
Abstract
Mechanobiology investigates how mechanical forces influence cellular behavior across scales, from molecular interactions to tissue-level responses. Advances in force measurement, high-resolution imaging, and multiomic profiling have generated datasets of increasing dimensionality, modality, and scale. Artificial intelligence (AI) and machine learning (ML) methods are well suited to analyzing such data, predicting cellular responses to mechanical stimuli, and identifying candidate mechanistic relationships. Here we review key applications of AI and ML in mechanobiology, including force inference from imaging data, tissue stiffness mapping, cell phenotype classification, chromatin-based prediction of protein localization, and computational modeling of mechanosensitive protein dynamics. We discuss emerging tools such as foundation models for cell segmentation and protein structure prediction, as well as the integration of AI-predicted mechanical properties with spatial transcriptomics. We also examine current limitations—including data scarcity, the gap between proof-of-concept and validated tools, and the challenge of causal interpretation—and identify opportunities for real-time closed-loop systems, therapeutic translation, and multiscale modeling.
Introduction
Mechanobiology is an interdisciplinary field that investigates how physical forces shape biological processes across scales, from single molecules to entire organisms. 1 Cells sense and respond to mechanical cues through specialized molecular machinery, including integrin-based adhesions, mechanosensitive ion channels, cytoskeletal networks, and mechanotransduction pathways such as the LINC complex and YAP/TAZ nuclear–cytoplasmic shuttling.2,3 Through these pathways, mechanical forces regulate critical processes including stem cell differentiation, tumor progression, wound healing, and tissue morphogenesis.4–6
Over the past two decades, advances in force measurement techniques, high-resolution imaging, and multiomic profiling have expanded both the scope and scale of mechanobiological research. At the same time, the resulting datasets have become increasingly high-dimensional, multimodal, and voluminous, frequently exceeding the practical limits of conventional statistical and manual analysis methods. Force measurements from atomic force microscopy (AFM), traction force microscopy (TFM), and molecular tension sensors can generate thousands to millions of spatially resolved data points per experiment. High-content imaging produces large volumes of morphological and spatiotemporal data, while single-cell and bulk omic approaches yield detailed molecular profiles. Together, these developments have created an acute need for computational methods capable of handling such complexity.
AI and ML methods are increasingly being applied to address this need. Whereas conventional approaches often depend on user-defined features and assume relatively simple relationships between variables, ML algorithms can learn complex, nonlinear mappings directly from data. These methods are therefore well suited to integrating heterogeneous data types, automating labor-intensive analyses, and identifying previously unrecognized relationships between mechanical stimuli and biological responses. Concurrently, the emergence of foundation models for microscopy7–9 and protein structure prediction,10,11 along with deep learning methods for single-molecule localization microscopy (SMLM),
12
is expanding the toolkit available to mechanobiologists, as has also been highlighted by recent perspectives on ML-driven force measurement and molecular tension probe analysis.
13
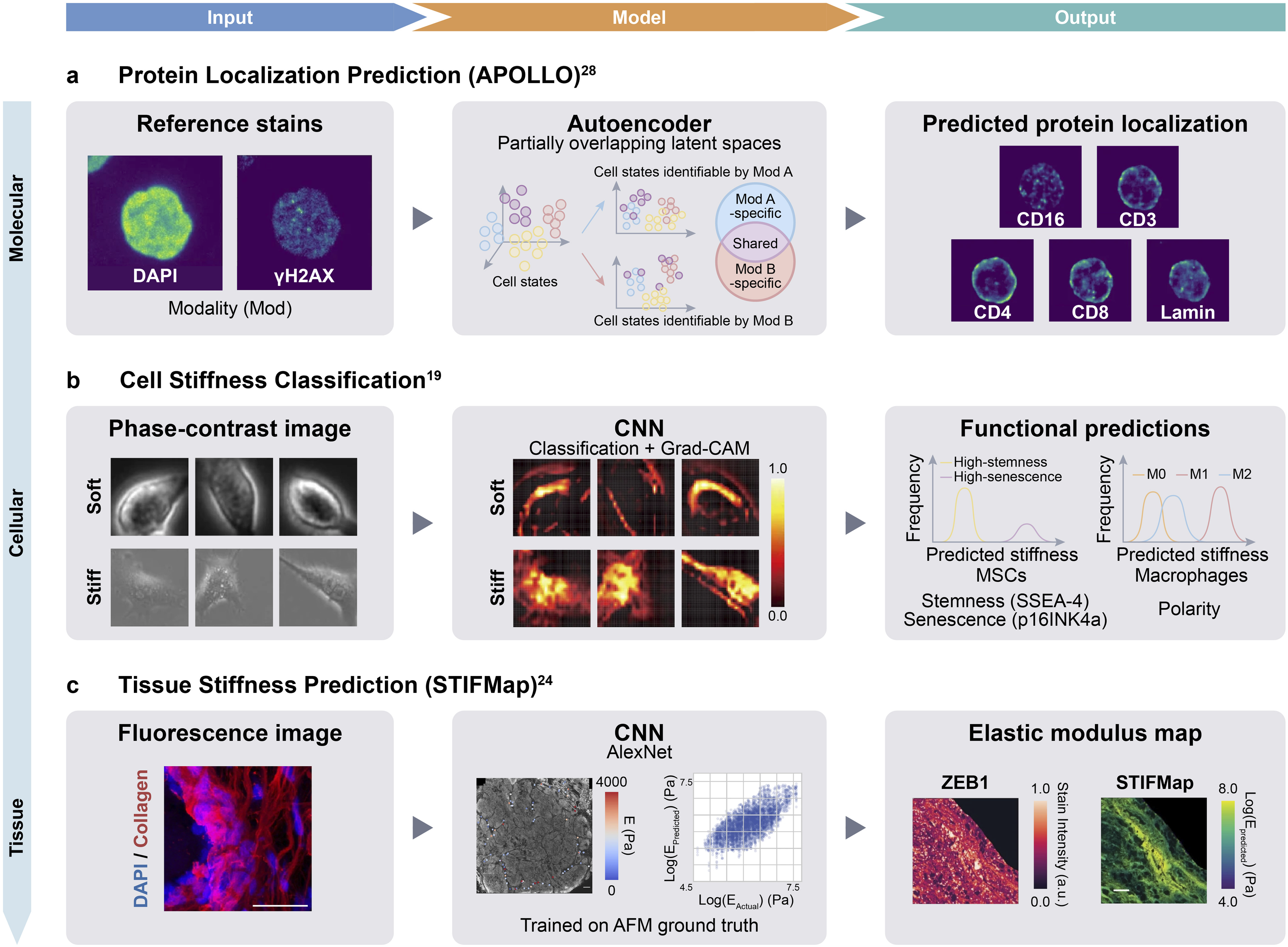
This review examines current AI and ML applications in mechanobiology, discusses key challenges, and identifies opportunities for future development and clinical translation (Figure 1). AI pipeline for mechanobiology research. Schematic illustrating the integration of mechanobiology and AI research workflows. Mechanical forces (shear flow, compression/stretching, substrate stiffness, stress relaxation, spatial confinement, and external stimuli) are applied to study platforms (2D/3D cultures, organoids/spheroids, and in vivo/ex vivo tissues). These systems generate input data (proteomic, transcriptomic, epigenomic, imaging, and force measurements), which are processed through AI/ML analysis methods including convolutional neural networks (CNNs), generative adversarial networks (GANs), foundation models, and physics-informed neural networks. Outputs include predictions of cell behaviors, cell fate, disease diagnosis, and personalized treatment strategies. The dashed feedback loop indicates the future direction of real-time closed-loop mechanobiology (‘Real-time and closed-loop mechanobiology’ section).
AI-driven force measurement and prediction
Machine learning for traction force microscopy
Quantifying cellular traction forces is central to mechanobiology, yet conventional TFM workflows remain limited by low throughput, the requirement for fluorescent fiducial markers, and computationally intensive displacement field analysis. Moreover, the underlying inverse problem is ill-posed, requiring careful regularization to balance accuracy against noise amplification. 14 Recent ML approaches have begun to address these limitations at multiple stages of the TFM pipeline.
Wang and Lin demonstrated that a modified U-Net with 3D convolutions, trained entirely on simulated stress–displacement pairs, can predict traction stress fields directly from substrate displacement maps, bypassing the regularization-dependent inverse problem. 14 On simulated cells, the model achieved a normalized root mean square error (RMSE) of ∼10.8%, significantly lower than ∼14.1% for Fourier transform traction cytometry (FTTC) with optimized regularization. This advantage persisted on experimental NIH 3T3 cells (∼27.7% vs ∼36.8%), although FTTC showed lower error in reconstituted displacement fields, indicating complementary strengths. The approach generalized across cell shapes, substrate stiffnesses, and magnifications, though it still requires bead-derived displacement fields as input.
Subsequent approaches have gone further by eliminating the need for fluorescent fiducial markers altogether. One notable example is wrinkle force microscopy, introduced by Li et al., which uses generative adversarial networks (GANs) to predict traction forces from substrate wrinkle patterns visible in phase-contrast images. 15 The method proceeds in three stages: first, cells are cultured on a plasma-irradiated silicone membrane substrate that deforms into visible wrinkle patterns under cellular traction, and conventional TFM data are acquired simultaneously as ground truth; second, wrinkle patterns are segmented from phase-contrast images using a convolutional neural network (CNN); and third, a conditional GAN is trained to map either raw images or segmented wrinkle patterns to traction force distributions. The GAN-based model showed moderate-to-good agreement with conventional TFM measurements, with correlation coefficients of 0.84–0.88, force magnitude errors of 38–41%, and directional errors of 19–23°. While these correlations are encouraging, they explain only ∼70–77% of variance (R2). Nonetheless, the results demonstrate that learned image-to-force mappings can approximate physics-based traction reconstruction. A key advantage of this approach is the elimination of fluorescent fiducial beads, cell-free reference images, and displacement field computation. In a complementary strategy, Schmitt et al. trained U-Net architectures on fluorescence images of focal adhesion proteins—particularly zyxin, whose localization to focal adhesions correlates with applied force—to predict traction force distributions without fiducial markers. 16 Notably, networks trained on fibroblasts generalized to unseen cell types and biochemical perturbations without retraining, and physics-constrained variants of the model revealed that cellular traction is encoded across two distinct length scales corresponding to individual adhesion sites and whole-cell morphology. Together, these approaches illustrate how ML can increase throughput and reduce experimental complexity in traction force analysis.
Beyond force estimation itself, upstream displacement mapping has also benefited from deep learning. DEFORM-Net employs a U-Net trained on synthetic displacement fields, generated via fractal Perlin noise with microscopy-informed image noise, to predict full-field displacement maps from live microscopy without experimental ground truth. 17 By combining supervised similarity losses with an unsupervised image-correlation-based “rewarp” loss, the model outperformed digital image correlation and optical flow across multiple cell types and imaging modalities, including beating neonatal mouse cardiomyocytes and pulsatile Drosophila larval epithelial cells, while achieving near-video-rate inference. Trained entirely on synthetic displacement fields, DEFORM-Net was evaluated on experimental data; however, systematic validation of its transferability across diverse experimental conditions remains an open question. As a further complementary strategy not yet demonstrated for TFM specifically, physics-informed neural networks—which embed governing equations such as force balance and constitutive laws directly into the training loss—have shown promise in related continuum mechanics problems and could, in principle, enforce physical consistency in traction force predictions while retaining the flexibility of data-driven learning. 18
Cell stiffness mapping
Deep learning approaches have also been applied to predict cellular mechanical properties from optical microscopy images, offering a label-free, non-contact alternative to direct mechanical measurement. Although AFM provides spatially resolved stiffness measurements at sub-micrometer resolution, it is inherently low-throughput, requires physical contact with the cell, and is limited to surface-accessible regions. To address these constraints, Wu et al. developed CNN-based classification models trained on phase-contrast images of cell subpopulations with experimentally modulated stiffness.
19
For human mesenchymal stem cells (MSCs), softened populations were generated using cytoskeletal inhibitors (cytochalasin D and blebbistatin), while stiffened populations were produced by treatment with glucose and hydrogen peroxide (H2O2), with a parallel model constructed for RAW264.7 macrophages. Both models achieved classification accuracy >97% (area under the receiver operating characteristic curve [AUC] = 1.00) on balanced datasets of chemically softened and stiffened cells. Gradient-weighted class activation mapping (Grad-CAM) analysis revealed that the models attended to bright peripheral regions in soft cells and heterogeneous intracellular intensity patterns in stiff cells. The classification framework was further adapted to estimate continuous stiffness values by mapping softmax output probabilities to AFM-derived Young’s modulus, yielding a mean absolute percentage error (MAPE) of ∼45% for MSCs (R2 = 0.56) and ∼57% for the cross-cell-type regression model. The substantial gap between classification and regression performance likely reflects the difficulty of predicting continuous mechanical values from morphology alone, a challenge compounded by the limited dataset size for the regression task (∼3,300 cells vs >120,000 for classification). This approach highlights a practical advantage of classification-based strategies: chemical perturbations can be used to generate large, labeled training datasets under controlled conditions, thereby circumventing the throughput limitations of direct mechanical measurement (Figure 2(b)). AI/ML approaches for imaging-based prediction of molecular, cellular, and tissue properties. (a) APOLLO: autoencoder with partially overlapping latent spaces predicts subcellular protein localization (Lamin, CD3, etc.) from reference stains (DAPI, γH2AX), achieving ∼95% prediction accuracy. Adapted from
28
. (b) Phase-contrast stiffness classification: CNN trained on chemically perturbed populations (cytochalasin D, blebbistatin for soft; glucose, H2O2 for stiff) classifies cell stiffness with >97% accuracy. Downstream analyses link predicted stiffness to stemness markers, macrophage polarization, and senescence. Adapted from
19
. (c) STIFMap: CNN-based prediction of tissue elastic modulus from collagen fluorescence images, trained on AFM-derived ground truth. Enables spatial stiffness mapping on formalin-fixed paraffin-embedded (FFPE) clinical specimens and identified correlations between stromal stiffness and epithelial-to-mesenchymal transition (EMT) markers. Adapted from
24
.
Cell phenotype and state prediction
In vitro applications
ML methods have been applied to predict cellular states and behaviors from morphological features in microscopy images, with growing relevance to mechanobiology. A central capability of deep learning in this context is the extraction of information from transmitted-light images that would otherwise require fluorescent labeling, potentially enabling longitudinal, label-free monitoring of cellular phenotypes. Underpinning these efforts, foundation models for cell segmentation—including CellSAM 7 and μSAM 8 —have substantially improved the scalability and accuracy of cell detection across imaging modalities, providing essential upstream components for mechanophenotyping pipelines.
Morphology-based classification
In silico labeling (ISL) uses transmitted-light z-stacks—acquired in brightfield, phase-contrast, or differential interference contrast—to computationally predict fluorescent labels without physical staining. 20 By training a custom multiscale deep neural network on paired transmitted-light z-stacks and corresponding fluorescence images, ISL models can predict nuclear labels (DAPI/Hoechst) with pixel-wise correlation coefficients >0.87, detect dead cells with 98% precision and 97% recall, and identify neurons in mixed cultures with prediction fidelity comparable to inter-annotator agreement. These capabilities support longitudinal, label-free monitoring of cell phenotypes by avoiding phototoxicity, potential alteration of cellular mechanics by fluorescent reporters, and the need for fixation. For mechanobiology specifically, ISL is particularly relevant because it enables tracking of nuclear morphological changes in response to mechanical stimuli without introducing exogenous labels that may perturb cellular mechanics. Deep learning applied to DAPI-stained nuclear images has also been shown to predict cellular senescence—a mechanically relevant state associated with increased cell stiffness—with ∼95% accuracy for radiation- and replication-induced senescence in human fibroblasts, relying primarily on nuclear shape features such as area, convexity, and aspect ratio. 21 The predictor generalized to mouse astrocytes, neurons, and progeria cell lines, though drug-induced senescence (for example, doxorubicin, antimycin A) required retraining with lower accuracy—raising the question of whether the model captures universal senescence-associated nuclear morphology or stimulus-specific signatures, a distinction with implications for generalizability.
Nanoscale phenotyping
While the approaches described above operate at diffraction-limited resolution, deep learning has also been applied to super-resolution imaging data to extract phenotypic information at the nanoscale. The Artificial Intelligence of the Nucleus (AINU) framework trains a DenseNet-121 CNN on stochastic optical reconstruction microscopy (STORM) super-resolution images of nuclear Pol II and histone H3 to classify cell phenotypes, achieving ∼94% accuracy (AUC = 0.98) in distinguishing human induced pluripotent stem cells (hiPSCs) from somatic cells. 22 Interpretable AI analysis revealed that Pol II localizations within nucleoli serve as the primary discriminative feature, a finding mechanistically validated by the observation that nucleolar Pol II transcribes antisense intergenic non-coding RNAs from rDNA intergenic spacer regions, with significantly higher expression in hiPSCs than in somatic cells. With appropriate retraining, AINU further detected HeLa cancer cells (∼84% accuracy) and identified HSV-1-infected cells as early as 1 h post-infection, before visible chromatin remodeling. The accuracy of such nanoscale analyses depends critically on the underlying localization algorithm; in this regard, DECODE—a deep learning framework trained on simulated SMLM data—demonstrated leading performance on public SMLM benchmarks by integrating temporal context across frames to achieve high-accuracy 3D localization at high emitter densities, while providing well-calibrated per-molecule uncertainty estimates that enable faster acquisition and denser labeling, facilitating live-cell super-resolution imaging with reduced phototoxicity. 12
In vivo and histological applications
Tissue-level stiffness prediction
ML approaches have been developed to predict tissue mechanical properties from histological images of extracellular matrix (ECM) architecture. Collagen fiber organization in tumors—characterized by tumor-associated collagen signatures (TACS) that correlate with cancer prognosis 23 —provides a rich set of morphological features for such predictions. Stashko et al. developed STIFMap (Spatially Transformed Inferential Force Map), a CNN-based framework that predicts stromal elastic modulus at micrometer-scale resolution from fluorescence images of collagen and nuclear (DAPI) staining. 24 The model was trained on automated AFM measurements collected along spatially distributed paths across tissue sections (∼200 measurements per sample), with an ensemble of 25 models achieving a Pearson correlation of r = 0.69 against AFM ground truth. The model generates stiffness maps across entire tissue sections, including formalin-fixed paraffin-embedded (FFPE) clinical specimens. Predicted stiffness correlated significantly with mechanosignaling markers (activated β1 integrin, phospho-MLC2; n = 60 fields of view, 10 patients), providing biological validation beyond AFM comparison alone. Using this approach, the authors identified spatial correlations between regions of elevated stromal stiffness and markers of epithelial-to-mesenchymal transition (EMT) (Figure 2(c)).
Spatial mechanomics integration
A proposed but as yet undemonstrated direction is the integration of AI-predicted tissue stiffness maps with spatial transcriptomics data, which could in principle enable spatial correlation of mechanical properties with gene expression patterns at comparable resolution scales. 24 For example, aligning STIFMap-derived stiffness maps with spatial transcriptomics platforms such as Visium could identify candidate stiffness-associated gene signatures within distinct tissue microenvironments. By correlating predicted mechanical properties with spatially resolved molecular profiles, these approaches could generate hypotheses about how ECM mechanics influence cellular phenotypes and disease progression in situ. From a clinical perspective, regions of elevated stromal stiffness that colocalize with specific transcriptional programs may indicate aggressive disease phenotypes or treatment-resistant cell populations.
Clinical translation
Several groups have begun exploring the clinical potential of image-based mechanical and morphological biomarkers. Among the most developed examples is the use of chromatin organization in peripheral blood mononuclear cells (PBMCs) as a liquid biopsy biomarker. Challa et al. reported that a Random Forest classifier trained on 3D chromatin features—including nuclear volume, heterochromatin content, and nuclear boundary curvature—distinguished PBMCs from healthy donors and cancer patients with an average single-cell accuracy of 74%; majority voting across ∼185 cells per patient (n = 10 healthy, 10 tumor) yielded perfect patient-level classification. 25 A separate three-way classifier further differentiated glioma, meningioma, and head and neck cancers with 85% patient-level accuracy (17/20 patients), although single-cell accuracy was more variable (70 ± 29%). In a proof-of-concept application, chromatin features tracked changes in PBMC nuclear organization during proton therapy, with chromometric signatures partially recovering toward healthy donor levels over the course of treatment across all tumor groups. Nuclear morphology-based senescence classification has similarly been translated to in vivo tissue: applied to H&E-stained human dermal biopsies from 169 individuals (cross-sectional), the predictor detected a modest age-dependent increase in predicted senescence (Pearson r = 0.23–0.35). 21 In an exploratory analysis of ICD-10 diagnosis codes, higher predicted senescence was associated with decreased rates of malignant neoplasms (P = 0.002, the only association surviving multiple-testing correction), consistent with the tumor-suppressive role of senescence. If validated in larger cohorts, such approaches could contribute to early cancer detection, treatment selection, and longitudinal monitoring of therapeutic response using nuclear mechanical and morphological biomarkers. Complementing these imaging-based chromatin biomarkers, DNA methylation-based predictors have emerged as robust molecular indicators of biological aging, disease risk, and environmental exposures, with established statistical frameworks for addressing data leakage and class imbalance that could inform mechanobiological biomarker development. 26 More broadly, computational approaches for predictive biomarker discovery in oncology increasingly integrate molecular, radiomic, and pathomic features to predict treatment response, yet translating these multimodal predictors from preclinical models to clinical practice remains a critical bottleneck even for well-resourced cancer programs.26,27 These challenges are likely to be shared by mechanobiology-derived biomarkers as they advance toward clinical use.
Genomic and molecular prediction
Chromatin-based prediction of protein localization
Chromatin organization can serve as a predictor of protein localization patterns, including those altered by mechanical stimuli. The APOLLO framework (Autoencoder with Partially Overlapping Latent space through Latent Optimization) exemplifies this approach by enabling prediction of protein distribution from reference cellular stains. 28 APOLLO is a general multimodal integration framework that has been applied to paired sequencing data (SHARE-seq and CITE-seq) and multiplexed imaging data, including from the Human Protein Atlas. In the imaging application, the framework uses reference stains—such as chromatin (DAPI), microtubule, and endoplasmic reticulum (ER) channels—to predict protein subcellular localization in held-out cells without directly imaging the target proteins (Figure 2(a)).
Methodologically, APOLLO employs a neural network architecture with partially overlapping latent spaces that explicitly separate shared information from modality-specific information, achieving more precise single-cell cross-modality predictions than standard multimodal autoencoders or image inpainting approaches. When tested on cells from patients excluded from the training set, the model generated protein localization predictions that performed comparably to actual protein images in downstream phenotype classification tasks. Complementing this image-only approach, Prediction of Unseen Proteins’ Subcellular localization (PUPS) integrates protein language model embeddings (ESM-2) with cellular landmark stains (nucleus, microtubule, and ER) to predict subcellular localization, enabling generalization to unseen proteins and cell lines while capturing single-cell variability. 29
The predictive relationship between chromatin and protein localization is particularly relevant to mechanobiology, where mechanical forces influence both chromatin organization 30 and nuclear protein transport. 31 Diverse mechanical inputs—including compressive forces, cyclic stretch, cell geometry, and matrix stiffness—converge on chromatin remodeling through interconnected mechanisms. These mechanisms involve actomyosin contractility, actin-dependent nuclear transport of transcriptional regulators (such as HDAC3, MRTF, and histone acetyltransferases), and downstream histone modifications.32–36
Multimodal integration of chromatin accessibility and gene expression
Beyond imaging applications, APOLLO has also been applied to paired sequencing data, demonstrating that the partially overlapping latent space framework generalizes across data modalities. When applied to paired scRNA-seq and scATAC-seq data from SHARE-seq, APOLLO partitions information into shared and modality-specific latent spaces that are biologically interpretable. Gene ontology enrichment analysis revealed that transcriptional regulation terms were enriched in both the shared and ATAC-specific latent spaces but not in the RNA-specific space, while post-transcriptional regulation terms were enriched in the shared space. The authors suggest this pattern may reflect a temporal lag between changes in chromatin accessibility and gene expression—consistent with established models in which chromatin remodeling precedes transcriptional output. While this application does not directly address mechanobiology, it establishes that disentangled multimodal integration can recover biologically meaningful latent structure from paired measurements—a capability that could prove valuable if paired datasets linking chromatin organization to mechanical properties (such as cell stiffness or nuclear deformability) were generated. Such applications remain unrealized and would require large-scale paired training data and validation of generalizability across cell types and conditions.
Computational and data-driven prediction of YAP/TAZ mechanosensing
YAP and TAZ are transcriptional coactivators whose nuclear–cytoplasmic distribution shifts in response to mechanical cues, serving as a widely used readout of the mechanical state of the cell and its microenvironment. Computational efforts to model this mechanostat span a range of approaches, from mechanistic biophysical simulations to data-driven machine learning.
Mechanistic mathematical models—including ordinary differential equation (ODE), partial differential equation (PDE), and stochastic frameworks—have been developed to simulate how substrate stiffness, cytoskeletal dynamics, and Hippo pathway signaling converge to regulate YAP/TAZ nuclear translocation. 37 At the cell–material interface, Zhang et al. developed a motor-clutch model coupling integrin–ECM adhesion, actomyosin contractility, and nuclear deformation to predict YAP nuclear-to-cytoplasmic ratios. 38 The model reproduced experimentally observed double-sigmoidal relationship between YAP N/C ratio and substrate stiffness (2–41 kPa), and was independently validated against traction force, F-actin anisotropy, and nuclear flattening measurements not used in model calibration. Notably, the model explained how N-cadherin ligation (via HAVDI peptides) reverses mechanics-driven YAP nuclear accumulation in MSCs at intermediate stiffnesses by reducing integrin binding rate and traction force.
On the data-driven side, Bonnevie et al. trained an artificial neural network on cell and nuclear shape descriptors—including cell area, cell aspect ratio, and nuclear area—and showed that morphological features alone explain over 50% of the single-cell variance in YAP/TAZ nuclear localization across multiple cell lineages in fibrous microenvironments. 39 Moving beyond imaging-derived features, Pham et al. developed an ensemble machine learning framework that uses baseline transcriptomic profiles to predict cancer cell line dependency on the YAP/TAZ pathway, leveraging gene expression signatures derived from YAP1/WWTR1 knockdown experiments across 42 cancer cell lines. 40 Using weighted gene coexpression network analysis, they identified gene clusters associated with YAP/TAZ transcriptional activity, one of which was strongly correlated with a KRAS dependency signature, revealing cross-talk between the Hippo and MAPK pathways. Chemical genetic screening confirmed that MEK inhibitors synergize with YAP depletion in YAP-amplified cancers, both in vitro and in vivo.
Together, these studies illustrate a progression from physics-based models of YAP/TAZ mechanotransduction to data-driven prediction of pathway activity from morphological and transcriptomic features. Extending these approaches—for example, by training image-level deep learning models on raw microscopy data or integrating multimodal datasets linking chromatin organization, gene expression, and mechanical readouts—could enable high-throughput, label-free assessment of mechanosensitive cell states.
Stiffness-based prediction of cellular function
Cellular stiffness is a biophysical marker that correlates with cell phenotype and function. Using the stiffness classification framework (‘Cell stiffness mapping’ section), Wu et al. further investigated whether image-predicted stiffness relates to functional cellular properties. 19 The authors showed that predicted MSC stiffness increases with passage number, paralleling a decline in stemness markers (SSEA-4) and an increase in senescence markers (p16INK4a). Notably, the CNN model detected stiffness differences between early and late passages that deformability cytometry could not resolve. This stiffness–passage relationship was consistent across MSCs from different donors and tissue sources (adipose, umbilical cord, and dental pulp), supporting stiffness as a potential quality indicator for MSC manufacturing. Extending this analysis to immune cells, the authors found that LPS-induced M1 macrophages (RAW264.7) exhibited higher stiffness than both resting M0 and IL-4-induced M2 macrophages, which showed similar stiffness levels to each other, consistent with previous reports linking macrophage polarization state to mechanical properties.
The study further examined whether MSC stiffness relates to immunomodulatory function. Conditioned media from stiffened MSCs (H2O2-treated) promoted M1 polarization in macrophages, while conditioned media from softened MSCs (blebbistatin-treated) suppressed M1 polarization. However, because these agents have effects beyond stiffness modulation—H2O2 induces oxidative stress and senescence-associated secretory changes, while blebbistatin disrupts actomyosin contractility and alters paracrine signaling—the observed effects on macrophage polarization cannot be attributed solely to changes in MSC stiffness. Experimental designs that decouple stiffness from these off-target effects would be needed to isolate the contribution of stiffness per se.
Toward in silico prediction of mechanosensitive protein dynamics
AI-driven structure prediction tools have begun to intersect with mechanobiology. The release of AlphaFold 310 and RoseTTAFold All-Atom 11 has substantially expanded the scope of computational structural biology, enabling prediction of protein–protein, protein–nucleic acid, and protein–small molecule complexes with unprecedented accuracy. For mechanobiology, these tools raise the question of whether computational structural prediction is yet sufficiently accurate to model conformational changes in mechanoactive proteins under force. To evaluate this prospect, Gomes et al. assessed whether AlphaFold-predicted structures are sufficiently accurate for in silico force spectroscopy (steered molecular dynamics simulations that model force-dependent dissociation of protein complexes). 41 Their analysis showed that AlphaFold produces reliable monomer folds but often lacks the accuracy needed for high-resolution complex prediction, which is critical for modeling force-dependent protein–protein interactions. Nonetheless, the ability to generate structures for proteins lacking experimental data could enable high-throughput computational screening of mechanical properties across protein families. More broadly, the differentiable programming paradigm—which embeds physical equations such as energy functions and molecular dynamics equations into trainable neural network architectures—provides a conceptual framework for integrating structure prediction with physics-based simulations in an end-to-end fashion. 42 This convergence of deep learning with physics-based simulation has already proven productive in structurally analogous problems involving force-dependent protein conformational changes. For example, in CRISPR genome editing, graph neural networks trained on molecular dynamics trajectories predict Cas protein conformational dynamics, and generative models enable de novo design of editors with tailored specificities. 43 The methodological parallel is direct: just as Cas proteins undergo functionally critical conformational rearrangements during DNA engagement, mechanosensitive proteins such as talin, vinculin, and Piezo channels undergo force-dependent unfolding or gating transitions that could be modeled using the same hybrid frameworks.
Challenges and solutions
Data scarcity and augmentation
Challenges and solutions in AI-driven mechanobiology.

Abbreviations: AFM, atomic force microscopy; APOLLO, Autoencoder with Partially Overlapping Latent space through Latent Optimization; Grad-CAM, gradient-weighted class activation mapping; LLM, large language model; METL, mutational effect transfer learning.
Several strategies have been employed to mitigate these constraints. Transfer learning, in which models pre-trained on large-scale imaging datasets are fine-tuned on smaller mechanobiology-specific data, has been applied in ISL 20 and could be extended using emerging foundation models for microscopy.7,8 Data augmentation through geometric transformations and noise addition can artificially expand dataset sizes, though its effectiveness depends on whether the transformations respect the underlying physics—for instance, force directionality is lost under random rotation of traction force maps. Classification-based experimental designs, in which chemical perturbations generate binary-labeled populations, 19 offer a practical route to larger training sets. Few-shot learning approaches, which aim to generalize from very small datasets, have shown promise in other biomedical imaging domains but remain largely untested in mechanobiology. In the protein domain, pretraining on biophysics-based simulated attributes has proven effective: the mutational effect transfer learning (METL) framework uses Rosetta-generated structural and energetic features to pretrain transformers that, after fine-tuning on as few as 64 experimental measurements, can design functional protein variants and extrapolate to unseen sequence positions. 45 Advances in computational microscopy may help address these data limitations. For example, physics-based background filtering for structured illumination microscopy now achieves sub-70 nm live-cell resolution of cytoskeletal dynamics, 46 and such higher-fidelity imaging data could, in principle, improve the quality of ML training sets.
Multimodal integration
Mechanobiology inherently generates heterogeneous data types—force measurements, morphological imaging, and molecular profiles—that capture complementary aspects of cellular mechanical behavior. Integrating these modalities is essential for a comprehensive understanding of mechanotransduction but raises substantial technical challenges (Table 1b). These include differences in spatial and temporal resolution across modalities (for example, AFM at sub-micrometer resolution vs Visium spatial transcriptomics at 55 μm spot size), the lack of standardized data formats and annotation conventions, and the difficulty of establishing ground-truth labels for integrated predictions.
Several approaches have begun to address these challenges. The APOLLO framework (‘Chromatin-based prediction of protein localization’ section) uses partially overlapping latent spaces to model shared and modality-specific information across paired imaging or sequencing datasets. The alignment of AI-predicted stiffness maps with spatial transcriptomics data (‘In vivo and histological applications’ section) represents another emerging direction. Beyond pairing molecular modalities, CellWhisperer has demonstrated that contrastive learning can align single-cell transcriptomes with natural language descriptions, enabling chat-based interrogation of gene expression profiles and zero-shot cell type classification across tissues. 47 Strategies including early fusion, late fusion, and hybrid architectures have been applied in related biomedical domains, but systematic comparison of these approaches on mechanobiological data is lacking. Dictionary learning-based bridge integration, which leverages multiomic datasets to map across disparate modalities without requiring shared features, offers one scalable framework that could be adapted for joint analysis of mechanical and molecular measurements. 48 Additionally, large language model (LLM)-based approaches that encode single-cell transcriptomic profiles as natural language sentences offer a complementary paradigm in which transcriptomic profiles, experimental metadata, and free-text queries are jointly processed within a unified language model framework, providing a flexible interface that could in principle extend to mechanical and imaging readouts. 49
Interpretability and validation
For ML models to contribute meaningfully to mechanobiology, their predictions must be both interpretable and experimentally validated (Table 1c). Several studies reviewed here have employed interpretability techniques to address the former requirement: Grad-CAM revealed that stiffness classification models attend to peripheral cell regions and intracellular intensity patterns 19 ; the AINU framework’s Grad-CAM analysis identified nucleolar structural features as key discriminators of cell identity at nanoscale resolution 22 ; and gene ontology enrichment of APOLLO’s latent spaces provided biological context for learned representations. 28 Each of these approaches generates testable hypotheses about the features linking cell morphology to mechanical state. An alternative strategy is to embed mechanistic priors directly into model architectures through differentiable programming, yielding parameters with physical meaning rather than relying solely on post hoc attribution. 42 Uncertainty quantification—through Bayesian neural networks, ensemble methods, or conformal prediction—will be essential for clinical and translational applications where prediction confidence must accompany point estimates.
Interpretability alone is insufficient without experimental validation. A recurring limitation across the reviewed studies is reliance on correlative evidence—for example, spatial colocalization of predicted stiffness with EMT markers 24 or associations between predicted YAP/TAZ localization and substrate stiffness.37,38 Establishing causal relationships requires interventional experiments that perturb the predicted variable and measure downstream effects, an approach that most current studies have not undertaken. Robust validation also requires testing model generalizability across cell types, culture conditions, and laboratories—a standard that few mechanobiology ML studies have met to date. Notably, few of the methods reviewed here have been independently reproduced by groups other than the original developers, and the availability of code, trained models, and benchmark datasets varies considerably across studies. Establishing community standards for model evaluation, including standardized test datasets and reporting requirements, would accelerate the translation of these approaches from proof-of-concept demonstrations to validated tools. It is also worth noting that for well-posed problems with established analytical solutions, ML approaches may offer marginal gains over conventional methods while introducing interpretability and reproducibility challenges.
Outlook
Real-time and closed-loop mechanobiology
Current ML applications in mechanobiology operate primarily on pre-collected datasets. An important future direction is the integration of ML inference into real-time experimental workflows, enabling closed-loop systems in which mechanical stimuli are adjusted on the basis of predicted cellular responses. For example, coupling image-based stiffness prediction models with programmable substrate systems—such as platforms that apply defined strain regimes to 3D tissue constructs 50 or nanotopographical cues that direct cell fate 51 —could enable stimulation protocols that adapt dynamically to observed changes in cell phenotype. The development of intelligent microscopy platforms that integrate data-driven acquisition with real-time feedback loops 52 suggests that such closed-loop systems are technically feasible, though they have not yet been demonstrated in mechanobiology. In the context of super-resolution imaging, deep learning-based localization methods such as DECODE already approach real-time processing speeds, offering a foundation for integrating nanoscale structural analysis into live-cell mechanobiology workflows. 12 ML-assisted structured illumination microscopy, which uses deep neural networks trained on synthetic data to reconstruct super-resolution images in real time, illustrates how ML can be embedded directly into the imaging pipeline to accelerate live-cell visualization of subcellular dynamics. 53 Neural operators such as Fourier Neural Operators, which learn solution mappings between function spaces and achieve orders-of-magnitude speedups over conventional PDE solvers, could provide the throughput required for real-time force field estimation in such closed-loop settings. 54
Therapeutic applications
Several of the approaches reviewed here have potential translational implications. The growing recognition that altered tissue mechanics—including ECM viscoelasticity, cell contractility, and nuclear deformability—drives pathogenesis across cancer, fibrosis, 55 diabetes, 56 cardiovascular disease, and neurodegeneration 57 broadens the therapeutic scope for AI-driven mechanobiological tools.
In cell therapy manufacturing, image-based mechanical phenotyping could complement conventional potency assays, as suggested by the association between MSC stiffness and passage-dependent changes in stemness and senescence markers. 19 Nuclear morphology-based senescence classification has been translated to in vivo tissue sections and correlated with age-related disease outcomes in human dermis, 21 further supporting image-based biomarkers as clinically actionable tools. Similarly, chromatin-based liquid biopsy biomarkers, if validated in larger cohorts, could contribute to cancer detection and treatment monitoring. 25 The emerging field of mechanopharmacology—which seeks to integrate mechanical cues into drug discovery pipelines 58 —could benefit substantially from ML-driven mechanical phenotyping for high-throughput compound screening.
Related advances in AI-driven drug discovery further illustrate this potential. Saar et al. demonstrated that ML models trained on physics-based descriptors from high-throughput crystallographic protein–ligand structures can prospectively identify potent inhibitors with over 10-fold potency improvement, an approach that could be extended to structure-guided design of compounds targeting mechanosensitive proteins. 59 Beyond protein targets, SMRTnet integrates RNA and chemical language models with convolutional and graph attention networks to predict small molecule–RNA interactions from secondary structure alone, eliminating the need for RNA tertiary structures 60 —an approach potentially relevant to mechanosensitive non-coding RNAs and riboswitches. Virtual screening of 7,350 compounds against ten disease-associated RNA targets yielded 40 experimentally validated binders with nanomolar-to-micromolar affinities, including a compound that downregulated MYC oncogene expression and inhibited proliferation in cancer cell lines. LLM-based single-cell foundation models have demonstrated virtual screening capabilities, identifying candidate compounds through in silico perturbation prediction on transcriptomic data, 49 an approach that could be adapted to prioritize mechanopharmacological targets. A systematic review of 90 AI-based biomarker studies in immuno-oncology, however, found that none incorporated AI within a prospective study design. 61 This translational gap is likely shared by mechanobiology. Translating these capabilities into clinical tools will require standardized measurement protocols, prospective clinical validation studies, and regulatory frameworks for AI-derived mechanical biomarkers.
Computational genomics and multiscale integration
A largely unrealized opportunity lies in adapting general-purpose computational genomics tools to mechanobiology. Deep learning models for predicting soluble protein expression from amino acid sequence have achieved promising accuracy across 88 species and experimentally validated mutant designs for enhancing heterologous expression, 62 and controlled sequence diversity in training data has been shown to improve both accuracy and generalization in DNA sequence-to-expression models even with limited datasets. 63 Incorporation of mechanistic sequence features—such as mRNA secondary structure stability, codon usage, and nucleotide composition—has been shown to substantially enhance generalization to unseen sequences across multiple organisms and construct types, compensating for the poor out-of-distribution performance of standard one-hot and language model-based sequence encodings. 64 If adapted to predict expression of mechanosensitive proteins under defined loading conditions, such models could complement experimental characterization of force-responsive gene programs. Deep learning peak callers such as LanceOtron, 65 which combines CNN-based peak shape recognition with enrichment scoring to outperform statistical methods across ATAC-seq, ChIP-seq, and DNase-seq benchmarks, could be applied to chromatin accessibility datasets from mechanically stimulated cells to identify force-responsive regulatory elements. Additionally, LLM-based foundation models such as C2S-Scale, trained on over 50 million single-cell transcriptomes, have demonstrated the ability to perform cell type annotation, perturbation prediction, and virtual drug screening by converting gene expression profiles into natural language representations 49 ; if extended to include mechanosensitive gene programs, such models could enable rapid in silico characterization of cellular responses to mechanical stimuli.
More broadly, integrating ML predictions across molecular, cellular, and tissue scales remains a fundamental challenge. Current models operate at single scales—predicting protein localization from chromatin images, traction forces from substrate deformation, or stiffness from phase-contrast microscopy—but do not bridge these levels. CellChat exemplifies one approach to bridging molecular and cellular scales, using a curated ligand–receptor database and a mass-action-based model incorporating receptor complex structure and cofactor modulation to quantify intercellular communication probabilities from single-cell transcriptomes, with social network analysis, pattern recognition, and manifold learning to identify signaling roles and coordinated communication patterns at the tissue level. 66 Multiscale modeling frameworks that connect molecular mechanotransduction events to tissue-level mechanical behavior through learned representations could provide a more integrated understanding of how forces regulate biological systems. Achieving such integration will likely require both architectural innovations in ML and the generation of training datasets that span multiple scales within the same experimental system. Data-driven methods for discovering governing PDEs from experimental observations, including sparse regression and symbolic approaches, could further aid this effort by identifying the effective equations of mechanotransduction at each scale without requiring a priori mechanistic assumptions. 67
Conclusions
This review has examined the emerging applications of AI and ML across several domains of mechanobiology, from traction force prediction and stiffness mapping to cell phenotype classification, chromatin-based protein localization, and spatial mechanomics.
First, the most successful applications to date share a common structure: they use ML to bypass experimentally costly steps by learning mappings between readily acquired data (for example, brightfield or phase-contrast images) and measurements that otherwise require specialized techniques (for example, AFM, fluorescent labeling, or TFM). The wrinkle force microscopy approach, 15 ISL, 20 and image-based stiffness classification 19 all follow this paradigm. The practical value of these methods lies not in replacing direct measurements but in enabling higher throughput and reduced experimental complexity in contexts where the speed and scalability outweigh the precision of direct measurement.
Second, a gap persists between proof-of-concept demonstrations and validated, generalizable tools. Most studies reviewed here were conducted on single cell types under controlled in vitro conditions, with limited assessment of cross-cell-type or cross-laboratory generalizability. Performance metrics, while encouraging, are reported without the independent validation cohorts that would be needed for clinical or industrial adoption. Closing this gap will require standardized benchmarking datasets, transparent reporting of model limitations, and prospective validation studies.
Third, the integration of ML-predicted mechanical properties with molecular profiling data—exemplified by the emerging combination of stiffness mapping with spatial transcriptomics—represents a promising but largely prospective direction. Realizing this potential will depend on the generation of paired datasets that link mechanical measurements to multiomic readouts within the same experimental systems, and on analytical frameworks capable of distinguishing causal relationships from spatial correlations.
Fourth, translating these capabilities toward clinical and therapeutic applications—including image-based mechanical biomarkers, mechanopharmacological screening, and real-time closed-loop experimental systems—will require prospective validation and regulatory frameworks for AI-derived mechanical readouts.
Finally, achieving these goals will depend on sustained collaboration between mechanobiologists and computational scientists, supported by open data practices and community standards for model evaluation and reproducibility. As mechanobiological datasets grow in scale and complexity, AI and ML methods are well positioned to extract insights that would be inaccessible through conventional analysis—provided they are applied with appropriate rigor and validated against the biological systems they aim to describe.
Footnotes
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study is supported by National Research Foundation of Korea; RS-2021-NR060095.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.