
Abstract
Arrhythmogenic right ventricular cardiomyopathy is an autosomal dominant genetic disease which leads to fatty replacement of the right ventricular myocardium, leading to the occurrence of ventricular arrhythmia. We present the case of a 19-year-old male who had recurrent episodes of syncope and was diagnosed to have arrhythmogenic cardiomyopathy with biventricular involvement, secondary to a heterozygous mutation in the desmoplakin gene.
Case Report
A 19-year-old young man was presented to our hospital with recurrent episodes of exertional syncope. There was no history of involuntary movements, spontaneous passage of urine or stool, angina, dyspnea, bipedal edema, or family history of sudden cardiac death.
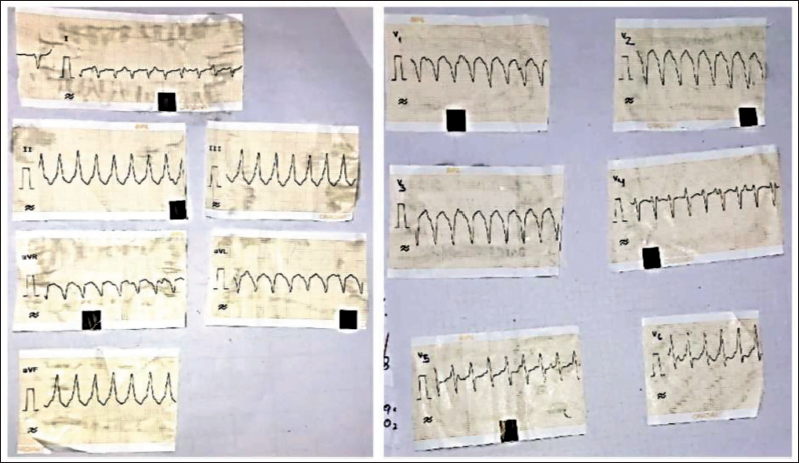
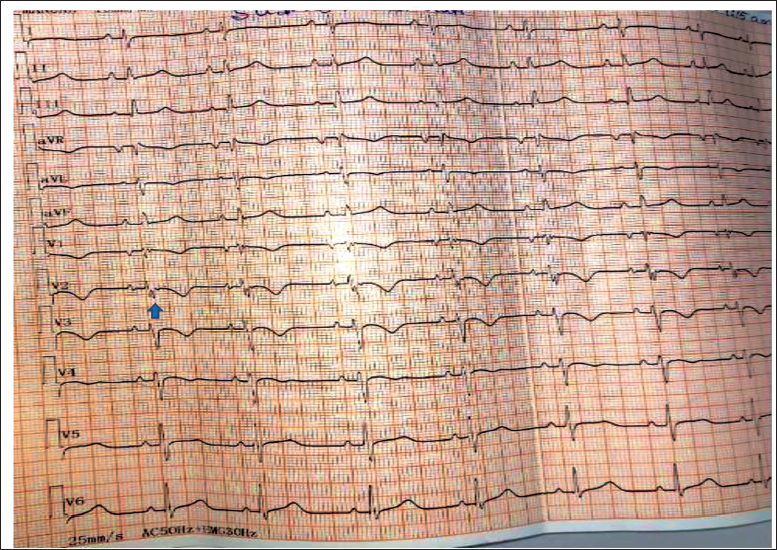
At the time of presentation, he had tachycardia with a heart rate of 160/min which was regular, and a blood pressure of 90/60 mm Hg. Twelve-lead electrocardiogram showed wide complex tachycardia with a left bundle branch block (LBBB) pattern with inferior axis (Figure 1), corresponding to ventricular tachycardia with a right ventricular outflow tract (RVOT) point of exit. He was immediately cardioverted, and the post cardioversion electrocardiogram showed sinus rhythm with low voltage complexes in limb leads, T wave inversion in leads V1-4 and R wave transition at V5, with a prominent epsilon wave (Figure 2).
Twelve-lead Electrocardiogram Showing Wide Complex Tachycardia of Left Bundle Branch Block Morphology with Inferior Axis.
Twelve-lead Electrocardiogram Showing Sinus Rhythm with Low Voltage QRS in Limb Leads, T Wave Inversion in V1-3 and a Prominent Epsilon Wave (Arrow).
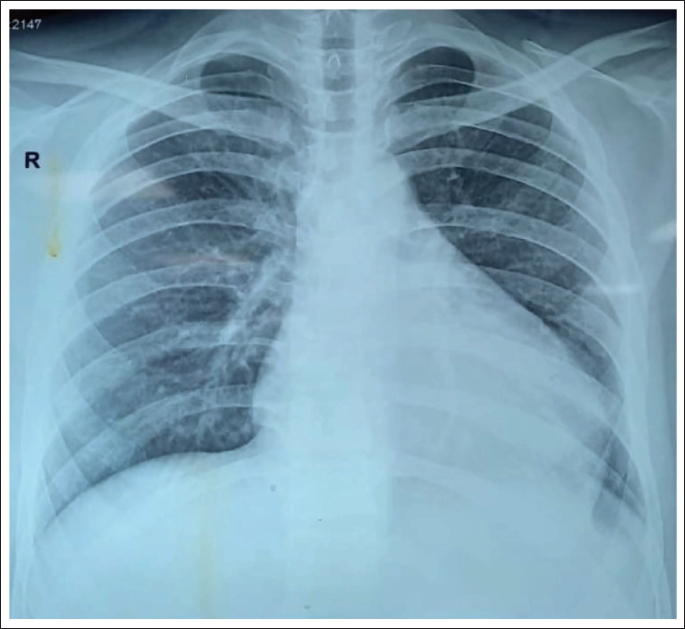
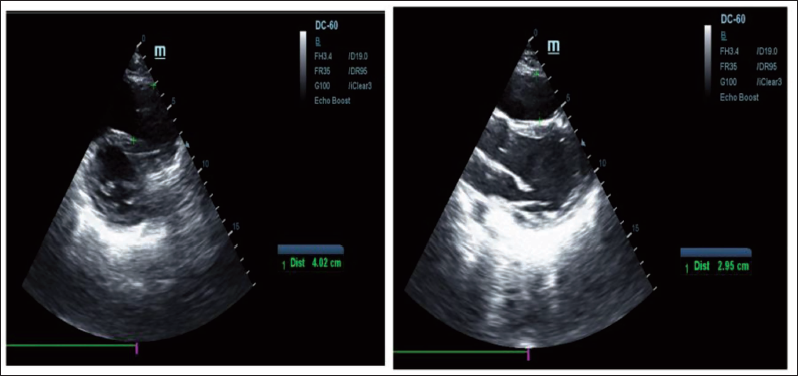
The chest X-ray showed cardiomegaly with no pulmonary arterial or venous engorgement (Figure 3). The transthoracic echocardiogram showed moderate left ventricular dysfunction and dilated RVOT (2.95 cm in the long axis view and 4.02 cm in the short axis view) (Figure 4).
Chest X-ray Showing Cardiomegaly.
Transthoracic Echocardiogram Showing Dilated Right Ventricular Outflow Tract (4.02 cm in Short Axis View [Left] and 2.95 cm in Long Axis View [Right]).
He started on oral metoprolol, amiodarone, and mexiletene, along with furosemide, spironolactone, ARNI, and dapagliflozin.
Keeping a possibility of arrhythmogenic right ventricular cardiomyopathy, he underwent a cardiac MRI which showed increased right ventricular end diastolic volume (RVEDVi – 114 mL/m2 BSA), dyskinesia of septum and inferior wall of right ventricle, with an ejection fraction of 14.7%. The left ventricle also showed global hypokinesia with ejection fraction of 34%. Patchy late Gadolinum enhancement was seen in the interventricular septum on the right ventricular free wall, and anteroseptal and inferoseptal walls of left ventricle at mid and base (Figures 5 and 6).
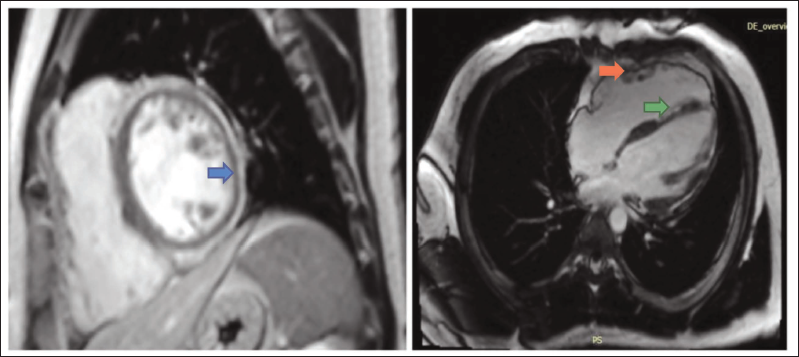
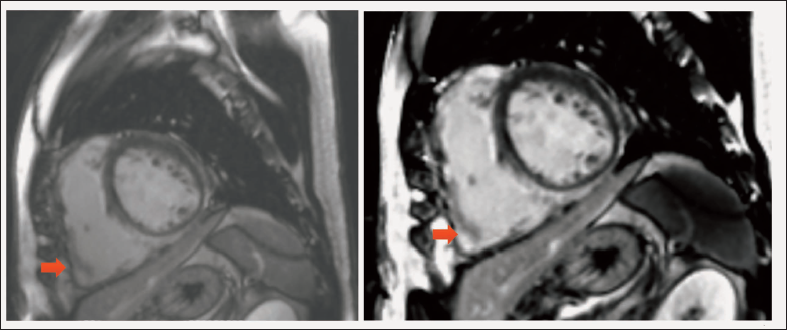
A diagnosis of arrhythmogenic cardiomyopathy with biventricular involvement was made, and he underwent a dual chamber automated intracardiac defibrillator implantation. The VT-1 zone was kept at 150/min, the VT-2 zone at 180/min, and the VF zone at 200/min.
Genetic sequencing showed a heterozygous mutation (c.5779C>T) in exon 24 of desmoplakin gene, leading to a stop codon and premature truncation of the protein at codon 1927 (p.Gln1927Ter; ENST00000379802.8). This was consistent with arrhythmogenic right ventricular cardiomyopathy (ARVC)––type 8.
Subsequently, he has been on follow-up for 12 months on oral medications (metoprolol, amiodarone, mexiletene, furosemide, spironolactone, ARNI, and dapagliflozin), with no recurrence of arrhythmia or syncope, and V-sensed rhythm.
Discussion
Arrhythmogenic right ventricular cardiomyopathy, first described by Guy Fontaine in 1977, is a genetic disease where fatty infiltration is seen in the free wall. 1 It is inherited in an autosomally dominant manner with variable penetrance and incomplete expression. 2
The prevalence of this disease is 1:2500 to 1:5000 and nearly half have mutations in the desmosomal proteins. 3 Patients may be asymptomatic or present with palpitations, fatigue, syncope, cardiac arrest, or sudden cardiac death.
Though classically described to involve the right ventricle, left ventricular involvement has recently been described in around 70% of cases. 4 Manifestations include left ventricular systolic dysfunction, T wave inversions in left precordial leads and low QRS voltage in limb leads. 5 The recently published Padua criteria provides the diagnostic criteria for left ventricular involvement in ARVC, and classifies the disease into arrhythmogenic cardiomyopathy with right ventricular involvement, left ventricular involvement or biventricular involvement. 6
Patients with left ventricular involvement have significantly lower five-year event free survival. 7 It is significantly associated with sudden cardiac death, implantable cardioverter-defibrillator placement, and aborted cardiac arrest. Significant predictors include presence of wall motion abnormality, fat infiltration, and late Gadolinum enhancement of the left ventricle. 4
Our patient had ventricular tachycardia of LBBB morphology, corresponding to a midseptal exit, and the left ventricular systolic dysfunction and low voltage QRS complexes on limb leads were tell-tale signs of left ventricular involvement. The biventricular involvement was established by cardiac MRI.
Footnotes
Acknowledgement
This case was presented in ACC Asia 2023 conference held in Manila.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Ethical Approval Statement
Not applicable.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Patient Consent
Informed written consent was obtained from the patient about publication.