
Abstract
Mechanical forces are fundamental regulators of cell behavior, tissue architecture and disease. The Hippo–YAP/TAZ pathway has emerged as a key mechanosensitive system that integrates extracellular matrix stiffness, cytoskeletal tension, cell–cell contact, and shear stress into transcriptional responses. Mechanical regulation of Hippo signaling operates across multiple cellular scales—from membrane-associated scaffolds and cytoskeletal dynamics to nuclear transport, chromatin organization and enhancer activation—shaping proliferation, differentiation, regeneration and tissue repair. Dysregulation of this mechanotransductive axis contributes to fibrosis, cancer progression, vascular remodeling and metabolic disorders. Recent advances reveal that Hippo signaling is structurally organized through spatially confined signaling assemblies, providing a physical framework for coupling mechanical inputs to biochemical activation. These assemblies modulate MST1/2–LATS activity, influence YAP/TAZ nuclear localization and support the formation of transcriptionally active TEAD complexes. Mechanical cues, osmotic adaptation and metabolic states converge on these regulatory architectures, enabling cells to translate physical forces into sustained gene-expression programs. Viewing Hippo signaling as a mechanochemical system unifies diverse observations across physiology, disease and emerging structural biology. This framework highlights new therapeutic opportunities, including modulation of YAP/TAZ activity through targeting upstream mechanosensors, nuclear transport mechanisms or TEAD-associated regulatory complexes.
Keywords
Introduction
Mechanotransduction refers to the cellular process by which mechanical stimuli are converted into biochemical signals that regulate cell behavior and gene expression.1,2 At the molecular level, mechanotransduction is achieved through mechanochemical signaling, in which mechanical inputs are converted into biochemical reactions such as protein phosphorylation, conformational changes, and regulated protein–protein interactions. Importantly, this force-to-signal conversion does not occur diffusely within the cell but is tightly dependent on the spatial organization of signaling components across distinct cellular compartments. Increasing evidence suggests that the localization, assembly, and compartmentalization of signaling molecules critically determine how mechanical information is processed and transmitted to downstream transcriptional programs.3,4 Through these mechanisms, cells and tissues generate appropriate physiological responses to external forces. Various types of mechanical forces are continuously exerted on the human body, including cardiovascular functions, cyclic stretch, and muscle tensile stress. Despite several studies on cellular mechanosensing, the underlying molecular mechanisms that integrate diverse mechanical loading into biological functions remain incompletely understood. Mechanobiology reveals how physical forces and mechanical properties of cells and tissue influence biological function, development, disease, and healing. 5 Among the cellular signaling, the Hippo signaling pathway has emerged as a center of mechanosensing that translate mechanical cues into gene expression changes, linking mechanical environment and cellular behavior. 6 The Hippo signaling senses the delicate physical movements and interaction of cells and ultimately connects cellular mechanics to gene expression programs.7,8 A comprehensive understanding of the Hippo signaling within the context of mechanobiology is essential to establish insights into development, disease, and searching therapeutic strategies. 9
The Hippo signaling is an evolutionally conserved signaling pathway that plays a fundamental role in cellular processes, including organ size control, regeneration, stem cell biology, and tumor suppression.10–12 The Hippo pathway is regulated by various regulators, including intercellular signaling and extrinsic factors such as cell-cell contact, mechanical cues, cell adhesion, cell morphology, and cell polarity.13,14 Cell-cell contact was discovered as the first signal regulating the Hippo pathway and several physiological regulators were reported.
15
The central effectors of the Hippo pathway in response to mechanical cues are the Yes-associated protein (YAP, also known as YAP1) and the transcriptional co-activator with PDZ binding motif (TAZ, also known as WWTR1). These proteins are particularly important in mediating cellular responses to mechanical signals. YAP/TAZ are regulated by a phosphorylation cascade consisting of Mammalian STE20-like protein kinase 1 (MST1, also known as STK4), MST2 (also known as STK3), large tumor suppressor 1/2 (LATS1/2)
16
(Figure 1). MST1/2 and their binding partner Salvador family WW domain-containing 1 (SAV1, also known as WW45) phosphorylate and activate the LATS1/2-MOB complex, thereby stimulating LATS kinase activity.17–19 LATS1/2 directly phosphorylate YAP/TAZ, triggering cytoplasmic sequestration and resulting in destabilization and degradation.
20
Scaffolding proteins coordinate the spatial organization of the MST1/2-LATS1/2 cascade and their partners. Key scaffold molecules such as SAV1, MOB1, Merlin, and Angiomotin (AMOT) family facilitate efficient interactions between the kinase and its substrate
21
(Figure 1). The Hippo signaling pathway in mammal. When Hippo signaling is “off” (Left), YAP/TAZ is activated and translocated to nucleus, binding with TEAD1-4 to induce the expression of target gene. Stiff ECM activates GPCR- and integrin-dependent mechanotransduction, leading actin polymerization and increased cytoskeletal tension, which in turn suppress LATS1/2 activity. When Hippo signaling is “on” (Right), the kinases of the hippo pathway, MST and LATS, phosphorylate YAP/TAZ and trigger cytoplasmic sequestration, resulting the destabilization and degradation of YAP/TAZ. Under Hippo on conditions, reduced actin polymerization and cytoskeletal tension allow AMOT to associate with YAP and LATS kinases, thereby promoting YAP phosphorylation and preventing its nuclear accumulation.
Various extracellular stimuli, including mechanical signals, cell adhesion, GPCR ligands, and cellular stresses, promote the nuclear translocation of YAP/TAZ. This subsequently initiates transcription, leading to conditions conducive to cell growth (Figure 2(a)). The subcellular distribution of YAP/TAZ indicates the activation of Hippo signaling, which affects the regulation of the target gene expression, including connective tissue growth factor (CTGF), Cysteine-rich angiogenic inducer 61 (CYR61), and AMOTL2. The E-cadherin/catenin complex inhibits cell growth that accrued at embryonic development and the maintenance of tissue architecture in adult organisms via control of YAP subcellular localization22–24. Taken together, YAP/TAZ are sensitive to mechanical forces and regulate tissue growth. Cell adhesion and tight junction proteins such as AMOT, protein tyrosine phosphatase non-receptor type 14 (PTPN14), and α-catenin, repress YAP/TAZ transcriptional activities through direct interaction.25–28 The Hippo signaling functions as mechano-transducer through nuclear-cytoplasm translocation of YAP/TAZ. (a) Schematic representations of diverse mechanical stimuli of the Hippo signaling. (b) Multiple mechanosensing pathways concerge on LATS1/2 to regulate the Hippo signaling. Distinct mechanosensors including Merlin, AMOT and RAP2 detect diverse mechanical cues and transmit them to LATS1/2. And then, activated LATS1/2 phosphorylates YAP/TAZ, inhibiting the expression of target gene.
Several excellent reviews have summarized the role of YAP/TAZ as mechanosensitive transcriptional regulators in development and disease.8,12 These studies have primarily focused on upstream mechanical inputs, cytoskeletal tension, and transcriptional outcomes of YAP/TAZ signaling.7,8 In contrast, the present review emphasizes how mechanical cues are processed through spatial organization and mechanochemical signaling within the Hippo pathway. By integrating recent advances in biomolecular condensation, kinase compartmentalization, and nuclear mechanotransduction, we propose a hierarchical framework in which mechanical information is encoded and transmitted through phase-organized Hippo signaling assemblies.29–31 This perspective highlights how spatial and physical properties of signaling components shape context-dependent YAP/TAZ responses across tissues and disease states.
Despite substantial progress in delineating Hippo signaling components, how this pathway integrates mechanical information across cellular compartments and translates it into context-dependent transcriptional outcomes remains incompletely understood. Recent advances in mechanobiology, nuclear mechanics, and spatial signal organization now provide an opportunity to revisit Hippo signaling beyond a linear kinase cascade. In this review, we synthesize current knowledge on mechanosensitive regulation of Hippo signaling from membrane-associated mechanosensors to nuclear YAP/TAZ-dependent transcription, highlight tissue-specific and disease-relevant mechanobiological contexts, and discuss emerging concepts that redefine Hippo signaling as an integrated mechanochemical system. By bridging molecular mechanisms with physiological and pathological mechanics, this review aims to provide a conceptual framework for understanding Hippo-mediated mechanotransduction and to identify future directions for therapeutic intervention.
Regulatory mechanisms and interaction with mechanical signals
Insight into the mechanisms linking the cell morphology to intracellular signals provides a clear view of mechano-response switching. The interaction between MST kinases and c-Raf (RAF1), together with the tumor suppressor RASSF1A, functions as a cytoskeleton-sensitive regulatory module that links cytoskeletal integrity to stress- and cell cycle–related signaling pathways, including JNK/SAPK signaling.32,33 In this context, RAF1 can form an inhibitory complex with MST kinases, whereas RASSF1A competitively disrupts this interaction, enabling MST activation under stress conditions. 32 Consistent with this model, actin cytoskeleton integrity is closely coupled to MST activity. Experimental studies have shown that MST1/2 colocalize with filamentous actin, and pharmacological or genetic disruption of the actin cytoskeleton leads to MST1/2 activation.33,34 Merlin, encoded by the tumor suppressor Neurofibromatosis 2 (NF2), localizes to the apical membrane and organizes membrane domains by linking junctional complexes to the actin cytoskeleton. Through this spatial organization, Merlin converts mechanical cues such as cell density and membrane tension into Hippo pathway activation. 35 Merlin cooperates with the tumor suppressor Expanded and, together with Kibra, accumulates at the apical cortex to promote phosphorylation of Warts, the Drosophila homolog of LATS.36–38 Downstream, LATS1/2 directly interact with F-actin and phosphorylate Angiomotin (AMOT) family proteins at HXRXXS motifs, thereby modulating actin dynamics and reinforcing Hippo pathway activation.39,40 In addition to cytoskeletal cues, extracellular matrix (ECM) rigidity regulates Hippo signaling through activation of the Ras-related GTPase RAP2, which promotes LATS1/2 activity in response to matrix stiffness 41 (Figure 2(b)).
These associations also link other signaling pathways to mechanical forces including Wnt and mTOR pathway.42,43 The nuclear YAP directly interacts with β-catenin to promote the expression of genes necessary for heart development. 44 Moreover, a deficient SAV1 in the developing mouse heart model causes nuclear accumulation of β‐catenin and increases expression of Wnt target genes. 45 Furthermore, cytoplasmic YAP inhibits the disheveled (DVL), independently of Axin. 46 These findings suggest that the Hippo signaling negatively regulates Wnt/β‐catenin signaling. Recent studies have suggested that the connection between mTOR and Hippo signaling is bidirectional and context-dependent. LATS1/2 directly interact with and phosphorylate RAPTOR, thereby inhibiting the mTORC1 activator Rheb. 47 Under nutrient-rich conditions, mTOR upstream insulin/insulin-like growth factor 1 (IGF-1) activates P13K/AKT signaling, and suppresses the Hippo pathway, subsequently activating YAP/TAZ. 47 Conversely, the Hippo signaling components assemble mechanical cues and modulate the mTOR signaling activity. Overall, the interplay between other signaling pathway and Hippo signaling pathway contributes to the adjustment of cellular processes, which has implications for development, tissue homeostasis, and disease, including cancer.
Mechanotransduction through the hippo–YAP/TAZ pathway
Cells are exposed to diverse forms of mechanical inputs that originate from both extracellular and intracellular sources. These forces include extracellular matrix (ECM) stiffness and composition, cell–cell contact and crowding, shear stress generated by fluid flow, tensile and compressive forces arising from tissue deformation, and intracellular contractile forces produced by the actomyosin cytoskeleton. Such mechanical inputs vary in magnitude, direction, and temporal dynamics across tissues and physiological contexts. Importantly, these forces are not sensed uniformly but are interpreted through specialized mechanosensitive structures that convert physical cues into biochemical signals. Understanding the origin and nature of mechanical inputs provides essential context for dissecting how mechanosensory proteins engage Hippo signaling.1,2
Hierarchical mechanosensing in the hippo pathway
The mechanosensitive regulation of the Hippo pathway operates through a hierarchical signal transduction architecture. 48 Specific mechanosensory proteins, including junctional and cytoskeleton-associated regulators such as Merlin, AMOT family proteins, and Kibra, detect mechanical stimuli and modulate the activity of the core Hippo kinase cascade (MST1/2–LATS1/2), thereby influencing YAP/TAZ activity.29,49 This mechanosensitive regulation can be categorized into two distinct functional layers: (1) upstream mechanosensors that directly sense diverse mechanical signals and perform initial processing, and (2) the core kinase cascade that receives signals from mechanosensors and directly regulates YAP/TAZ activities. 50 Upstream mechanosensors play specialized roles in different mechanical contexts, including cell-cell contact, matrix stiffness, and mechanical stress. While they converge their signals by activating MST1/2 and LATS1/2. 51 This functional distinction reflects spatial organization, with the upstream sensors predominantly associated with junction and membrane while the core kinases operate as a cytoplasmic cascade. 29 Additionally, the temporal sequence shows upstream sensors are activated first, followed by core kinase responses. The convergence of diverse upstream inputs into a common core pathway represents a fundamental principle of mechanotransduction in the Hippo signaling network. 51
Upstream mechanical sensors and signal initiation
Networks cells have developed specialized mechanosensor proteins to detect mechanical changes in diverse contexts, each sensing unique mechanical inputs while converging signals into common downstream pathways. 48 These mechanosensors employ distinct molecular mechanisms to detect specific types of mechanical perturbations and transmit signals to the core kinase cascade. 48 Under conditions of increased cell-cell contact, Merlin functions as the primary mechanosensor. 29 As cell density increases and adherens junctions and tight junctions mature, Merlin detects these changes and recruits LATS1/2 to the plasma membrane. 29 Merlin directly binds LATS1/2 through its FERM domain and positions them for phosphorylation by membrane-associated MST1/2, thereby promoting LATS1/2 activation independently of enhancing MST1/2 intrinsic kinase activity. 29 In situations where tight junction integrity and barrier function are critical, AMOT family proteins (AMOT, AMOTL1, and AMOTL2) play pivotal roles. 52 These proteins operate through a unique dual mechanism: they directly bind to YAP/TAZ and sequester them at tight junctions, while simultaneously enhancing LATS1/2 kinase activity.52,53 This dual regulatory system enables immediate detection and response to epithelial barrier perturbations. To address broader mechanical stress and environmental changes, KIBRA and the MAP4K family form a third sensing network. 48 KIBRA functions as a mechanical stress-responsive regulator protein working with Merlin, responding to physical changes. 54 RAP2 GTPase has been identified as a key mechanotransducer that senses ECM stiffness, establishing a mechanosignaling pathway from matrix stiffness to the nucleus. 41 Under low substrate stiffness, GTP-bound RAP2 stimulates MAP4K4/6/7, which in turn augments LATS1/2 kinase activity, consequently leading to the inhibition of YAP/TAZ. These systems possess distinct characteristics, notably MAP4Ks kinase ability to directly activate LATS1/2 without requiring MST1/2.41,55 This parallel pathway provides a redundant mechanosensing mechanism that maintains Hippo signaling even when MST1/2 function is compromised. These three sensing systems operating in different mechanical contexts all converge at LATS1/2 as a common integration point. 48 Cell-cell contact sensing through Merlin, junction integrity monitoring through the AMOT family, and direct mechanical stress detection through MAP4K all culminate in LATS1/2 activation. Additional mechanosensitive regulators such as PTPN14 have been identified within this upstream sensor network, directly interacting with YAP under contact inhibition conditions to induce cytoplasmic sequestration. 56 This convergent architecture enables cells to generate integrated responses even in complex mechanical environments while ensuring system stability through mechanosensor redundancy.
Integration of mechanical inputs into the core kinase cascade
The diverse mechanical information collected from upstream mechanosensors undergoes integrated processing in the MST1/2-LATS1/2 core kinase cascade, ultimately leading to YAP/TAZ activity regulation. 51 This signal integration process represents the core mechanism by which cells convert complex and potentially conflicting mechanical cues into coherent biological responses. 48 Rather than functioning as simple binary switches, the core kinases exhibit graded responses that integrate multiple inputs with different weights and temporal dynamics. MST1/2 functions as the primary integration hub for multiple upstream signals. 29 Merlin-mediated cell-cell contact signals, junction integrity information from the AMOT family, and direct cellular stress signals such as osmotic stress or growth factor withdrawal all converge on MST1/2 activation. MST1/2 is activated through homodimerization and trans-autophosphorylation, whereby simultaneous input from multiple upstream signals promotes MST1/2 dimer formation and amplifies kinase activity. 29 Subcellular localization changes of MST1/2 represent a crucial aspect of signal integration. Under mechanical stress conditions, MST1/2 translocates to the membrane, enabling efficient interactions with downstream targets. 57 This localization-dependent activation provides a mechanism by which MST1/2 converts spatial information into temporal dynamics, with signal specificity further enhanced through scaffold complex formation with SAV1. 29
LATS1/2 serves as the central processing unit receiving signals from both MST1/2 and the MAP4K family. 55 LATS1/2 activity is subjected to opposing regulation depending on mechanical context, exhibiting high activity in soft matrix environments that potently suppresses YAP/TAZ, while activity decreases in stiff matrices, permitting YAP/TAZ activation. 51 This matrix stiffness-dependent regulation is closely associated with focal adhesion formation through integrin signaling and is mediated by membrane recruitment and cytoskeletal anchoring of LATS1/2. 58 Membrane localization of LATS1/2 by AMOT and Merlin promotes MST1/2-mediated phosphorylation and fine-tunes kinase activity through complex formation with MOB1.29,59 LATS1/2 exhibits differential activation patterns through various phosphorylation sites, enabling selective responses to different upstream inputs. 48 Signal integration in the core kinase cascade encompasses decision-making processes for conflicting mechanical inputs. 55 For instance, when YAP/TAZ activation signals through cell spreading in stiff matrix environments coexist with contact inhibition signals due to high cell density, cells process this conflicting information across temporal and spatial dimensions. 51 The MST1/2-LATS1/2 cascade encodes these complex input patterns into the amplitude and duration of YAP/TAZ phosphorylation, generating graded responses that enable appropriate cellular reactions to environmental changes. 48
Additional regulatory mechanisms maintain balance between system stability and responsiveness. 55 Phosphatase activities, particularly the PP2A-STRIPAK complex, provide negative feedback by dephosphorylating MST1/2, thereby antagonizing Hippo pathway activation and maintaining homeostatic control. 60 This creates regulatory loops where MST1/2 autophosphorylation recruits STRIPAK through SLMAP binding to phosphorylated MST2 linker, leading to subsequent MST1/2 inactivation. The dynamic assembly of the STRIPAK complex in response to cell density and other upstream signals enables it to function as a biosensor that integrates various cellular inputs. Recent studies have further revealed that STRIPAK integrates upstream signals through a RhoA-rhophilin-NF2/Kibra axis to control the activities of both MST1/2 and MAP4Ks, thus serving as a master regulator for initiating Hippo signaling. 61 This STRIPAK-mediated regulation extends to innate immune and inflammatory signals, with Tak1 activating the Hippo pathway through STRIPAK-Tao signaling. The core of signal integration relies on principles where the intensity and duration of each mechanical input differentially contribute to final kinase activity. 48 The kinase cascade forms signal hierarchies that enable priority systems where survival-critical signals such as severe mechanical stress can override relatively weaker signals such as moderate matrix stiffness changes. 55 This hierarchical processing enables appropriate balance between emergency responses and routine homeostasis. 51
Consequently, the MST1/2-LATS1/2 core kinase cascade functions as an integration platform that processes diverse mechanical inputs to generate context-appropriate YAP/TAZ activity regulation. 48 However, for this signal integration to ultimately influence cellular function, the subcellular localization of YAP/TAZ must be precisely regulated. 51 The mechanical information processed by the core kinase cascade is converted into nuclear-cytoplasmic shuttling of YAP/TAZ through mechanisms that extend beyond LATS1/2-mediated phosphorylation alone. 62 Recent discoveries demonstrate that changes in cytoskeletal architecture and nuclear mechanics themselves directly influence YAP/TAZ translocation, revealing additional dimensions of mechanotransduction. 30
Nuclear mechanotransduction governing YAP/TAZ localization
Mechanical cues are interpreted through coordinated mechanosensors and kinase cascades, providing the biochemical basis for YAP/TAZ regulation. Accumulating evidence shows that mechanical forces can directly control the nuclear–cytoplasmic transport of YAP/TAZ through mechanisms that bypass or complement classical phosphorylation-dependent regulation. 30 This direct mechanotransduction establishes an additional regulatory tier by which physical forces shape transcriptional outcomes.
Nuclear mechanics and regulation of nuclear pore transport
Mechanical loading rapidly remodels nuclear architecture. On stiff substrates, actomyosin contractility transmits tension to the nucleus through the linker of nucleoskeleton and cytoskeleton (LINC) complex, composed of KASH- and SUN-domain proteins that bridge the cytoskeleton and nucleoskeleton. 63 This tensile force flattens the nucleus, decreases its volume, and increases surface area. These geometric changes restructure nuclear pore complexes (NPCs), widening their central channels and elevating nuclear permeability. 30 Experimental studies demonstrate that mechanical tension applied to the nuclear envelope causes reversible dilation of NPCs, revealing a core mechanosensitive property governing nucleocytoplasmic transport. Large cargos such as YAP/TAZ (∼65 kDa) are particularly sensitive to mechanically induced shifts in pore geometry. 30 The transport machinery itself is mechanosensitive: nuclear envelope tension modulates the binding dynamics of importins and exportins, altering transport efficiency. The LINC complex, particularly SUN1, directly interacts with NPC components including Nup153, forming a conduit through which cytoskeletal forces are transmitted to the pore structure. Through this mechanism, NPC deformation provides a rapid, phosphorylation-independent pathway linking mechanical forces to transcription factor localization.
Cytoskeletal architecture and force transmission to the nucleus
The actin cytoskeleton is a major determinant of YAP/TAZ localization. On rigid matrices, RhoA–ROCK signaling promotes formation of contractile stress fibers that deform the nucleus and enhance YAP/TAZ nuclear entry.30,51 In contrast, soft substrates or confined environments favor Arp2/3-dependent branched cortical actin, promoting YAP/TAZ cytoplasmic retention. Intriguingly, sparse-cell studies show that YAP can enter the nucleus in an F-actin–dependent manner even without myosin II contractility and independently of phosphorylation at Ser112, 58 underscoring that actin integrity can override contractile or phosphoregulatory inputs. Nuclear actin polymerization further contributes to transcriptional control. Nuclear F-actin engages chromatin remodeling complexes such as BAF, improving accessibility to TEAD target loci. 64 Myosin II–dependent cytoskeletal tension additionally influences nuclear shape and chromatin organization. 30 Recent work shows that nuclear compression generated by contractile work is a strong predictor of YAP dynamics, surpassing substrate stiffness in explanatory power. YAP also participates in mechanical feedback by promoting focal adhesion assembly and transcription of adhesion-related genes, reinforcing contractile force generation. Conversely, YAP/TAZ can suppress excessive cytoskeletal maturation through negative feedback to maintain motility, establishing a dynamic equilibrium.
Integration of cytoskeletal and nuclear mechanotransduction
Direct mechanical regulation of YAP/TAZ follows a hierarchical sequence: ECM stiffness is sensed through integrins, initiating focal adhesion maturation and actomyosin stress fiber formation. Cytoskeletal tension is then transmitted via the LINC complex to deform the nucleus and expand NPCs, thereby facilitating YAP/TAZ nuclear import. Within the nucleus, actin polymerization enhances chromatin remodeling and amplifies transcriptional output. 65 Nuclear compression has been shown to promote YAP/TAZ nuclear entry and is associated with changes in nuclear architecture that may influence chromatin organization and transcriptional responsiveness. Chromatin mechanics exhibit bidirectionality: tensile loading induces EZH2-dependent compaction, contributing to mechanical memory in mesenchymal stem cells, while nanopillar-induced deformation can cause acute decompaction accompanied by nuclear stiffening—an effect diminished in highly malignant cancer cells. During confined migration, deformation-induced chromatin compaction reorganizes nuclear bodies and modulates their phase behavior. This generates a reverse mechanotransduction axis in which chromatin state influences adhesion strength by regulating transcription of mechanoregulatory genes such as RhoA, Rock, and actin polymerization factors. Activated YAP/TAZ further enhance ECM synthesis and CTGF expression, reinforcing cytoskeletal tension and matrix stiffness to sustain mechanically driven signaling. 66
To ensure robust adaptation to the mechanical environment, cells utilize two parallel regulatory mechanisms: the canonical Hippo signaling pathway and direct mechanical control. The Hippo-dependent arm is primarily regulated by upstream components such as junctional AMOT or Merlin, and the KIBRA–MAP4K axis linking energy state to mechanical stress. These factors modulate MST1/2–LATS1/2 kinase activity, leading to the phosphorylation of YAP/TAZ at Ser127 and Ser397, which dictates cytoplasmic sequestration (via 14-3-3 binding) and subsequent degradation. 51 This pathway operates on slower timescales (minutes to hours), governing long-term adaptation and cell-fate specification. In contrast, Hippo-independent mechanical regulation bypasses phosphorylation control and enables immediate adaptation on fast timescales (seconds to minutes). This direct pathway is initiated by actomyosin contractility on stiff substrates, where tension is transmitted to the nucleus via the LINC complex to deform the nucleus and expand Nuclear Pore Complexes (NPCs). 30 This mechanism allows even LATS-phosphorylated, 14-3-3–bound YAP/TAZ to enter the nucleus. 62 Once nuclear, mechanical signals are amplified: nuclear compression modifies chromatin structure, and nuclear F-actin polymerization recruits SWI/SNF complexes to potentiate TEAD accessibility and transcription. 67 This cooperative regulation is spatially (junction vs nucleus) and temporally partitioned, with epithelial cells preferentially relying on junction-based Hippo regulation, while mesenchymal cells primarily engage direct mechanotransduction. Ultimately, this extensive crosstalk facilitates robust adaptation to dynamically changing environmental conditions by synchronizing rapid mechanical responses with long-term transcriptional programs.
YAP/TAZ-dependent mechanosensitive transcriptional programs
Nuclear translocation of YAP/TAZ represents only the initial step in mechanotransduction; the subsequent conversion of mechanical signals into specific transcriptional outputs requires sophisticated regulatory mechanisms that operate at multiple levels. YAP/TAZ, as transcriptional co-activators lacking intrinsic DNA-binding activity, must interact with transcription factors to regulate target gene expression. 51 The mechanical environment profoundly influences not only YAP/TAZ nuclear accumulation but also their transcriptional activity through chromatin-level regulation and temporally coordinated gene activation patterns.
YAP/TAZ–TEAD complex formation and chromatin accessibility
The transcriptional activity of nuclear YAP/TAZ is regulated by competitive interactions with chromatin remodeling complexes in a mechanically-dependent manner. A critical regulatory mechanism involves the mechanical context-dependent interaction between YAP/TAZ and the ARID1A-SWI/SNF chromatin remodeling complex. 64 Under soft substrate conditions or low mechanical signaling, YAP/TAZ is sequestered by the ARID1A-SWI/SNF complex through interactions between the PPXY motifs of ARID1A and the WW domains of YAP/TAZ. In this sequestered state, YAP/TAZ cannot bind to TEAD transcription factors, resulting in transcriptional repression despite nuclear localization.
Mechanical forces reverse this inhibitory interaction through nuclear actin polymerization.
64
On stiff substrates or under high mechanical stress, increased nuclear F-actin directly binds to the ARID1A-SWI/SNF complex, releasing YAP/TAZ from sequestration. Liberated YAP/TAZ can now form active transcriptional complexes with TEAD1-4, enabling target gene activation. This mechanical switch provides a molecular basis for how identical YAP/TAZ nuclear concentrations can yield entirely different transcriptional activities depending on mechanical context. Nuclear actin polymerization additionally promotes chromatin remodeling by recruiting chromatin-modifying complexes to YAP/TAZ-TEAD target loci.
64
The interaction between nuclear F-actin and chromatin remodeling complexes such as BAF enhances accessibility to TEAD binding sites, amplifying transcriptional output. This multi-layered regulation ensures that mechanical signals are faithfully translated into appropriate gene expression programs (Figure 3(a)). Mechanical contexts orchestrate YAP/TAZ activity through nuclear dynamics and tissue-specific signaling hierarchies. The schematic depicts how mechanical contexts dictate YAP/TAZ activity through nuclear chromatin remodeling and tissue-specific signaling hierarchies. (a) Mechanical control of YAP/TAZ–TEAD complex formation via nuclear actin. Under low mechanical signaling (soft substrates), the ARID1A-SWI/SNF complex sequesters YAP/TAZ, preventing TEAD binding. On stiff substrates, increased nuclear F-actin binds to the ARID1A-SWI/SNF complex, releasing YAP/TAZ from sequestration to form active transcriptional complexes with TEAD. (b) Diversity of tissue-specific mechanosensor networks and transcriptional outputs. In high cell-cell contact tissues, NF2 serves as the predominant mechanosensor for cell–cell contact and polarity, promoting cytoplasmic retention of YAP/TAZ. In flow-reponsive tissues, hemodynamic forces differentially regulate signaling: laminar flow suppresses YAP/TAZ to maintain homeostasis, while oscillatory flow activates YAP/TAZ via AMOT and the Thrombospondin-1–RAP2 axis, promoting inflammation and remodeling. In stiffness-sensitive tissues, integrin–focal adhesion complexes sense high matrix stiffness, driving TAZ–RUNX2 interactions for osteogenic differentiation. Each tissue establishes a self-reinforcing feedback loop to maintain its specific mechanical identity.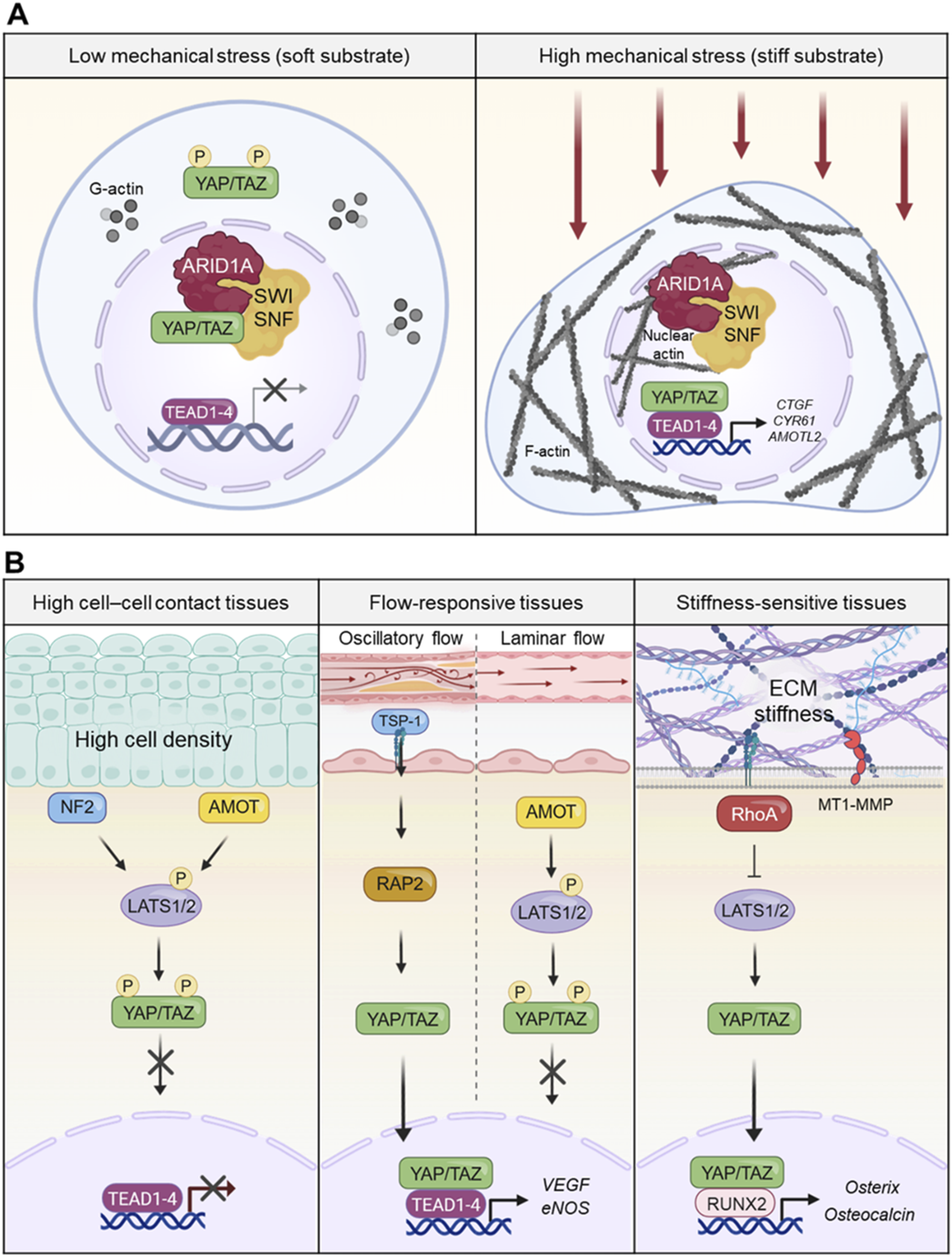
Temporal control of mechanosensitive gene expression
Mechanotransduction through YAP/TAZ operates as a temporally orchestrated process rather than a simple on-off switch. Following mechanical stimulation, YAP/TAZ-dependent gene expression proceeds through sequential waves, each characterized by distinct kinetics and functional roles. 48 Immediate-early genes including c-Fos, c-Jun, and EGR1 exhibit rapid induction within minutes of mechanical stimulation, reflecting their location at constitutively accessible chromatin regions. 68 These transcription factors, functioning as members of the AP-1 complex, establish a framework for subsequent gene expression and are required for full activation of YAP/TAZ target genes. A second wave emerges within 30 min to 2 h, encompassing classical YAP/TAZ direct targets such as CTGF, CYR61, and ANKRD1. 51 These genes encode proteins that modulate cell-matrix interactions and enhance mechanosensitivity, thereby establishing positive feedback circuits. CTGF mediates extracellular matrix protein binding, CYR61 activates integrin signaling, and ANKRD1 regulates cytoskeletal organization. 48 Later phases, occurring over several hours, involve expression of structural extracellular matrix proteins including collagen I, fibronectin, and laminin, alongside integrin subunits and focal adhesion proteins. This delayed response enables long-term tissue remodeling and sustained mechanosignaling. The temporal coordination of gene expression allows cells to mount both immediate adaptive responses and sustained architectural changes appropriate to their mechanical environment.
Signal amplification through transcriptional feedback loops
YAP/TAZ-activated genes establish multiple interconnected positive feedback loops that amplify mechanical signals and stabilize cellular mechanical states. 48 These feedback systems operate at three principal levels: enhancement of mechanosensing capacity, augmentation of cellular contractility, and remodeling of the extracellular matrix environment. Enhanced mechanosensing results from YAP/TAZ-driven upregulation of integrin subunits (β1, α5) and focal adhesion proteins including talin, vinculin, and paxillin. 69 YAP transcriptionally activates genes encoding these focal adhesion components, thereby tuning cell mechanics, force development, and adhesion strength. 69 Increased expression strengthens cell-matrix adhesion and enhances sensitivity to mechanical cues, amplifying the initial mechanical stimulus. 67 Mechanosensitive kinases such as FAK and Src are similarly upregulated, further sensitizing signaling pathways. Contractility augmentation occurs through increased expression of contractile machinery components including myosin heavy chain IIA/B, α-actinin, and tropomyosin. Enhanced cytoskeletal contractility sustains nuclear deformation, thereby maintaining conditions favorable for YAP/TAZ nuclear retention. 30 Simultaneously, upregulation of RhoA and ROCK reinforces actomyosin contractility, creating a self-sustaining mechanical state.
Extracellular matrix remodeling represents the most far-reaching feedback mechanism. YAP/TAZ activate expression of lysyl oxidase (LOX) and LOXL2, enzymes that catalyze collagen and elastin cross-linking, thereby increasing matrix stiffness.70,71 Concurrently, YAP/TAZ regulate matrix metalloproteinases (MMP2, MMP9, MMP14) that degrade existing matrix and enable deposition of newly synthesized, stiffer matrix components. This active matrix remodeling perpetuates the mechanical conditions that favor YAP/TAZ activation, effectively creating a self-reinforcing mechanical niche. 72 In vascular and pulmonary systems, this constitutes a forward feedback loop where ECM stiffening activates YAP/TAZ, which then drive transcription programs promoting further ECM deposition and crosslinking. 48 These three feedback layers are tightly interconnected. Enhanced mechanosensing drives stronger contractility, which promotes matrix remodeling; remodeled matrix in turn strengthens mechanosensing and contractility. This multi-layered feedback architecture enables cells not merely to respond to their mechanical environment but to actively construct and maintain their mechanical niche, with profound implications for development, homeostasis, and disease progression.
Tissue-specific mechanobiology of YAP/TAZ signaling
While YAP/TAZ function as universal mechanotransducers across diverse tissues, their regulation and transcriptional outputs exhibit remarkable tissue specificity. Understanding how identical core machinery generates tissue-appropriate responses requires examination of tissue-specific mechanosensor hierarchies, context-dependent transcriptional programs, and specialized feedback mechanisms that reinforce each tissue’s mechanical identity (Figure 3(b)).
Diversity of tissue-specific mechanosensor networks
Each tissue’s unique mechanical environment establishes a distinct hierarchy among upstream mechanosensors, reflecting the predominant mechanical challenges faced by that tissue. 48 Consequently, even broadly expressed mechanosensors such as Merlin, AMOT proteins, and integrin–focal adhesion complexes shape mechanotransduction in a context-dependent manner, with their relative influence differing from tissue to tissue.
Epithelial tissues: Contact-dominant mechanoregulation
Epithelial tissues prioritize cell–cell contact sensing and polarity maintenance as primary mechanical inputs. Merlin serves as the predominant mechanosensor in this context, detecting changes in the circumferential actin belt underlying adherens junctions. 73 As cell density increases and junctions mature, Merlin activates the MST1/2–LATS1/2 cascade, promoting YAP/TAZ phosphorylation and cytoplasmic retention. The AMOT family provides complementary regulation at tight junctions, directly sequestering YAP/TAZ while simultaneously enhancing LATS1/2 activity 53 (Figure 3(b)).
Cardiovascular tissues: Flow-responsive hippo signaling
The cardiovascular system confronts continuous hemodynamic forces, with shear stress from blood flow and pulsatile pressure as dominant mechanical inputs. AMOT family proteins and the Thrombospondin-1–RAP2 axis function as primary mechanosensors under these conditions. 74 Endothelial cells discriminate between laminar and oscillatory flow patterns; laminar flow suppresses YAP/TAZ to maintain quiescence and inhibit inflammation, whereas oscillatory flow activates YAP/TAZ to promote proliferation and inflammatory responses. Thrombospondin-1, through interactions with integrin αvβ1, modulates RAP2 GTPase activity in response to mechanical forces, thereby controlling YAP nuclear translocation and vascular remodeling programs 74 (Figure 3(b)).
Musculoskeletal tissues: Stiffness-dependent YAP/TAZ control
Bone and other musculoskeletal tissues operate in extremely stiff mechanical environments (>40 kPa) where substrate rigidity and mechanical loading dominate. The integrin–focal adhesion complex assumes primacy as the mechanosensor in these contexts. 75 High matrix stiffness promotes integrin clustering and focal adhesion maturation, activating FAK and establishing stress fibers that transmit force to the nucleus. Matrix metalloproteinase-14 (MT1-MMP)–mediated ECM remodeling plays a particularly important role, with mesenchymal stem cells using MT1-MMP to reshape their local matrix, activating the integrin β1/RhoA axis and promoting YAP/TAZ nuclear accumulation to drive osteogenic differentiation 76 (Figure 3(b)).
Tissue-specific transcriptional outputs and feedback mechanisms
Following nuclear translocation, YAP/TAZ activation of distinct transcriptional programs is dictated by tissue context through differential transcriptional partnerships and tissue-specific chromatin accessibility. While canonical YAP/TAZ signaling proceeds through TEAD transcription factors, tissue-specific transcriptional partnerships can override this default program. In bone, TAZ preferentially interacts with RUNX2 rather than TEAD, forming osteogenic transcriptional complexes that activate bone-specific genes including Osterix, osteocalcin, and alkaline phosphatase. 77 Conversely, in cardiovascular tissues, TEAD1/2/4–YAP/TAZ complexes remain dominant, activating angiogenic programs and regulating vascular morphogenesis. 78 Mechanically induced YAP/TAZ activation is further filtered through each tissue’s chromatin architecture. Bone-specific super-enhancer clusters near osteogenic genes (e.g., RUNX2, Osterix) are preferentially accessible in mesenchymal cells, enabling efficient activation by YAP/TAZ–RUNX2 complexes. In endothelial cells, angiogenic enhancer regions remain constitutively accessible, allowing rapid transcriptional responses to hemodynamic changes. Epithelial tissues maintain accessible chromatin at loci governing cell–cell adhesion and barrier function. This chromatin-level regulation ensures that YAP/TAZ direct transcriptional resources toward tissue-appropriate gene programs. These transcriptional outputs establish specialized, self-reinforcing feedback systems that perpetuate each tissue’s mechanical identity. In epithelial tissues, Merlin-mediated contact sensing activates homeostatic programs that strengthen cell–cell junctions through increased E-cadherin, claudin, and occludin expression.79,80 Enhanced junction proteins and polarity complexes stabilize adherens and tight junctions, reinforcing contact inhibition and maintaining barrier integrity.
In cardiovascular tissues, flow-sensing through AMOT and Thrombospondin-1–RAP2 activates angiogenic programs. Upregulation of VEGF, angiopoietins, and eNOS drives vascular remodeling, while increased AMOT expression and Thrombospondin-1 signaling enhance flow-sensing fidelity. 74 This flow-responsive feedback network enables dynamic adaptation to hemodynamic changes.
In bone, the integrin–focal adhesion mechanosensor system activates RUNX2-mediated osteogenic programs These programs upregulate collagen I, fibronectin, LOX, and LOXL2, increasing matrix stiffness through enhanced cross-linking. 81 Simultaneously, increased expression of integrin β1/α5 and focal adhesion proteins such as talin, vinculin, and paxillin augments stiffness-sensing capacity. 82 This stiffness-amplifying feedback loop enables bone to continuously adapt to mechanical loading.
These specialized feedback mechanisms illustrate how tissue-specific mechanosensor hierarchies and transcriptional programs integrate to establish self-reinforcing mechanical identities optimized for each tissue’s physiological requirements—barrier integrity in epithelia, hemodynamic responsiveness in vasculature, and structural adaptation in bone.
Mechanobiological dysregulation of hippo signaling in disease
A defining feature of Hippo–YAP/TAZ signaling in disease is its strong context dependence. Depending on tissue type, mechanical environment, and cellular state, YAP/TAZ activity can exert either protective or pathological effects. Transient YAP/TAZ activation under physiological mechanical conditions contributes to tissue repair, regeneration, and adaptive remodeling, whereas sustained activation in mechanically altered or chronically stressed environments promotes malignant transformation, fibrosis, and inflammatory pathology. 7 This duality highlights the importance of interpreting YAP/TAZ function within specific mechanobiological contexts rather than as a uniformly pathogenic signaling pathway.83,84 In the following sections, we examine how mechanobiological dysregulation of Hippo–YAP/TAZ signaling manifests across distinct disease settings, with particular emphasis on how tissue-specific mechanical environments shape pathological outcomes.
Mechanobiological control of malignant cell behaviors through hippo–YAP/TAZ signaling
Cancer metastasis refers to the complex process by which cancer cells detach from the primary tumor and spread to other organs. This process is largely driven by the ability of cells to alter their shape and migrate, and it is influenced by mechanical signals from both the cells themselves and the surrounding microenvironment. These mechanical signals play a crucial role in tumor progression and malignancy. 7 In the early stages, tumor cells undergo genetic transformations and biochemical communications, leading to changes in their migratory and invasive behaviors.85,86
The role of hippo signaling in cancer cell migration
Metastasis, the leading cause of cancer mortality, requires tumor cells to undergo EMT, invade surrounding tissues, intravasate into circulation, and evade immune surveillance.
87
The Hippo signaling pathway, beyond its canonical function in organ size control, has emerged as a key regulator of cancer cell dissemination. Central to this pathway are the transcriptional co-activators YAP and TAZ.
88
Their activation, or dysregulation of upstream kinases MST1/2 and LATS1/2, promotes EMT by inducing transcription factors such as Snail, Slug, ZEB1, and Twist, while repressing epithelial markers like E-cadherin89–92 (Figure 4(a)). Notably, knockdown of YAP/TAZ reverses mesenchymal morphology, restoring epithelial characteristics.92,93 YAP can also substitute for KRAS in colon cancer, promoting survival and EMT through cooperation with FOS and suppression of junctional proteins.
91
Mechanobiology is deeply intertwined with Hippo signaling. YAP/TAZ are mechanosensors that respond to ECM stiffness and fluid shear stress. For example, YAP is activated by fluid flow in microfluidic models, enhancing cancer cell motility.
94
Mechanistically, YAP transcriptionally upregulates ARHGAP29, an inhibitor of RhoA, leading to increased actin turnover and cytoskeletal flexibility—key for cell migration and invasion
95
(Figure 4(a)). This mechanism is especially relevant for circulating tumor cells and metastatic colonization.96,97 Collective migration models, such as Drosophila border cell migration, further illustrate Hippo pathway control of actin dynamics. Here, core kinases polarize F-actin to form protrusions, while Warts kinase inhibits actin assembly at cell-cell contacts.
98
The small GTPase Rap1 interacts with Hippo components, suppressing pathway activity and promoting persistent migration, while its homolog Rap2 activates LATS1/2 in response to low stiffness, highlighting complex context-dependent regulation.41,99,100 Mechanobiological dysregulation of Hippo signaling in disease pathologies. The schematic illustrates how aberrant mechanical cues disrupt Hippo signaling and activate YAP/TAZ, driving the progression of various diseases. (a) Stiffened extracellular matrix (ECM) within the tumor microenvironment promotes integrin-mediated focal adhesion assembly and actomyosin tension. This mechanical stress overrides Hippo pathway inhibition, facilitating YAP/TAZ nuclear translocation to drive tumor proliferation, metastasis, and chemoresistance. (b) In lung fibrosis, alveolar injury leads to pathological tissue stiffening, which triggers the fibroblast-to-myofibroblast transition (FMT). Nuclear YAP/TAZ induces the expression of profibrotic genes and ECM components, establishing a positive feedback loop that perpetuates tissue hardening. (c) Hemodynamic forces regulate endothelial cell (EC) homeostasis. While laminar flow suppresses YAP/TAZ, disturbed flow at arterial branches increases cytoskeletal tension and activates YAP/TAZ, leading to endothelial inflammation and atherosclerosis. (d) In neurological disorders, physical changes such as brain tissue stiffening or traumatic injury modulate glial activation. Dysregulated YAP/TAZ activity contributes to neuroinflammation and neurodegeneration, as observed in conditions like Alzheimer’s disease.
In summary, the Hippo pathway integrates biochemical and mechanical cues to regulate EMT, cytoskeletal remodeling, and cancer cell migration. These insights underscore the therapeutic potential of targeting Hippo pathway components and their mechanobiological interactions to inhibit metastasis.
Tumor microenvironment and mechanical forces
Tumor progression is accompanied by profound biochemical and mechanical changes within the tumor microenvironment (TME). Notably, the stiffness of the ECM and the mechanical properties of tumor and stromal cells are dynamically altered during tumor growth, directly impacting cancer cell migration, invasion, and metastatic potential.101–103 These mechanical changes are not merely passive consequences of tumor expansion but actively contribute to tumor progression by promoting invasive phenotypes and resistance to therapy. During tissue morphogenesis and tumor development, cells constantly sense and respond to mechanical stress from neighboring cells, the ECM, and shear forces encountered during migration. Mechanical cues—such as tension generated by tissue geometry and varying matrix stiffness—are transmitted through membrane receptors, the actin cytoskeleton, and the nuclear envelope, ultimately influencing gene expression, cell cycle progression, and cell fate decisions.104,105 These mechanotransduction pathways are central to shaping tissue morphology and tumor cell behavior. Recent research has elucidated how the Hippo signaling pathway integrates mechanical signals within the TME. Hippo pathway components localize at cellular junctions and interact with the actin cytoskeleton, acting as sensors and transducers of mechanical cues.33,106,107 For instance, disruption of actin stress fibers activates MST1/2, while increased F-actin polymerization—such as that caused by mutations in capping proteins—leads to upregulation of YAP/TAZ target genes and tissue overgrowth.33,106,108 Furthermore, mechanical stress or changes in cell morphology can promote YAP nuclear localization and activity in a LATS-dependent manner.26,51,109 Upstream regulators such as Merlin and Expanded coordinate the transmission of mechanical signals from the membrane to the Hippo core kinases, further linking cell morphology and cytoskeletal dynamics to Hippo pathway activity. 36 Rho GTPase activity, which is essential for the formation of contractile actin networks in response to a stiff matrix, also modulates YAP/TAZ activity.26,104,105 AMOT acts as a molecular bridge, with LATS-mediated phosphorylation switching AMOT’s binding preference from F-actin to YAP, thereby sequestering YAP in the cytoplasm and inhibiting its transcriptional activity. 26 Clinically, tumor stiffness can be measured using in vivo MRI elastography or ex vivo atomic force microscopy, and these mechanical properties have been correlated with disease progression and metastatic risk.102,103 As tumors grow, compressive forces generated by proliferation further perturb the ECM and blood flow, creating a microenvironment conducive to tumor invasion and metastasis. 103
In summary, the dynamic interplay between mechanical forces in the TME and the Hippo signaling pathway is critical for regulating cancer cell migration, invasion, and metastatic dissemination. Understanding these mechanotransduction mechanisms offers promising avenues for therapeutic intervention targeting the physical and signaling landscape of tumors.
Hippo pathway and contact inhibition in cancer
Cancer cells gradually lose the ability to respond to cell-cell contact inhibition, a process that is tightly regulated by mechanical signals such as ECM stiffness and cell shape. The Hippo pathway is a critical regulator of contact inhibition in cancer cells. High cell density and strong cell-cell adhesion activate the Hippo pathway, leading to phosphorylation and cytoplasmic retention of YAP/TAZ, which causes cell cycle arrest and prevents excessive cell proliferation. In contrast, in cancer, this response is often disrupted, resulting in YAP/TAZ activation, leading to over-proliferation and migration. 110 Mechanistically, Spectrin, a cytoskeletal protein, plays a critical role in restraining YAP/TAZ activity in response to cell-cell contact inhibition. Loss of Spectrin proteins leads to hyperactivation of YAP/TAZ, which facilitates excessive cell proliferation and migration, even under conditions of high cell confluence 48 (Figure 4(a)).
Mechanobiological remodeling and fibrotic activation of hippo pathway in organ fibrosis
Lung fibrosis
Myofibroblasts are responsible for fibrogenesis and produce a series of ECM components such as collagens, laminins, and fibronectins to promote their contractile force. 111 Recently, the Hippo pathway was shown to contribute to the pathogenesis of fibrosis, in which hyperactive YAP/TAZ accumulate in both the epithelial and stromal tissue compartments of fibrotic tissues. 51 The basic unit of the lungs is the pulmonary alveoli, which are comprised of epithelial and ECM layers surrounded by capillaries. 112 During lung development and repair, respiratory epithelial and mesenchymal progenitors undergo significant cellular behavior changes to maintain appropriate cell type composition and structural organization through re-epithelialization. 113 Alveolar epithelial cells serve as the main source of fibroblasts and myofibroblasts, which convert to a mesenchymal cell phenotype following injury, potentially triggering fibrosis via EMT.114,115 The Hippo pathway regulates epithelial progenitor cell proliferation, migration, and differentiation in both developing and mature lungs. Cell detachment activates the Hippo pathway via cytoskeleton reorganization to induce anoikis. TAZ functions as a co-activator of transcription factors by binding the WW domain to the (L/P)PXY motif of these factors. 77 During fetal lung development, TAZ and thyroid transcription factor 1 (TTF-1) co-express in respiratory epithelial cells, where TAZ directly interacts with the (L/P)PXY motif of TTF-1 to activate transcription of target genes, including surfactant protein C (SP-C), which maintains lung morphogenesis. 116 TAZ knockout mice exhibit abnormal lung alveolarization, and microarray analysis of wild-type and TAZ-deficient mouse lungs has identified CTGF as a direct TAZ-TEAD target gene crucial for peripheral epithelial cell differentiation and lung development. 117 CTGF is also implicated in EMT, lung fibrosis, and lung development, linking TAZ to fibrosis.118,119
Moreover, the matrix mechanical environment influences the subcellular localization of YAP/TAZ in lung fibroblasts. 120 An active YAP/TAZ feed-forward mechanism amplifies a profibrotic response, inducing fibrosis in the lungs of mice injected with YAP5SA/TAZ4SA-overexpressing fibroblasts. 118 Similarly, YAP/TAZ are highly expressed in the fibroblastic foci of idiopathic pulmonary fibrosis (IPF) patients, with nuclear accumulation indicating their role in fibrosis. 121 Culturing lung fibroblasts on a stiff matrix induces YAP/TAZ nuclear translocation, leading to increased proliferation, contraction, and ECM production. 120 Silencing YAP/TAZ via small interfering RNAs (siRNAs) reduces fibroblast responses induced by a stiff matrix, highlighting the necessity of YAP/TAZ activation for myofibroblast phenotypic features 118 (Figure 4(b)). Further evidence from murine models of pulmonary fibrosis supports the link between YAP/TAZ and fibrosis. Deletion of a single copy of Taz (WWTR1) attenuates bleomycin-induced pulmonary fibrosis, reducing collagen deposition and lung elastance. 116 Additionally, microRNA (miRNA) involvement in IPF progression has been identified; for example, miR-15a blocks YAP1/Twist-induced fibroblast activation and fibrogenesis. 122 IPF is characterized by fibroblast proliferation, ECM remodeling, and increased tissue stiffness, which leads to irreversible lung architecture distortion. 123 Single-cell RNA sequencing of normal and IPF lung epithelial cells has revealed abnormal activation of multiple pathways, including TGF-β, Hippo, PI3K/AKT, p53, and Wnt signaling cascades. 124 Increased nuclear YAP localization in IPF lung epithelial cells suggests that YAP cooperates with mTOR-PI3K-AKT signaling to drive aberrant cell proliferation and migration while inhibiting epithelial differentiation. 121 Thus, YAP/TAZ activity contributes to IPF pathogenesis in both lung fibroblasts and epithelial cells (Figure 4(b)).
Cardiac fibrosis
Cardiac fibrosis is a critical contributor to heart failure (HF), occurring in various cardiac diseases, including ischemia, myocardial infarction (MI), and hypertrophy. 125 Hippo signaling components play significant roles in cardiac fibrosis development and progression. 126 While YAP/TAZ activation promotes cardiac fibrosis, their suppression mitigates angiotensin II (AngII)- or MI-induced fibrosis. 127 Cardiac fibroblasts are major sources of pathological ECM synthesis during cardiac remodeling. 128 Elevated YAP and downregulated LATS1, an upstream Hippo kinase, are observed in the left ventricular tissue of HF patients, correlating with fibroblast proliferation. 129 Additionally, preclinical MI models show YAP and TAZ activation in resident cardiac fibroblasts. 126 YAP/TAZ directly induce fibroblast differentiation into pathological myofibroblasts. 129 Fibroblast-specific deletion of Yap/Taz F/F ;Col1a2 Cre(ER)T mice or Yap F/F ;Tcf21 MCM mice reduce fibrosis and inflammation post-MI.130,131 Loss of YAP also attenuates myocardial fibrosis and cardiac dysfunction in response to chronic neuroendocrine stimulation by AngII. 126 In vitro, YAP/TAZ inhibition via siRNA or verteporfin abrogates TGF-β1-induced fibroblast-to-myofibroblast transition and ECM production. 132 Conversely, YAP overexpression promotes myocardial inflammation, fibrosis, and hypertrophy. 126 Mechanistically, RhoA regulates AngII-induced YAP activation, which mediates fibroblast transition into fibrotic myofibroblasts. 129 YAP also interacts with myocardin-related transcription factor A (MRTF-A) to facilitate α-smooth muscle actin (α-SMA)-positive myofibroblast formation and profibrotic gene expression. 44 YAP/TAZ further regulate interleukin-33 (IL-33) to promote cardiac myofibroblast formation. 127 Cardiac ECM homeostasis is disrupted post-MI, leading to pathological expansion of the interstitium, a process known as cardiac fibrosis. 125 The persistence of myofibroblasts in the infarcted heart results from a feed-forward loop in which increased ECM stiffness perpetuates fibroblast activation. 129 Understanding the complex interplay between mechanical signaling and YAP/TAZ activation is essential for developing therapeutic strategies for cardiac fibrosis and heart failure.
Hemodynamic and mechanical control of hippo–YAP/TAZ in cardiovascular pathology
The Hippo-YAP/TAZ axis serves as a central mechanotransduction pathway in the heart, converting biomechanical signals such as ECM stiffness, shear stress, and cytoskeletal tension into gene regulatory programs that drive disease when dysregulated.12,133
Cardiac hypertrophy and heart failure
Cardiac hypertrophy develops in response to chronic mechanical overload, where Hippo-YAP/TAZ signaling determines the balance between adaptive and maladaptive remodeling.134,135 Sustained mechanical stress leads to persistent YAP/TAZ activation in cardiomyocytes, increasing glycolytic flux and hypertrophic gene expression; excess activity, however, drives pathological fibrosis, apoptosis, and heart failure.134,136 Loss of YAP function impairs adaptive hypertrophy and increases myocardial fibrosis and cell death, showing that precise control over YAP activity is crucial for healthy cardiac adaptation 134
Myocardial infarction and ischemia-reperfusion injury
After myocardial infarction or ischemia-reperfusion (I/R) injury, Hippo-YAP/TAZ pathway activity is upregulated in responding cardiomyocytes and fibroblasts.135,137
YAP activation promotes cardiomyocyte survival and proliferation, suppresses apoptosis, and coordinates tissue repair; however, sustained Hippo inhibition may risk uncontrolled proliferation or impaired functional restoration. 126 Pharmacological manipulation of YAP/TAZ—using gene therapy, small molecules, or regulating upstream mechanics—shows promise to improve heart function after MI.126,137
Pulmonary arterial hypertension (PAH) and vascular remodeling
PAH pathogenesis involves chronic pulmonary vascular remodeling and hyperproliferation of vascular smooth muscle cells (VSMCs) and fibroblasts, primarily driven by ECM stiffening and abnormal mechanotransduction. 138 YAP/TAZ activation in these cells initiates gene programs that foster further ECM deposition and cell growth, forming a cycle of progressive vessel occlusion and stiffness. 135 Therapies targeting ECM crosslinking, YAP/TAZ activity, or upstream mechanics can help break the feedback loop and slow disease progression.135,139
Atherosclerosis and in-stent restenosis
Disrupted hemodynamics and matrix stiffening trigger chronic YAP/TAZ signaling in endothelial cells, promoting inflammation, lipid accumulation, and vascular plaque formation. 140 YAP/TAZ further induce phenotypic switches in VSMCs, supporting proliferation and migration seen in both atherosclerosis and restenosis following stent placement. 141 Targeting YAP and its mechanical signaling partners like integrin-FAK remains a promising strategy to limit pathological vascular remodeling and vessel re-narrowing 142 (Figure 4(c)).
Mechanical and metabolic modulation of hippo signaling in neurological disorders
Mechanical microenvironment and microglial sensing
Microglia reside as the primary immune effector cells in the central nervous system and are finely attuned to changes in mechanical properties, such as ECM composition and tissue stiffness, which shape their activation state.143,144 The Hippo signaling pathway critically handles these mechanosensory inputs, integrating cues from cell-cell contact, cytoskeletal tension, and metabolic state to modulate the activation and nuclear localization of its key effectors YAP and TAZ.8,145 Loss of the ECM protein Kindlin-3 in microglia impairs membrane tension, directly affecting nuclear YAP activity and mechanosensory responsiveness. 146 Indeed, increasing evidence shows YAP operates as a central tension sensor, influencing microglial reactivity thresholds in response to mechanical stimuli144,145
Hippo-YAP pathway in neuroinflammation and injury
Upon CNS injury or ischemia, upstream factors including Src, c-Abl, and Daxx activate MST1, a core Hippo pathway kinase, with downstream modulation of NF-κB signaling and enhanced inflammatory cytokine production in microglia. 147 Suppression or genetic deletion of MST1 is neuroprotective, reducing ischemia/reperfusion-induced inflammation and cell death. 147 Physical signals, such as increased tissue stiffness post-ischemia, are sensed by microglial mechanotransduction machinery, triggering Hippo pathway responses and shaping neuroinflammatory outcomes. 148 Although direct evidence in microglia remains limited, recent studies point to integrin-mediated Src regulation of YAP as a mechanosensitive axis affecting glial activation and survival 147 (Figure 4(d)).
Hippo pathway alterations in neurodegenerative disease
Global proteomic profiling of Alzheimer’s disease (AD) brains reveals decreased expression of Hippo pathway components, implicating this pathway in disease progression. 145 Microglial YAP expression is inversely related to proinflammatory activation: reductions in YAP levels, triggered by amyloid-beta (Aβ), enhance inflammatory cytokine secretion, while activation or overexpression of YAP reverses these effects. 149 YAP downregulation arises early in AD, found in experimental models and human tissue long before substantial amyloid or tau pathology manifests. 150 Functionally, YAP deficiency leads to increased Aβ1-42 accumulation, tau phosphorylation, and upregulation of amyloidogenic proteins including BACE1 and PSEN1/2, highlighting YAP’s protective role against neurodegeneration. 149 Furthermore, intracellular Aβ aggregates sequester YAP, disrupting its nuclear translocation and promoting neuronal necrosis, exemplifying the interplay between abnormal mechanical cues and Hippo pathway dysregulation 150 (Figure 4(d)).
Intercellular crosstalk and metabolic integration
Astrocytes display high levels of YAP, and targeted ablation of YAP in astrocytes results in microglial activation and blood–brain barrier disruption, likely via cytokine and chemokine signaling. 151 Microglial activation is also finely regulated by metabolic states, with AMPK serving as a master switch: energy stress and metabolic cues prompt AMPK to phosphorylate and inhibit YAP, thereby integrating metabolic and mechanical signals through the Hippo pathway.152,153 The LKB1–AMPK–E-cadherin complex underscores how cell-cell contact, cytoskeletal tension, and metabolic status converge to coordinate Hippo signaling output in glia.153,154 Taken together, the mechanobiology of the Hippo pathway offers a compelling framework for understanding how physical, metabolic, and inflammatory cues integrate to drive microglial and astrocyte responses in neurodegenerative disease.8,143 Altered ECM stiffness, increased cytoskeletal tension, and energy stress directly affect MST1/YAP activity, contributing to homeostasis breakdown, chronic neuroinflammation, protein aggregate pathology, cell loss, and CNS barrier dysfunction.8,155
Current advances in mechanobiological regulation of the hippo pathway
Condensation hierarchy in hippo signaling and mechanical coupling
Over the past few years, the Hippo signaling pathway—traditionally viewed as a kinase-driven cascade—has been redefined through the lens of biomolecular condensation. 156 Increasing evidence demonstrates that multiple Hippo components, from MST1/2–SAV1 at the apex to YAP/TAZ–TEAD at the transcriptional output, can undergo liquid–liquid phase separation (LLPS) to assemble transient, high-concentration microdomains that organize signaling reactions. 156 This emerging view bridges mechanobiology and phase separation, suggesting that the Hippo pathway functions as a mechanosensitive signaling network organized by biomolecular condensation. Such condensate-driven organization provides a plausible solution to several long-standing questions—how cells locally sense mechanical tension, convert it into biochemical activation, and preserve mechanical “memory” in transcriptional programs. Thus, the study of condensation within the Hippo pathway is not merely a structural curiosity but a mechanical–biochemical interface that may unlock previously unresolved aspects of tissue growth control and mechanotransduction.
Merlin, AMOT/KIBRA, and SLMAP: Multiphase coordination of hippo activation and inhibition
Upstream scaffolds including Merlin, AMOT, and KIBRA orchestrate Hippo activation by forming LLPS-driven condensates, while SLMAP forms counteracting inactivating assemblies. AMOT/KIBRA condensates emerge upon cell–cell contact or osmotic stress (e.g., NaCl, sorbitol) and concentrate MST and LATS kinases to stimulate the pathway, whereas SLMAP–STRIPAK condensates sequester these kinases and dampen Hippo signaling.
157
Such “multiphase coordination” likely reflects the spatial segregation between apical AMOT/KIBRA condensates and cytoplasmic SLMAP–STRIPAK assemblies, allowing reciprocal control of Hippo activation and inhibition. Although osmotic challenges rather than direct stretch were used to trigger condensation, the recent demonstration that mechanical stress elevates intracellular sorbitol, stabilizing LLPS droplets,
158
implies that these osmotic perturbations mimic a mechanically derived osmolyte response. Thus, Hippo regulatory condensates likely represent mechano-metabolic modules that integrate membrane tension with osmotic adaptation (Figure 5(a)). The function of the Hippo signaling condensates in mechanobiology. (a) In response to mechanical or osmotic stress, the scaffolding proteins spatially concentrate MST1/2 and LATS1/2, forming multiphase coalescence and facilitating their kinase activation. (b) Hippo signalosome operate as a context-dependent mechanochemical switch that integrates mechanical and metabolical cues. (c) DDR1 condensates sequester LATS1/2, physically isolating them from MST1/2 kinase and their coactivator MOB1 on stiff matrix. (d) Mechanical inputs are converted into biological gene responses through Hippo signaling condensates. YAP/TAZ-TEAD condensates stabilize gene expression programs with other transcription molecules. This condensate-driven organization provides a unified framework for mechanosensing, signal amplification, and mechanical memory in Hippo signaling.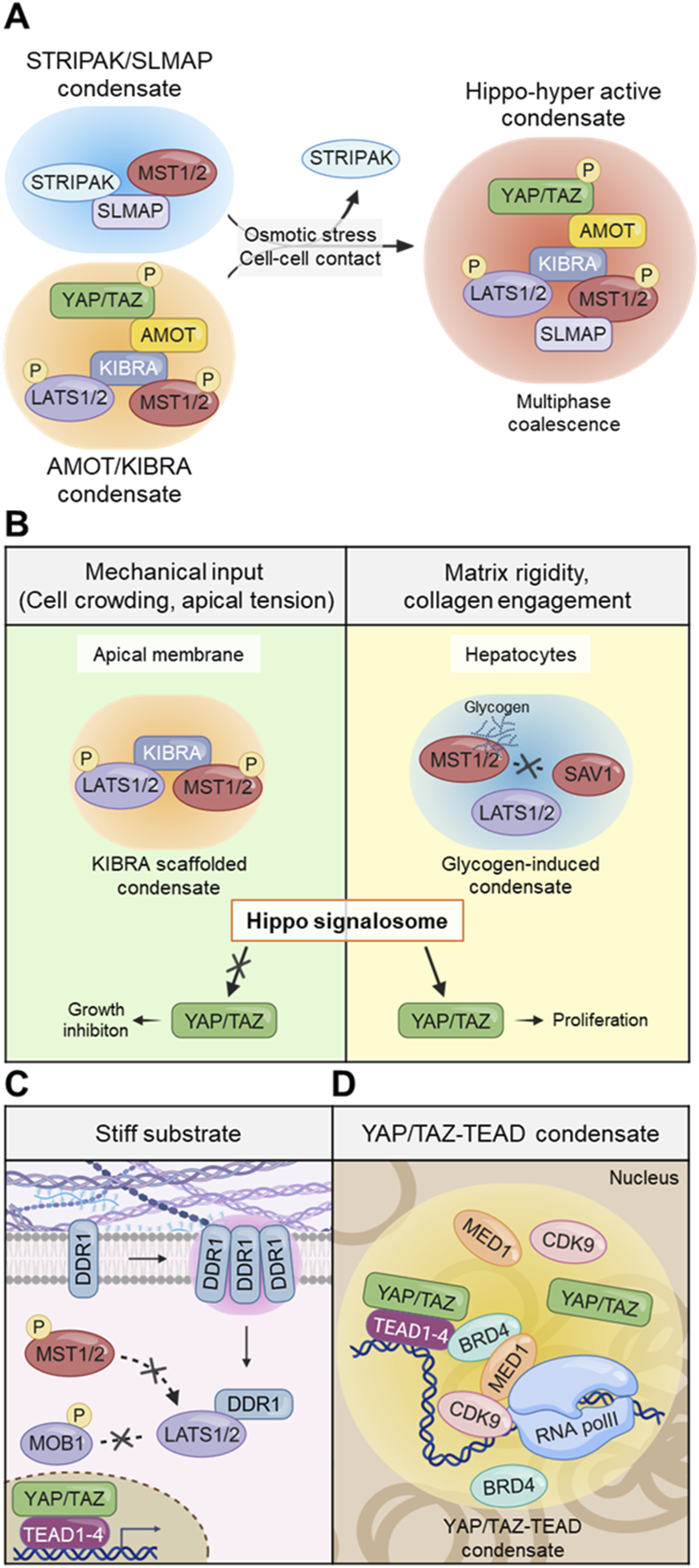
MST1/2–SAV1–LATS condensates: Mechanochemical signal integration
At the core of the Hippo cascade, the Hippo kinase (Hpo; MST1/2 in mammals) and its cofactors SAV1 and LATS/Wts organize into phase-separated condensates that integrate mechanical and metabolic cues into kinase activity. In Drosophila epithelia, Hpo itself localizes to apical puncta that coalesce in response to cell crowding and junctional tension, where it associates with Sav and phosphorylated Wts (pLATS). 84 Within these apical puncta, Kibra serves as the principal scaffold that nucleates LLPS through its intrinsically disordered region. The Kibra condensates selectively recruit pLATS, forming phase-separated “Hippo signalosomes” that spatially concentrate the kinase module and enhance Hippo signaling. This LLPS-driven compartmentalization provides a mechanical interface through which cell crowding and apical membrane tension are translated into Hippo kinase activation, thereby coupling mechanical compression to growth inhibition. In mammalian contexts, similar clustering of MST1/2–LATS complexes is observed under conditions of cytoskeletal tension or osmotic compression, although the exact scaffold components remain less defined. Conversely, during glycogen accumulation in hepatocytes, MST1/2 and LATS are sequestered into metabolically induced condensates, which suppress Hippo signaling and facilitate YAP/TAZ activation. 159 These models are supported by multiple experimental approaches, including live-cell imaging demonstrating stress- or crowding-induced clustering of Hippo kinases, perturbation experiments disrupting scaffold components such as Kibra or Sav, and biophysical analyses indicating liquid-like material properties of these assemblies.157,159,160 Collectively, these findings support the emerging view that Hippo kinase condensation functions as a context-dependent mechanochemical switch, whereby mechanical LLPS promotes pathway activation, whereas metabolic LLPS favors inhibition (Figure 5(b)).
LATS1/2–DDR1 condensates: Stiffness-responsive inhibitory hubs
Mechanical stiffening of the ECM provides one of the most direct physical cues regulating Hippo signaling. Recent work demonstrated that Discoidin Domain Receptor 1 (DDR1), a collagen-binding receptor tyrosine kinase, undergoes liquid–liquid phase separation (LLPS) in response to matrix rigidity and collagen engagement. 161 Under these conditions, DDR1 forms stiffness-induced condensates that co-sequester LATS1/2, physically isolating them from upstream MST1/2–SAV1 kinases and thereby preventing LATS activation. The trapped LATS1/2 lose access to activating phosphorylation, leading to Hippo pathway inhibition and YAP/TAZ nuclear accumulation. Mutation of the DDR1 cytoplasmic intrinsically disordered region (IDR) or culture on a compliant (soft) matrix disrupts DDR1 condensation and restores LATS phosphorylation, confirming that the material state of DDR1 assemblies acts as a mechanical off-switch for Hippo signaling. This mechanism effectively links ECM stiffness to YAP/TAZ-driven transcriptional programs associated with fibrosis and tumorigenesis. Notably, LATS condensation can have opposite outcomes depending on context. In Drosophila, phosphorylated Wts (LATS ortholog) condenses within Kibra-driven activation droplets, whereas in mammalian cells DDR1-mediated co-condensation traps and inhibits LATS 84 (Figure 5(c)).
YAP/TAZ–TEAD condensates: The biophysical output layer
At the terminus of the Hippo pathway, transcriptional activation is orchestrated by YAP and TAZ, which interact with TEAD transcription factors to form phase-separated nuclear condensates that convert mechanical and metabolic cues into gene expression. 156 These condensates represent the biophysical output layer of mechanotransduction. YAP contains a low-complexity transcriptional activation domain that drives liquid–liquid phase separation (LLPS) and coalesces with TEAD and RNA polymerase II, forming dynamic nuclear puncta that promote transcriptional activation. 31 TAZ, sharing similar modular organization, also undergoes actin-tension–dependent LLPS; its nuclear condensates compartmentalize TEAD4 and are enriched for the transcriptional elongation factor CDK9 and super-enhancer markers BRD4 and MED1, establishing a transcriptionally active microenvironment. 162 While Zhu et al. did not directly examine mechanical perturbations, TEAD condensation likely represents the transcriptional effector of upstream mechanical signaling, as YAP/TAZ nuclear translocation—known to be driven by cytoskeletal tension—promotes TEAD co-condensation and transcriptional activation. 163 Structural studies revealed that TEAD palmitoylation stabilizes its hydrophobic pocket and YAP/TAZ binding, maintaining condensate integrity. 164 Together, these findings depict a hierarchical architecture: mechanical forces transmitted through the cytoskeleton and nuclear envelope promote YAP/TAZ nuclear localization and condensate formation with TEAD, generating a spatially confined environment for transcriptional amplification. Mechanobiologically, the YAP/TAZ–TEAD condensate acts as a potential molecular memory unit, integrating prior mechanical and metabolic signals into persistent transcriptional programs controlling proliferation, migration, and differentiation. Its assembly marks the culmination of the Hippo signaling condensate cascade—from membrane-associated scaffolds and kinase signalosomes to a nuclear phase-separated transcriptional hub that defines the final mechanical output of the cell (Figure 5(d)).
Conclusion
Representative mechanical cues and their context-dependent regulation of YAP/TAZ signaling.

At the molecular scale, condensation provides the missing physical link that bridges these mechanical signals to transcriptional control. From Merlin–AMOT/KIBRA scaffolds at the membrane, to MST/LATS signalosomes in the cytoplasm, to YAP/TAZ–TEAD condensates in the nucleus, the Hippo pathway functions as a hierarchical condensate network. This organization enables force-induced molecular crowding, selective kinase activation, and persistent transcriptional “memory” of mechanical history. In this view, Hippo signaling represents a phase-organized continuum that couples mechanical perturbation with gene-regulatory feedback to maintain structural homeostasis. Therapeutically, this paradigm opens a new dimension of drug discovery—targeting not only the enzymatic activity of Hippo components but also their biophysical states. Modulating condensate assembly or TEAD palmitoylation, for instance, may reprogram YAP/TAZ activity and offer promising strategies against fibrosis, cancer, and regenerative disorders. Understanding how mechanical stress, osmotic adaptation, and metabolic flux reshape the phase behavior of Hippo molecules will further refine this framework.
Despite the therapeutic promise of targeting Hippo–YAP/TAZ signaling, several challenges remain. YAP/TAZ functions are highly pleiotropic and tissue-specific, raising concerns that global inhibition may disrupt physiological processes such as tissue repair, regeneration, and homeostasis. Moreover, prolonged or systemic modulation of Hippo signaling carries a risk of unintended toxicity due to its broad involvement in developmental and stress-response pathways. These considerations underscore the necessity of context-specific therapeutic strategies. Emerging approaches, including mechanically informed targeting, modulation of upstream mechanosensors, and tissue- or cell-type–restricted delivery systems, may enable more precise control of YAP/TAZ activity while minimizing off-target effects. Together, these strategies represent an important future direction for translating mechanobiological insights into safe and effective therapies.
In conclusion, the Hippo pathway stands as a model of mechanochemical communication, where mechanical forces are encoded, processed, and memorized through phase-separated architectures. Recognizing this pathway as a condensate-based mechanotransduction system not only deepens our understanding of tissue growth control but also establishes a conceptual foundation for future therapeutic interventions that harness the physical logic of cellular signaling.
Footnotes
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.