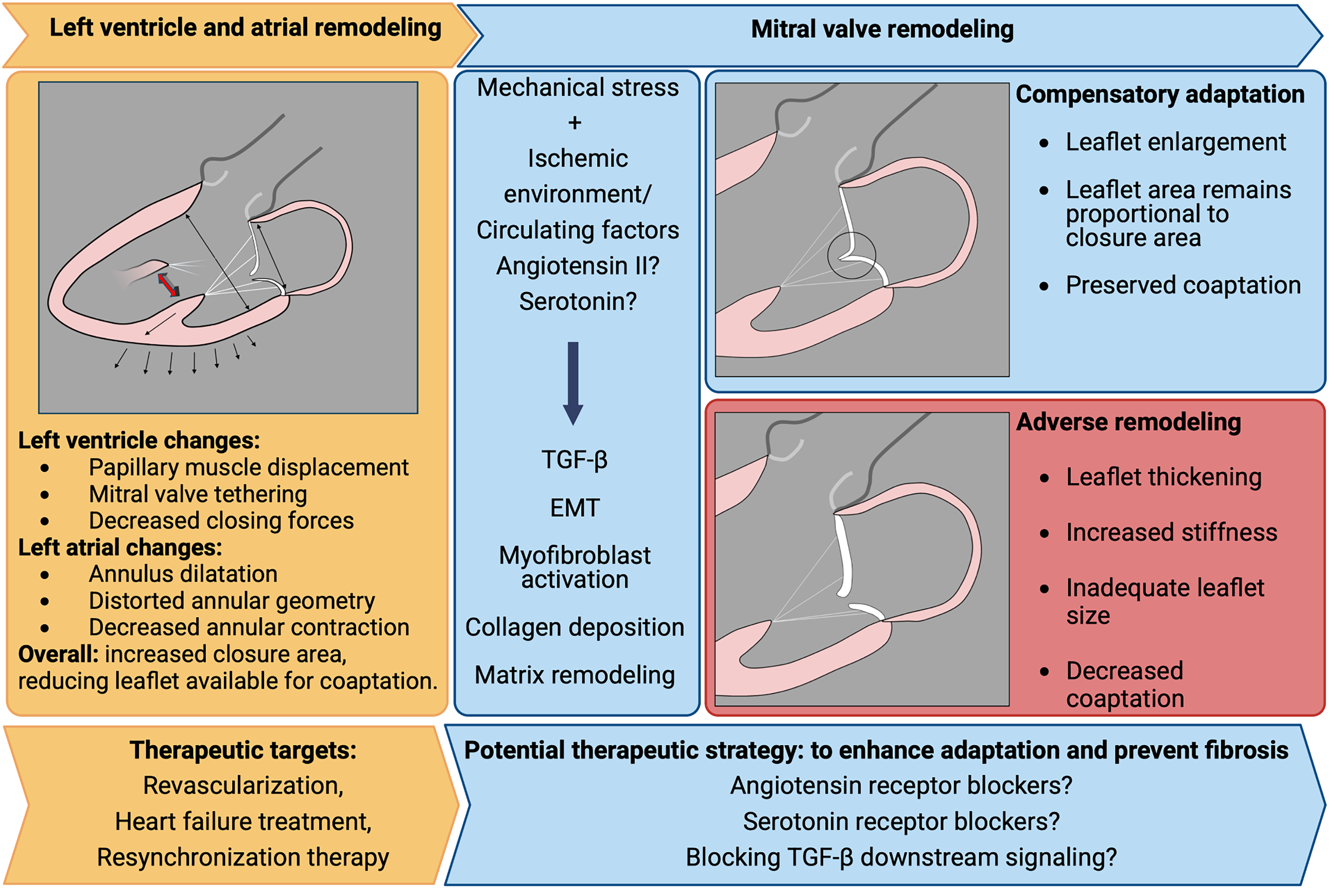
Mechanistic Understanding of Functional Mitral Regurgitation. Left: ventricular and atrial changes will cause mitral valve deformation. Center: valvular response is triggered by mechanical stretch and neurohumoral environment. Right: two potential outcomes for mitral valve remodeling: adequate leaflet enlargement preventing regurgitation, or fibrotic remodeling contributing to regurgitation. EMT: endothelial-to-mesenchymal transformation; TGF-β: transforming growth factor beta.
Conclusion
Significant progress has been made in recent years regarding our understanding of mitral valve biology in diseases where its role was previously unrecognized. Although FMR is initially triggered by LV or LA remodeling, subsequent valvular adaptation is induced and can be influenced by systemic biological processes that are still under investigation. Active valve remodeling is also seen in physiologic conditions (pregnancy) and other primary LV diseases such as hypertrophic cardiomyopathy. A better understanding of the molecular pathways driving valve remodeling and fibrosis in ischemic and non-ischemic myocardial disease is critical. While most of the research in this field has targeted mitral regurgitation, the findings could potentially be applicable for other valvular diseases. This novel knowledge has deep implications for future research, and pharmacological therapies targeting valvular remodeling could represent a major therapeutic advance.
Footnotes
Abbreviations
Acknowledgements
Figure 1 graphics were Created in BioRender. Rakotoarivelo, V. (2024) https://BioRender.com/u97j197. Figure 3 was created in BioRender. Rakotoarivelo, V. (2025) https://BioRender.com/k03e888.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: J. Beaudoin is supported by Fonds de Recherche Québec – Santé (311367), and his research program supported by Canadian Institute for Health Research (468726) and Fondation de l’Institut Universitaire de Cardiologie et de Pneumologie de Québec. RAL is supported in part by the National Institutes of Health (R01 HL141917 and R01 HL173930), the American Heart Association (22TPA963793), Leducq Foundation grant 22ARF02 for the Preventing Rheumatic Injury bioMarker Alliance, and the Ellison Foundation, Boston. J.Beaudoin has received research support from JAMP Pharma.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.