
Abstract
The extracellular matrix (ECM) is a dynamic mechanochemical network that integrates biochemical ligands with physical cues—such as stiffness, viscoelasticity, topography, and confinement—to regulate cell fate and tissue homeostasis. Cells sense and transmit ECM-derived forces through integrin-based adhesions, the actin cytoskeleton, mechanosensitive ion channels, and the LINC complex, coupling cytoplasmic tension to nuclear mechanics, chromatin organization, and transcriptional programs. In this review, we summarize key mechanobiological principles across major ECM contexts and outline how “mechanical niches” are established and remodeled during development and repair. We highlight the adhesion–nucleus–chromatin axis and central transcriptional hubs, including YAP/TAZ-mediated mechanotransduction, that translate extracellular mechanics into gene expression and epigenetic memory. We further discuss how ECM stiffening, altered viscoelastic relaxation, and matrix compositional switching drive disease progression, with emphasis on fibrosis, cancer, and vascular pathologies. Finally, we survey emerging experimental platforms—tunable hydrogels, engineered and decellularized ECM, and quantitative 3D culture/organoid systems—that enable controlled perturbation of ECM mechanics and multiscale readouts. These approaches provide a foundation for improved biomarkers and therapeutic strategies aimed at normalizing pathological microenvironments and restoring tissue function.
Keywords
Introduction
Tissues experience constant mechanical inputs, including tensile stress, compression, shear, and confinement. The extracellular matrix (ECM) constitutes the primary mechanical and structural scaffold that transmits and integrates these forces with biochemical signals.1–3 Classical views of the ECM as a static support have been replaced by a dynamic picture in which ECM composition, architecture, and mechanics continuously adapt to cellular activity and environmental cues, thereby feeding back on cell behavior.1–4
Representative ECM types, principal components, and key mechanical features relevant to mechanotransduction.

Mechanotransduction—the conversion of physical inputs into biochemical and gene-regulatory responses—underpins fundamental processes such as morphogenesis, stem cell fate decisions, immune responses, and tissue repair, and is increasingly recognized as a central driver of diseases including organ fibrosis, tumor progression and metastasis, cardiovascular and pulmonary disorders, and degenerative conditions.5–21 Within mechanobiology, the ECM occupies a privileged position as both the origin and target of mechanical signals: cells remodel their surrounding ECM by producing, degrading, and reorganizing matrix components, thereby creating feedback loops in which ECM mechanics and cellular responses co-evolve over time.3,4,8,9
Cell–matrix mechanotransduction: From adhesions to signaling networks
Integrin-mediated adhesions and focal adhesion maturation
Cells sense ECM mechanics largely through integrins, which bind ECM ligands such as fibronectin, collagen, laminin, and vitronectin, and organize into focal adhesions (FAs), multi-protein complexes containing talin, vinculin, paxillin, FAK, Src-family kinases, and scaffolding molecules.1–4,22,23 Force-dependent unfolding of talin and recruitment of vinculin link integrins to actomyosin stress fibers, enabling cells to apply contractile forces and “probe” ECM stiffness.5,24,25
On stiff substrates, resistance to cell-generated forces promotes FA growth and stabilization, leading to enhanced FAK/Src signaling, RhoA activation, elevated actomyosin contractility, and reinforcement of stress fibers; on soft matrices, limited resistance leads to smaller, more transient adhesions and diminished mechanotransductive signaling.4,5,8,16 In 3D interstitial and basement membrane–like matrices, integrins assemble distinct adhesion structures (e.g., fibrillar adhesions, 3D matrix adhesions, invadopodia-like protrusions) whose composition and lifetime depend on local ECM type, fiber orientation, and degradability (Figure 1).26–28 Stiffness-dependent maturation of focal adhesions. On soft substrates, adhesions remain small and dynamic with limited force transmission, whereas stiff substrates support reinforcement of integrin–talin–vinculin linkages, stress-fiber formation, and growth into stable force-bearing focal adhesions.
Cytoskeleton dynamics and mechano-sensitive machineries
The actin cytoskeleton, intermediate filaments, and microtubules form integrated networks that transmit mechanical forces from the plasma membrane to the nucleus.5,29,30 Myosin II–driven contractility generates tensile forces in actin stress fibers; cross-talk with microtubules and intermediate filaments such as vimentin and cytokeratins stabilizes cell shape and modulates load-bearing capacity.
Mechanosensitive machineries include stretch-activated ion channels (e.g., Piezo1), mechano-regulated small GTPases (RhoA, Rac1, Cdc42), and conformationally responsive cytoskeletal proteins that change binding affinities or enzymatic activity under force.5,6,8,31,32 The relative contributions of these systems depend on ECM type: cells embedded in viscoelastic, fibrillar interstitial matrices rely heavily on actomyosin contractility and proteolysis to create migration tracks, whereas cells on basement membranes or in pericellular matrices may be more sensitive to local tension, shear, and glycocalyx-mediated force filtering. Forces sensed at adhesions are distributed through actin, microtubules, and intermediate filaments and relayed to the nuclear envelope via the LINC complex, enabling mechanical regulation of nuclear shape and chromatin organization.33,34
YAP/TAZ and other transcriptional mechanotransducers
The transcriptional co-activators YAP and TAZ are central hubs converting mechanical cues into gene expression programs. ECM stiffness, cell spreading, and cytoskeletal tension promote nuclear localization and activation of YAP/TAZ, whereas soft matrices, reduced actomyosin contractility, or cell confinement drive their cytoplasmic retention.6,7,35–37 Multiple pathways couple ECM mechanics to YAP/TAZ, including integrin–FAK/Src–RhoA–ROCK–actomyosin signaling, interactions with the Hippo kinase cascade, and nuclear mechanics that influence nuclear pore transport.6,11,30,35,36,38–40
Computational and experimental studies have revealed that YAP/TAZ responses depend on cell shape, nuclear shape, ECM type (basement membrane vs interstitial vs 3D hydrogels), and dimensionality (2D vs 3D), and that nuclear deformation serves as a key integrator of mechanical cues upstream of YAP/TAZ activation.11,36,37,39,42,43 In fibroblasts, matrix stiffness–driven YAP/TAZ activation coordinates a positive feedback loop that sustains matrix deposition and fibrogenesis.7–9
ECM mechanics and cellular metabolism
Matrix stiffness and architecture modulate cellular metabolism, including glycolysis, oxidative phosphorylation, and biosynthetic pathways. ECM-induced cytoskeletal tension and YAP/TAZ activity intersect with metabolic regulators (e.g., mTOR, AMPK) and influence mitochondrial dynamics and reactive oxygen species, linking mechanical cues to metabolic reprogramming in development, regeneration, and disease.8,41,42 Recent work suggests that stiffness- and viscoelasticity-dependent metabolic states may help define “mechanotypes” of cells that predict their behavior in homeostasis and pathology.41,42 Tissue-specific ECM types impose distinct metabolic demands—for example, cells in highly crosslinked, stiff interstitial matrices versus cells on compliant basement membranes or within soft pericellular matrices—further diversifying mechano-metabolic coupling.
The nucleus as a mechanosensitive organelle
Force transmission to the nucleus via the LINC complex
The nucleus is coupled to the cytoskeleton through the LINC (linker of nucleoskeleton and cytoskeleton) complex, allowing external forces to deform nuclear shape, strain the nuclear lamina, and reorganize chromatin.29,30,33,34 Lamin A/C and B-type lamins form the nuclear lamina, which provides mechanical stability and participates in gene regulation through lamina-associated domains. Nuclear deformation alters the spatial organization of chromatin and nuclear bodies, thereby influencing transcriptional programs.29,30,43–46 These nuclear mechanotransduction pathways and associated epigenetic outcomes are discussed in Sections 3.2–4,47
Lamin a scaling with tissue stiffness
Landmark work demonstrated that lamin A levels scale with tissue stiffness: nuclei in cells derived from stiff tissues (e.g., bone, muscle) are lamin A–rich and mechanically robust, whereas those from soft tissues (e.g., brain, fat) are lamin A–poor and more deformable. 45 Matrix elasticity can upregulate lamin A levels and turnover, tuning nuclear mechanics to match the mechanical environment and contributing to lineage specification.30,45,46
More recent studies have shown coordinated increases in nuclear tension and lamin A with ECM stiffness and cytoskeletal prestress, providing a mechanistic basis for long-term nuclear adaptation to mechanical microenvironments, as well as protective roles of lamin A/C against mechanical damage.18,29,46 These adaptations occur in parallel with ECM remodeling, linking organ-specific ECM types and mechanics to nucleus-scale structural changes (Figure 2). Matrix stiffness regulates nuclear mechanics and Lamin A levels.
Nuclear deformation and gene regulation
ECM stiffening and cell-generated traction forces can flatten or elongate nuclei, leading to redistribution of nuclear stresses and nuclear lamina “unwrinkling.” Once the lamina becomes taut, further deformation can trigger changes in chromatin organization and gene expression.30,43,44
3D matrix confinement and anisotropic ECM architectures—such as aligned collagen fibers in interstitial tumor stroma or dense provisional matrices in wounds—can induce nuclear envelope folding, chromatin condensation or decondensation, and changes in accessibility of mechano-sensitive loci, thereby reprogramming gene expression patterns associated with lineage specification, migration, and invasion.30,38,43,44,48 Recent models describe nuclei as “drops” with surface tension regulated by lamin A/C, linking nuclear shape changes to YAP/TAZ nuclear localization and mechanosensitive transcription.11,38,43
Matrix stiffness and epigenetic reprogramming
Mechanical cues from the ECM modulate epigenetic states, including DNA methylation, histone modifications, and chromatin accessibility. Matrix stiffness can act as a biphasic regulator of epigenetic reprogramming efficiency and has been shown to influence chromatin-modifying enzymes and the accessibility of lineage-specific gene loci.41,48
In cancer and fibrosis, increased matrix stiffness and altered ECM type (e.g., replacement of compliant basement membranes with stiff, collagen-rich interstitial or provisional matrices) can epigenetically regulate oncogenic or pro-fibrotic programs (including YAP/TAZ target genes), and softening or “normalization” of the ECM can partially reverse such epigenetic changes, highlighting direct links between mechanical microenvironments and gene regulation.9,13,41,42,48
ECM mechanobiology in tissue homeostasis and disease
Fibrosis and wound healing
Fibrosis is characterized by excessive deposition and crosslinking of ECM, leading to progressive tissue stiffening, architectural distortion, and loss of organ function. Activated fibroblasts and myofibroblasts are central effectors that both sense and remodel matrix mechanics, integrating pro-fibrotic cytokines and growth factors with changes in stiffness, viscoelasticity, and topography.4,7–11,13,14,19,38,41,49 Fibrotic matrices typically show expansion of fibrillar collagens I and III, accumulation of fibronectin (including EDA-containing isoforms), periostin, thrombospondins, and other matricellular proteins, along with altered proteoglycan and glycosaminoglycan content that modify interstitial fluid flow and tissue swelling.4,8–11,13,14,19,21,49 Beyond bulk stiffening, changes in stress relaxation and plasticity create mechanical “memory” that stabilizes myofibroblast phenotypes even after the initial injury subsides, helping to explain why late-stage fibrosis is often refractory to therapy.7–9,14,38,41,49,50
Representative disease contexts in which ECM mechanics actively drive pathogenesis.

Cancer progression and metastasis
Tumors often exhibit pronounced stiffening and reorganization of the ECM due to collagen accumulation, lysyl oxidase (LOX)-mediated crosslinking, and fiber alignment, as well as basement membrane disruption and replacement by desmoplastic interstitial matrix.1,2,8,17,18,42,53–56 This altered mechanical landscape is highly heterogeneous: dense, aligned collagen tracks provide low-resistance paths that guide collective invasion of tumor cells and cancer-associated fibroblasts (CAFs), whereas softer, proteoglycan- and hyaluronan-rich niches can shelter stem-like, slow-cycling, or therapy-tolerant tumor cell populations.17,18,42,53,54,56 CAFs themselves behave as mechano-responsive stromal hubs that integrate stiffness, strain, and confinement cues, and in turn deposit, remodel, and crosslink ECM, thereby amplifying local stiffness gradients and generating physical barriers to drug penetration and immune cell entry.17,18,42,53,54,56
At the cellular level, stiff tumor ECM promotes malignant behaviors including enhanced proliferation, epithelial–mesenchymal transition, invasion, and metastasis, in part through integrin–FAK/Src–RhoA–ROCK signaling, YAP/TAZ activation, and mechanosensitive changes in chromatin organization and metabolism.6,8,17–19,41,48,53–56 Mechanotransductive signaling intersects with oncogenic and metabolic pathways to reprogram tumor cells and stromal cells, while epigenetic changes driven by ECM mechanics can sustain oncogenic programs even after removal from the stiff microenvironment.6,8,17–19,41,48,53–55 In parallel, tumor mechanobiology exerts profound effects on anti-tumor immunity: elevated matrix stiffness and aberrant topography impede cytotoxic T-cell and NK-cell infiltration, impair immune synapse formation, and skew myeloid cells toward immunosuppressive phenotypes, contributing to resistance to immune checkpoint blockade and other immunotherapies.17,42,53,54,56,57
These insights have motivated a new generation of mechano-focused therapeutic strategies. Targeting tumor ECM mechanics—by inhibiting crosslinking enzymes such as LOX, degrading specific matrix components, modulating tumor-associated basement membranes and interstitial matrices, or blocking mechanosensing pathways in tumor and stromal cells—is under active investigation as a strategy to normalize the tumor microenvironment and improve responses to therapy.17,18,42,53–56,58 In preclinical models, pharmacologic or biomaterial-based softening of the tumor stroma can enhance perfusion, facilitate drug delivery, and increase T-cell infiltration and effector function, thereby boosting responses to chemo-, radio-, and immunotherapies.17,18,42,53–58 Conceptually, these approaches shift the focus from treating tumors solely as biochemical entities to explicitly targeting the abnormal mechanical context in which malignant and immune cells reside.
Vascular and cardiovascular diseases
In the vasculature, ECM mechanics regulate endothelial and smooth muscle cell behavior, vascular development, and tone by distributing pressure, flow-derived shear, and cyclic stretch across the arterial wall.19,32,59–62 Arterial ECM comprises elastic lamellae rich in elastin and fibrillin, interspersed with collagen- and proteoglycan-rich interstitial matrices and endothelial basement membranes; together, these structures define arterial stiffness, compliance, and the pattern of wall strain.19,32,59–62 With aging and cardiometabolic disease, fragmentation of elastin, accumulation and crosslinking of collagen, proteoglycan deposition, and focal calcification progressively stiffen the arterial wall, alter pulse wave velocity, and modify the transmission of cyclic strain to endothelial and smooth muscle cells, contributing to atherosclerosis, vascular calcification, aneurysm formation, and pulmonary hypertension.11,14,19,32,59–62
Stiffened arterial ECM enhances smooth muscle cell contractility, promotes pro-inflammatory and synthetic phenotypes, and alters endothelial barrier function, while changes in basement membrane composition and elasticity impact angiogenesis, leukocyte trafficking, and vascular remodeling.19,32,59–62 Beyond static stiffness, complex combinations of pressure, shear stress, and cyclic stretch converge with ECM composition to regulate vascular mechanotransduction across arterial beds; regions of disturbed flow and locally stiffened matrix at branch points augment pro-atherogenic signaling, whereas compliant segments experiencing unidirectional shear promote anti-inflammatory, atheroprotective endothelial phenotypes.19,32,59–62 Mechanistic studies and comprehensive reviews of vascular mechanotransduction have begun to map how mechanosensitive ion channels, glycocalyx structures, and adhesion complexes integrate these forces to control vascular tone, permeability, and remodeling, and emphasize that strategies aimed at restoring physiological ECM mechanics may complement traditional blood pressure– and lipid-lowering therapies in preventing cardiovascular disease progression (Figure 3).19,32,59–62 Vascular ECM stiffening and calcification in vascular disease.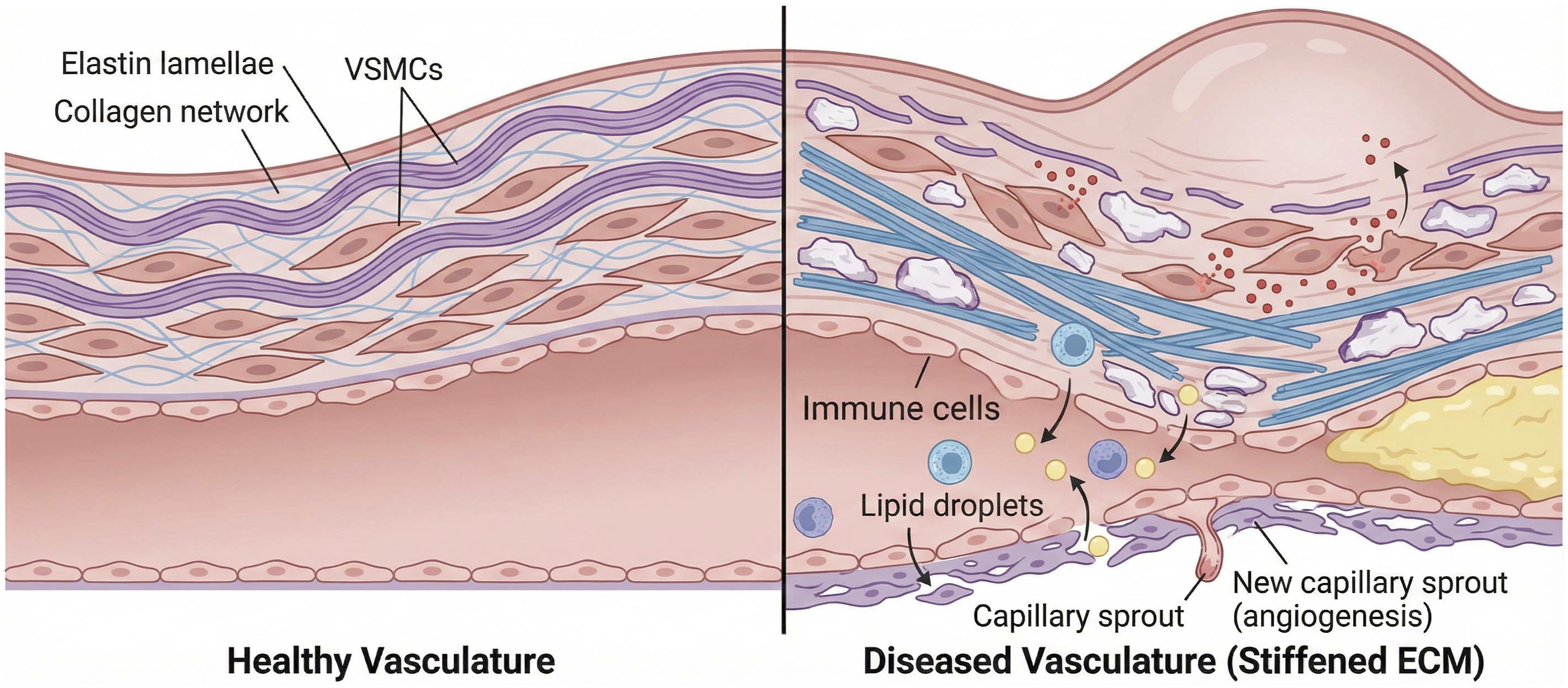
Conclusions
ECM mechanics reside at the critical intersection of external mechanical cues, intracellular mechanotransduction, and the balance between tissue homeostasis and pathology (Figures 3 and 4). ECM-derived forces and deformations are sensed and integrated through multi-scale, feed-forward networks that span integrin-based adhesions, cytoskeletal architecture, nuclear mechanics, and epigenetic landscapes, collectively shaping durable cell states and tissue phenotypes.1–6,8,9,29,30,35,36,38,41,45,46,48,61 Over the past decade, a series of conceptual advances has firmly established ECM stiffening, ECM type switching, and ECM remodeling as active drivers—rather than passive readouts—of fibrosis, cancer progression, vascular and pulmonary diseases, and defective wound repair.4,7–14,18,49,53,54 ECM-derived mechanical cues are transmitted through integrin adhesions to the cytoskeleton and nucleus.
Looking ahead, it will be important to decipher the mechanical “codebooks” that map specific combinations of stiffness, viscoelasticity, topology, ECM type, and spatial confinement onto defined cellular and tissue states, including their stability and reversibility.26–28,63,64 A central challenge is to integrate mechanobiology with multi-omics approaches so that mechanical inputs can be systematically linked to transcriptional, epigenetic, metabolic, and matrisome outputs at single-cell and spatial resolution, thereby revealing mechano-regulated molecular circuits that are amenable to therapeutic intervention. In parallel, more realistic models of dynamic cell–ECM feedback loops are needed to capture how mechanical cues and ECM types emerge, amplify, or dissipate during disease initiation, progression, and resolution across multiple temporal and spatial scales.
Mechanobiology-informed in vitro platforms—ranging from tunable synthetic and natural hydrogels to organoids cultured in engineered or decellularized ECM—provide tractable 3D testbeds to map ECM mechanical parameters to cell fate and to screen mechano-therapeutics under physiologically relevant conditions.14,26–28,38,48,55,61,64–68
At the translational level, these insights can inform the development of personalized mechano-therapies that target ECM composition, crosslinking, or mechanotransductive pathways in a patient- and organ-specific manner, guided by quantitative phenotyping of mechanical and matrisome signatures. In addition, continued advances in non-invasive mechanical and ECM diagnostics —including imaging modalities and circulating ECM biomarkers—will be crucial for monitoring ECM mechanics and remodeling in vivo, for stratifying patients according to their mechanical disease phenotype, and for tracking responses to mechano-focused interventions.4,8–10,13,14,42,49,55,58 By tightly integrating approaches from the physical sciences, materials engineering, molecular and cell biology, and clinical medicine, the mechanobiology community is well positioned to transform fundamental insights into ECM mechanics into a new generation of diagnostic tools and mechano-targeted therapeutics that directly address the physical dimension of disease.
Footnotes
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: National Research Foundation of Korea (RS-2023-00208339), Inha University (2025).
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.