
Abstract
The next generation of therapies is moving beyond the use of small molecules and proteins to using whole cells. Compared with the interactions of small-molecule drugs with biomolecules, which can largely be understood through chemistry, cell therapies act in a chemical and physical world and can actively adapt to that world, amplifying complexity but also the potential for truly breakthrough impact. Although there has been success in introducing targeting proteins into cells to achieve a therapeutic effect, for example, chimeric antigen receptor (CAR) T cells, our ability to engineer cells is generally limited to introducing proteins, but not modulating large-scale traits or structures of cellular “machines,” which play critical roles in disease. Example traits include the ability to secrete compounds, deform through tissue, adhere to surrounding cells, apply force to phagocytose targets, or move through extracellular matrix. There is an opportunity to increase the efficacy of cell therapies through the use of quantitative automation tools, to analyze, sort, and select rare cells with beneficial traits. Combined with methods of genetic or epigenetic mutagenesis to create diversity, such approaches can enable the directed cellular evolution of new therapeutically optimal populations of cells and uncover genetic underpinnings of these optimal traits.
Keywords
The Promise of Cell Therapies
In the future, engineered cell therapies 1 will be a pillar of medicine, along with molecular (drugs and proteins) and genetic (gene therapy) interventions ( Fig. 1 ). In the last few years, there has been particular success in the use of engineered immune cell-based therapies in treating cancer, 2 including recent FDA approvals of two chimeric antigen receptor (CAR) T-cell products. This class of cell therapy has shown significant clinical efficacy in treating hematological malignancies, but less success in killing solid tumors. 2 Cell therapies are also being applied to treat a variety of other pathologies, such as diabetes,3,4 neurodegenerative disease, 5 and graft versus host disease, 6 and general challenges exist in the field related to reproducible production of therapeutic products,7,8 significant costs for individualized treatments, and quality control. 9 I think it is instructive to look at the challenges overcome by the pharmaceutical industry, developing small-molecule therapeutics, as the fields of organic and analytical chemistry matured. 10 High-precision approaches to synthesize molecules, analyze their properties (e.g., nuclear magnetic resonance), and separate out closely related molecules (e.g., chromatography) were drivers of reproducible medicines. Cell therapies have similar needs for synthesis (genetic modification technologies), analysis tools, and separation approaches. However, cells cannot be as easily classified, given the significant heterogeneity even in genetically clonal cells, 11 and a lack of automated approaches to measure and select based on overall cell function. Here I focus on cell therapy challenges in oncology, specifically extending immunotherapies to solid tumors, as an example space where automation technologies that can drive evolution of cell populations with new integrative traits can play a significant role.
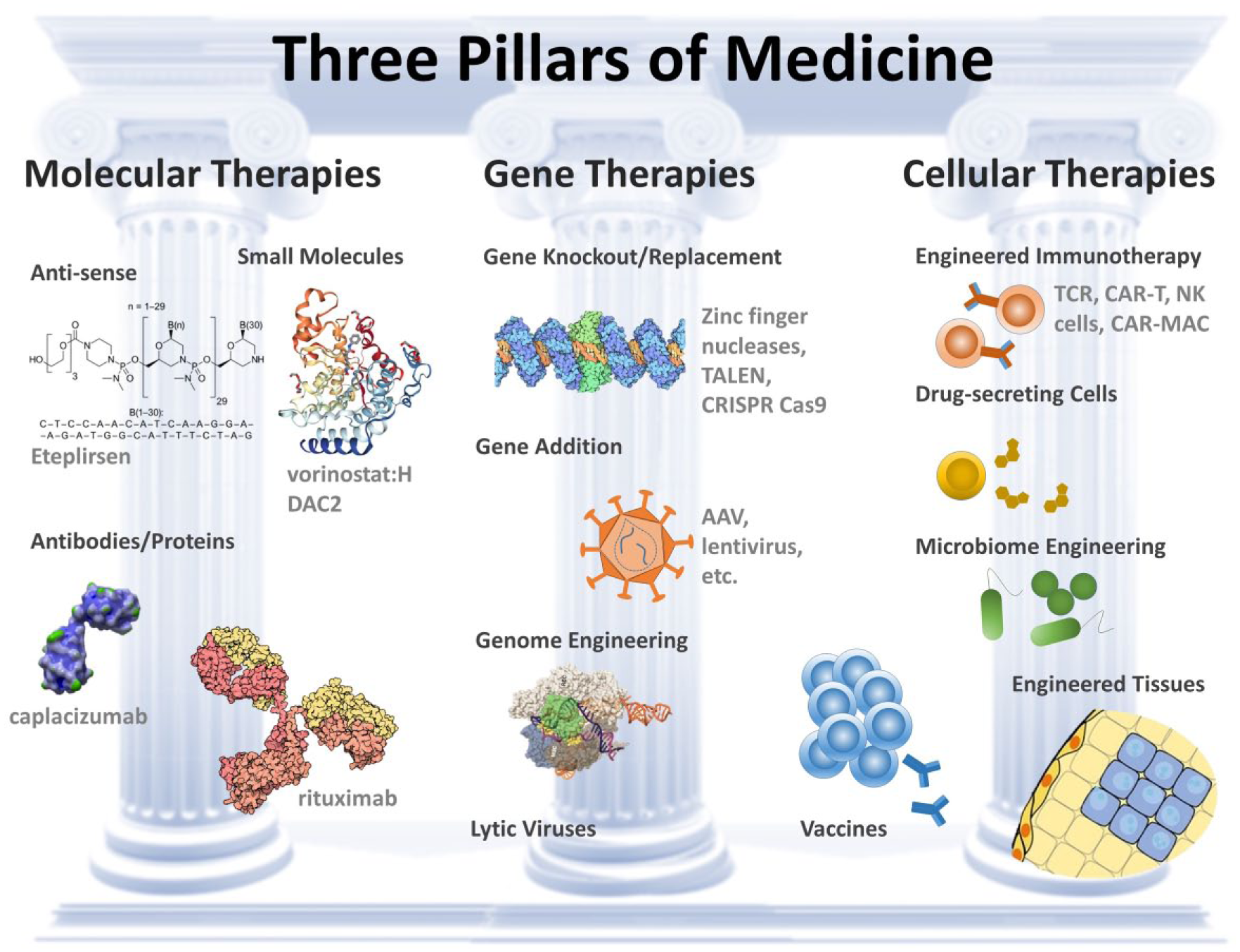
Three pillars of medicine. Molecular therapies, gene therapies, and cellular therapies allow intervening at the level of molecular interactions, generation and silencing of pathological biomolecules, and selective cell killing or long-term sensing and production of molecular therapies. Example therapies are listed in some categories. AAV = adeno-associated virus; TALEN = transcription activator-like effector nucleases; CRISPR = clustered regularly interspaced short palindromic repeats; TCR = T-cell receptor; NK = natural killer; MAC = macrophage.
Challenges in Extending Successes to Solid Tumors
Why do we care to engineer complex traits within cells? As we aim to solve critical problems limiting current therapies, such as (1) tissue penetration, (2) homing and trafficking, and (3) targeting, the physical environment in which cells operate becomes paramount. In a solid tumor, barriers to immune-mediated attack of tumor cells include modified vasculature endothelium that limits immune cell recruitment in concert with a pathological extracellular matrix (ECM) that can prevent invasion 12 as well as divert immune cells away from the tumor along ECM tracks. The confined tumor microenvironment with impaired perfusion 13 also allows malignant cells to better control the immunosuppressive soluble environment through secreted factors. 14
In expanding cancer immunotherapies to be efficacious in solid tumors, controlling complex traits of immune cells to penetrate the immunosuppressive tissue environment will be of similar importance as neo-antigen targeting. My reasoning is as follows: (1) Getting T cells into tumors improves outcomes. Regression and good prognosis are correlated with the presence of tumor-infiltrating lymphocytes. 15 In addition, CAR and engineered T-cell receptor therapy efficacy in solid tumors is cell dose dependent. A lower percentage of infused Teff cells reaching the tumor and longer expansion times lead to “senescent” phenotypes. 16 (2) Tumor vascular endothelium is a barrier for lymphocyte trafficking. Tumor endothelium downregulates traditionally recognized cell adhesion molecules leading to reduced lymphocyte trafficking compared with in neighboring vessels. 17 (3) A fibrotic capsule and dense ECM are a physical barrier to lymphocyte entry. Stroma has been shown to be a barrier for T-cell entry and cell killing in multiple studies. Proteolytic activity genes are differentially expressed in cured versus primary chemotherapy-refractory diffuse large B-cell lymphoma. 18 More directly, treatment of tumors with collagenase or heparanase enhances infiltration.19,20 Two recent papers published in Nature in 2018 further support the hypothesis that the dense ECM around tumors is a key impediment for infiltration by lymphocytes, which reduced the effectiveness of checkpoint inhibitor therapies.21,22 Even knowing these barriers exist, there is a critical gap in research worldwide in understanding how to engineer cells to overcome these barriers.
The ability to penetrate the tumor stroma is a critical issue that others have recognized and hope to address through approaches such as enzymes that can help open paths in ECM around a tumor. In fact, Caruana et al. found that engineering CAR T cells to overexpress heparanase promoted T-cell infiltration into solid tumors and antitumor activity. 20 Mariathasan et al. also showed that inhibition of transforming growth factor (TGF)-β signaling influenced peritumoral fibroblasts and enabled increased infiltration of CD8+ T cells into tumors along with increased response to checkpoint inhibitors. 22 Secretion of ECM-degrading enzymes should act synergistically with other complex traits such as augmented ability to adhere, squeeze, and migrate through peritumoral fibrotic tissue to achieve the most efficacious engineered cell therapies.
Trafficking
Importantly, freely circulating T cells must also be able to easily traffic through the bloodstream and lymphatics to local sites of tumor growth. Endothelial cells lining blood vessels in the tumor microenvironment can be modulated by surrounding tumor cells to downregulate normal “cell capture” ligands, such as intercellular adhesion molecule (ICAM)-1, and other chemokines, such as CXCL9 and 10, that collectively aid with local recruitment of T lymphocytes. 23 Recent work has also shown that the physical properties of leukocytes (i.e., deformability and size) critically affect margination, that is, the proximity of cells to blood vessel walls during flow, 24 and the subsequent ability for cells to adhere to vessel walls and extravasate. Modulation of engineered cell adhesion and mechanics may therefore play a role in enhancing trafficking and cell delivery and should be well controlled and optimized in therapeutic cells.
Targeting Tumor Cells
Once cells have the ability to adhere to the endothelium in the vicinity of a tumor, infiltration into the diseased tissue and specific activation should also be selected to enhance tumor cell killing, which may depend on a combination of various physical traits, such as deformability, contractility, and chemokine secretion. For example, two recent investigations implicated cytoskeletal dynamics and the force generated by T cells as critical for proper activation and cell killing. When engaging an antigen-presenting cell, force was observed to be critical to naïve T-cell activation, 25 and when T-effector cells recognized cognate antigen, force enhanced cell killing by perforin released by these cells. 26
Epigenetic Factors Driving Cell Therapy Efficacy
Work from Noonan et al. 27 provides orthogonal evidence that unique epigenetic traits can drive efficacy in engineered cell therapies. In the work, T cells that have a unique tropism for multiple myeloma in bone marrow, that is, marrow-infiltrating lymphocytes (MILs), were used as a base cell population to create CAR T cells and compared with genetically identical CAR T cells generated from peripheral blood. MIL-derived CAR T cells were found to have increased tumor specificity and improved patient outcomes. Therefore, a unique phenotype, which is independent of the CAR targeting moiety, selected for in the bone marrow was critical for optimal function. Interestingly, this phenotypic difference for MILs was maintained following genetic modification and suggests that in vitro selection for epigenetic enhancements that lead to unique phenotypes may be therapeutically useful in parallel to genomic changes.
Holistic Investigation of Cell Biology
Genetic or epigenetic engineering of complex traits in cells is challenging. First, it is difficult to define these emergent traits in terms of expression patterns of particular genes or a particular epigenetic state. Second, quantitative tools to measure these traits are needed at the single-cell level to be able to analyze or engineer such traits and connect to the molecular underpinnings. The current reductionist paradigm in cell biology would investigate the underlying genes and proteins that contribute to these phenotypes one gene at a time, which cannot easily inform on cases of network-level interactions that result in a complex phenotype. For example, it is clear that the nuclear-to-cytoplasmic ratio of a cell, which can control the overall deformability of a cell, 28 must depend on a range of genes, their regulated transcription, and downstream protein activity over time. There is no single gene controlling these critical phenotypes.
Evolution has the power to shape complex behaviors in cells as evidenced by the complexity of function throughout the diversity of cells in our body—from cells that produce materials (in bone, hair follicles, or cartilage) to cells that rapidly conduct electrical signals (e.g., throughout our nervous system) to cells that use fluid mechanical and adhesive differences to circulate and find areas of injury (neutrophils and other cells of our immune system). An alternative paradigm to identify the gene and protein networks underpinning complex phenotypes is to evolve cell populations with drastic changes and identify expression levels and epigenetic and genomic changes associated with these cells. This holistic approach will inevitably result in a list of changes correlated with a trait and will require statistical averaging across multiple cell lines evolved independently to yield a more complete picture of the landscape that should be engineered into a cell to recapitulate the evolutionary process. Standard reductionist tools can then be applied to refine this list further and move us closer to an era in which we can engineer such overarching traits ( Fig. 2 ).
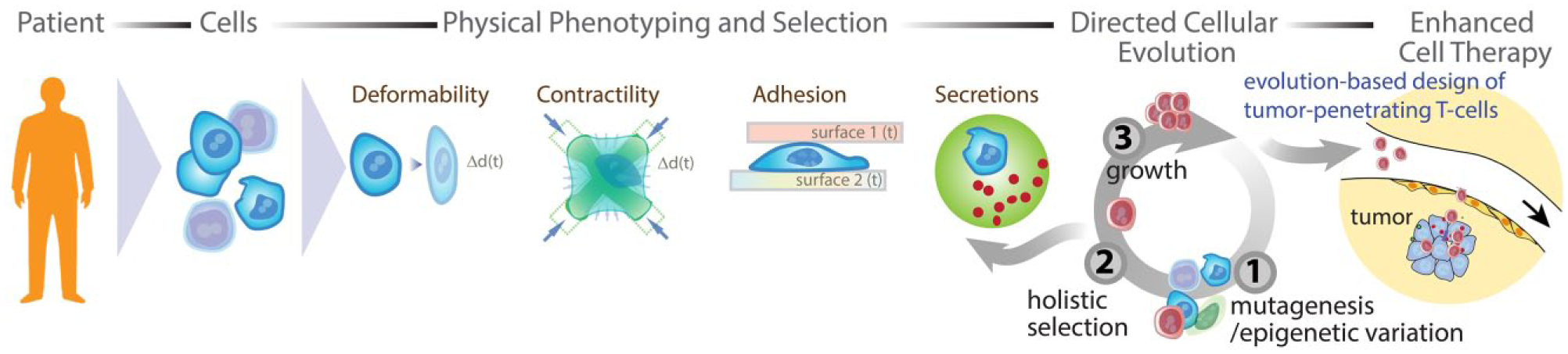
Potential workflow for the evolution of engineered cell therapies to penetrate and kill solid tumors and identify molecular underpinnings that can lead to the design of new genetic constructs for cell therapies. Cells extracted from a patient (e.g., T cells) are activated to promote growth and subjected to mutagenesis or methods of epigenetic diversity generation, selected using new automated sorting tools based on complex traits, and regrown to complete an evolutionary cycle. Final selected cells can be analyzed to identify key molecular changes to optimize therapy design. Alternatively, epigenetic “mutagenesis” can give rise to an enhanced therapeutic cell population without genetic modification that could be infused back into the patient.
Rate of Evolution
The evolution of complex traits does not require a prohibitive number of generations. Given the enormous sequence space for an individual protein, let alone the combination of proteins, nucleic acids, and other regulatory elements present within a single cell, one might expect that searching this space using a random process would take a prohibitively long time. However, the sequential nature of evolutionary cycles, assisted by “local” minima in the fitness landscape for each cycle, accelerates this process significantly. At the molecular level, only a few generations of evolution can lead to significantly improved function. For example, after only three cycles of mutagenesis and selection, subtilisin E was evolved to have >200-fold higher activity in 60% dimethylformamide solvent compared with the wild-type enzyme. 29 Similarly, directed evolution of a number of monomeric fluorescent proteins suitable for use as protein fusions was achieved in four to six cycles. 30 Even at the organism level, the evolution of complex traits occurs on practical timescales. An interesting example supporting this is the “farm-fox” experiment. Over 40 years, two populations of silver fox were bred based on selecting for behavior toward humans, one tame and one aggressive. 31 Behavioral changes were associated with physiological and biochemical changes in these two populations. Even after 10 generations, statistically significant reduction in glucocorticoid levels was observed in the tame population compared with unselected animals in response to handling and blood collection. Of course, we are also familiar with selective breeding processes that have resulted in a number of specialized dog breeds with unique shapes, sizes, fur colors/textures, and behaviors, processes that have occurred over tens of generations in recent human history. Selective breeding of crops has also been well documeted, leading to large-scale trait changes over a similar timescale.
Models of evolution also suggest that evolution-based design can proceed rapidly with large, diverse populations, along with methods to quickly quantify phenotypes and select cells with rare outlier phenotypes ( Fig. 3 ). The rate of evolution is generally reported to depend on mutation rates, population size and structure, and fixation probabilities and times for advantageous mutations, which depend largely on selective advantage. 32 For artificial selection processes, selective advantage is well defined (e.g., truncated at a cutoff value) ( Fig. 3 ), and in this case the breeder’s equation is often used to determine an expected rate of evolution, or response to selection (R) in an evolutionary cycle.
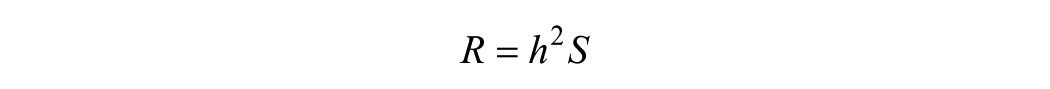
Here, h2 is the narrow-sense heritability of a trait that is selected for with a selection differential S quantifying the difference in a trait between the selected population and the starting population mean. 33 The narrow-sense heritability is a measure of the relationship between the phenotype and the heritable basis for that phenotype. For directed evolution of enzymes, we expect strong phenotype–genotype linkages (e.g., h2 → 1), and therefore accelerated rates of evolution. In the directed evolution of cells, h2 will be less than 1 as a result of nongenetic underpinnings of phenotype, such as microenvironment, cell cycle, and other epigenetic processes, but is likely bounded on the lower end by the narrow-sense heritability observed for organisms that also interact in complex ways with the environment to yield a phenotype. Any error introduced due to technical noise from automated selection instruments discussed herein also reduces h2 and slows the rate of evolution. The breeder’s equation also implies that with a fixed selection cutoff the population will asymptotically approach a mean at the selected value, and therefore a sliding selection criterion that ratchets up such that S is constant or increasing in subsequent selection cycles can maintain a consistent increase in the modulation of a trait.
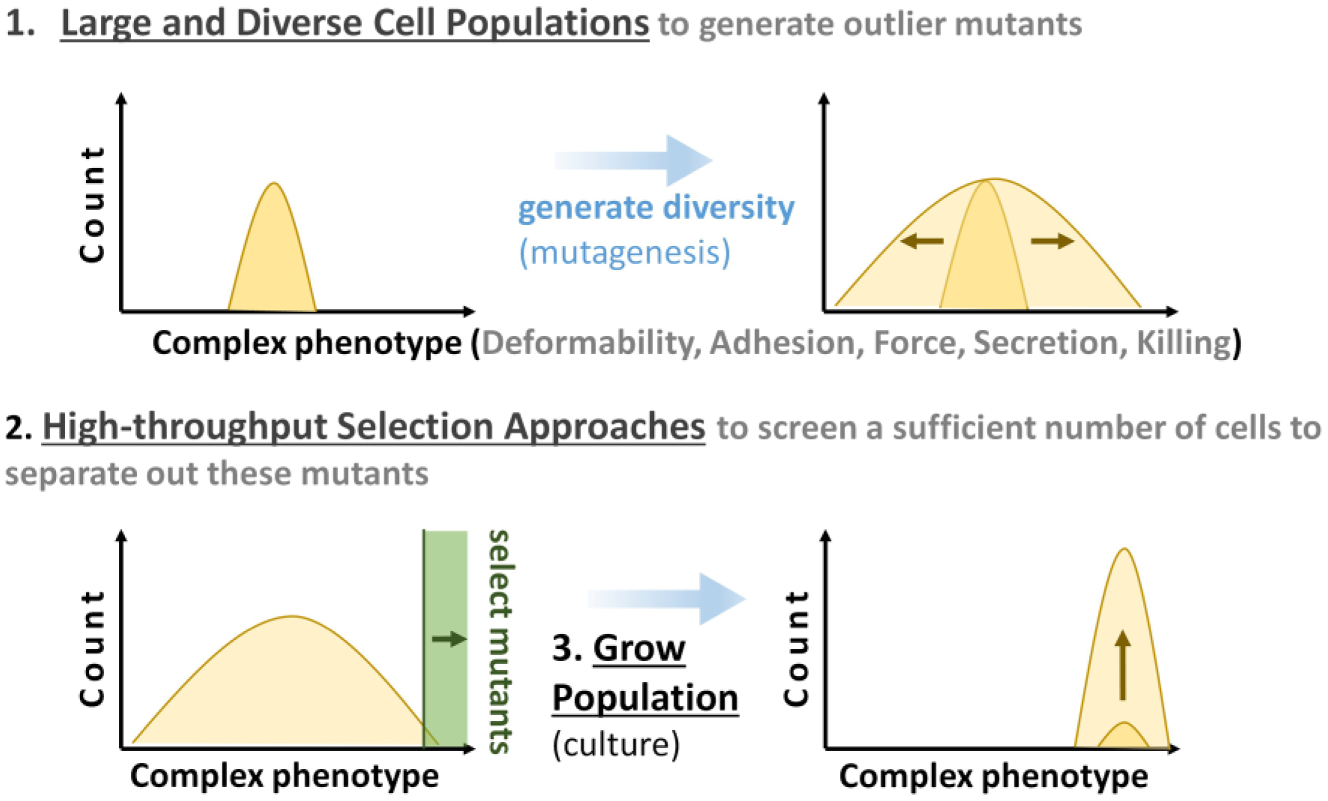
The evolution of cellular traits requires three key processes. (1) A mechanism to generate diversity. This diversity can arise from normal stochastic variation within a population or can be induced through an external process of mutagenesis (genetic diversity) or epigenetic “mutagenesis” (epigenetic diversity generation). (2) A mechanism to rapidly select outlier cells. The more rapid this selection/sorting method, the rarer a mutant can be while still selecting a sufficient number of cells in a practical time period. (3) Finally, the selected population is expanded and the process repeats.
The rate of evolution of organisms in the wild appears to be orders of magnitude lower than that in selective breeding experiments. These rates are often expressed in units of haldanes, which are normalized with regard to the lognormal variation in a population. 34 Interestingly, artificially selected populations have been found to have rates in haldanes >200,000-fold higher than natural evolutionary processes, for example, when selecting for animal size changes. 33 This large discrepancy may be due to a number of factors, including fluctuating selection pressures in natural environments that are not present during directed evolution processes. As discussed, selection pressure can be consistent or steadily ratcheted, which is encouraging for the feasibility of modulating large-scale traits in a reasonable time period.
Taking these data into account, it is expected that evolutionary processes can act on complex cellular processes over a tractable number of generations, with increased population diversity and size playing a role. Note that selective breeding processes in the past did not introduce external mutagenesis, therefore leading to a larger number of evolutionary cycles because of the lack of population diversity. Assuming that a population of cells has a normally distributed phenotype, selecting a sufficient number of cells with traits above a threshold difference from the initial mean value requires either increasing the variance of the population or increasing the rate at which cells are analyzed. Practically, the variation within a population is increased through mutagenesis (either genetic or epigenetic). High-throughput sorting technologies separately enable screening through larger populations of cells to identify rare mutants. The combination of mutagenesis and high-throughput sorting is ideal for each selection cycle as it can enable sorting of a sufficient number of cells with a quantitative difference in traits farther away from the mean value of the initial population (e.g., larger S), accelerating the evolutionary process. Leveraging new high-throughput tools, taking into account the high rates of directed evolution of enzymes, and given the >200,000-fold enhanced rates of evolution seen for selective breeding compared with natural selection, I would predict 5–20 cycles will be sufficient to evolve complex traits in cells.
Need for High-Throughput Tools to Select Cells Based on Complex Phenotypes
Flow cytometry and fluorescence-activated cell sorting (FACS) have been high-throughput and quantitative tools that allow the analysis and sorting of live cells based on molecular phenotypes (mainly based on staining with dyes to cell surface proteins). Although FACS has significant advantages in terms of throughput and commercial development, it is limited in the type of selection pressure that can be applied. FACS was used to evolve cells with a mutant scramblase that exposes phospholipids on the cell surface, for example. 35 Sorting cannot be performed based on arbitrary complex phenotypes like secreted proteins (e.g., proteases, nucleases, pore-forming compounds) in live cells, deformability, contractility, and adhesiveness, which are intrinsically linked to a number of internal proteins and structures in cells. Imaging flow cytometry can interrogate internal structural features or spatial locations of molecules, but sorting based on these properties has only recently become possible, 36 with no commercially available systems. Therefore, there is a critical need to augment FACS and employ imaging flow sorting or develop entirely new technologies to sort and select cells based on physiologically important phenotypes to enable the evolution of new therapeutic cell populations ( Fig. 2 ).
Development of New Cell Selection and Sorting Technologies
New cell sorting and selection approaches that can be sufficiently high throughput and automated to apply to an evolution workflow are needed. I discuss recent advances to develop new sorting approaches based on complex traits: (1) enzyme secretion, (2) deformability, (3) contractility, (4) adhesion, and (5) cell killing. I will briefly outline emerging automation technologies to sort in high throughput and quantitatively based on these traits, which are intimately linked to barriers in solid-tumor efficacy of engineered cell therapies ( Fig. 4 ).
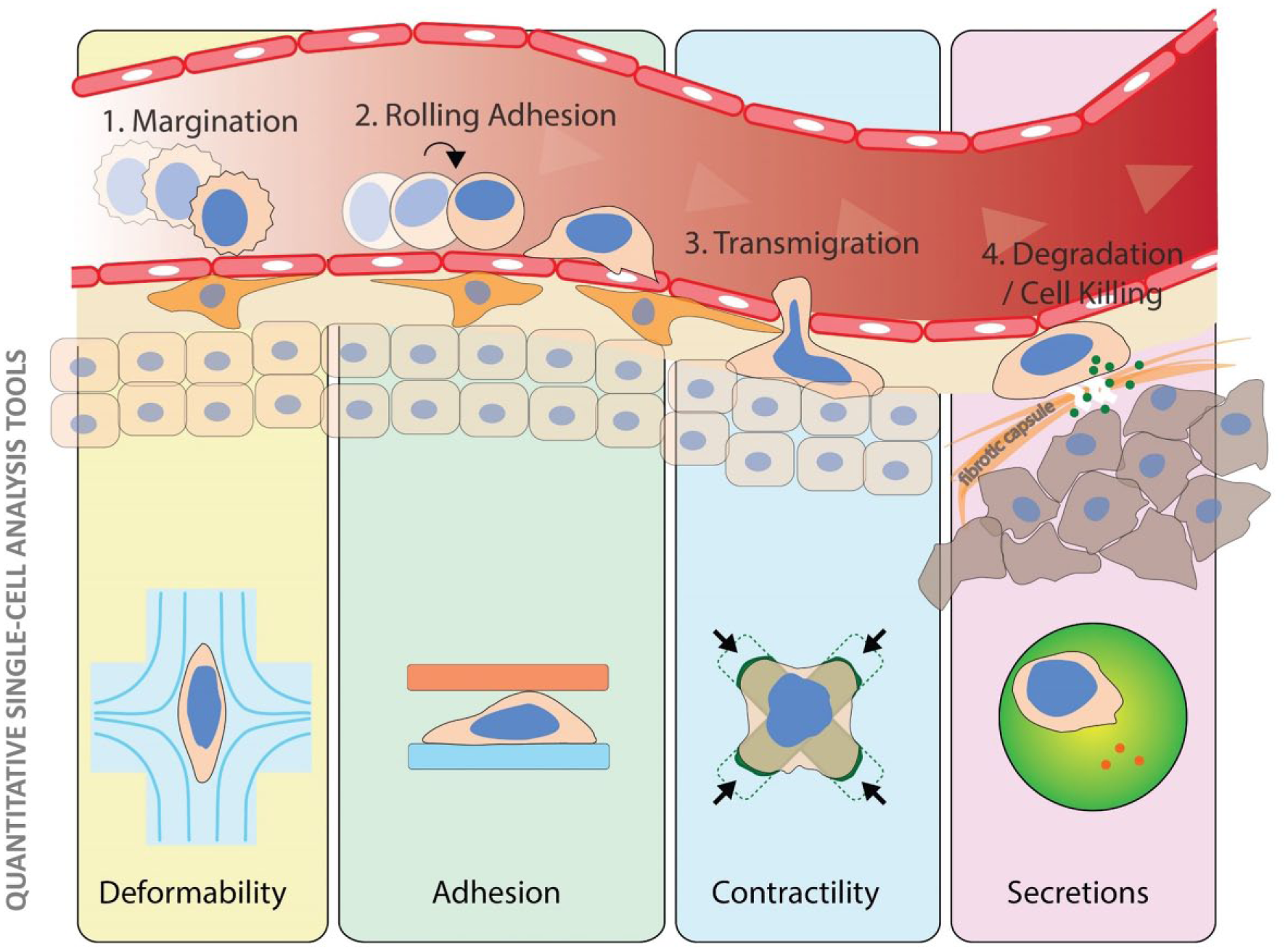
Techniques to quantify and sort based on complex physical properties of cells corresponding to important gateways of the cancer microenvironment to mount an effective cytotoxic T-lymphocyte-based defense.
Secretion
A key challenge to characterize and select cells based on enzyme secretion is that most methods of measurement (e.g., enzyme-linked immunosorbent assay [ELISA] or Western blotting) operate at the bulk level. Further challenges arise in characterizing single-cell secretions; background secretions of neighboring cells should be removed, and secretions should be concentrated into a small volume to allow the detection of small quantities of molecules. Miltenyi recently introduced a bivalent antibody to CD45 and several cytokines of interest that enable the capture of secreted cytokines on the surface of a cell. 37 These cytokines can then be labeled with a secondary antibody and cells can be sorted based on this signal. 38 The downsides of this approach are that not all cytokines secreted by a cell will be captured on its surface due to diffusion and background cytokines, and those secreted from neighboring cells can contaminate each cell’s signal when not used at very low cell dilutions. Alternative capture approaches using cholesterol-linked aptamers have also been proposed to analyze/sort single cells based on interferon-γ secretion. 39
To address the challenges of secretion capture, cross-talk, and background secretions, we have been developing a microfluidic vortex trapping platform that performs cell capture, washing, and solution exchange, followed by downstream encapsulation of single cells into droplets.40,41 By introducing a wash solution with a fluorescence resonance energy transfer (FRET) reporter on a peptide targeted by matrix metalloproteinases (MMPs) followed by encapsulation in this solution, we were able to fluorescently track the single-cell activity of MMPs from leukocytes and cancer cells ( Fig. 5 ). 41 The importance of washing away background secretions has also been highlighted in work from Jing et al., where they use an alternative microfluidic strategy, deterministic lateral displacement, to move cells into a new solution before encapsulation in droplets. 42 Following isolation and accumulation of secretions, droplets can be sorted based on accumulated fluorescence,43,44 such that viable leukocytes could be selected based on MMP levels or overall protease activity. Besides proteases, cells can be selected based on other therapeutically important secreted molecules (e.g., esterases, heparanases, DNAses, granzyme, perforin, interleukins) by using separate fluorogenic substrates or homogenous immunoassay platforms.45–48 Alternative approaches to capture secretions using monodisperse agarose or gelatin 49 phase-transitioned droplets can also play a role, 50 although improved approaches to sort these gelled drops at high throughput are needed.
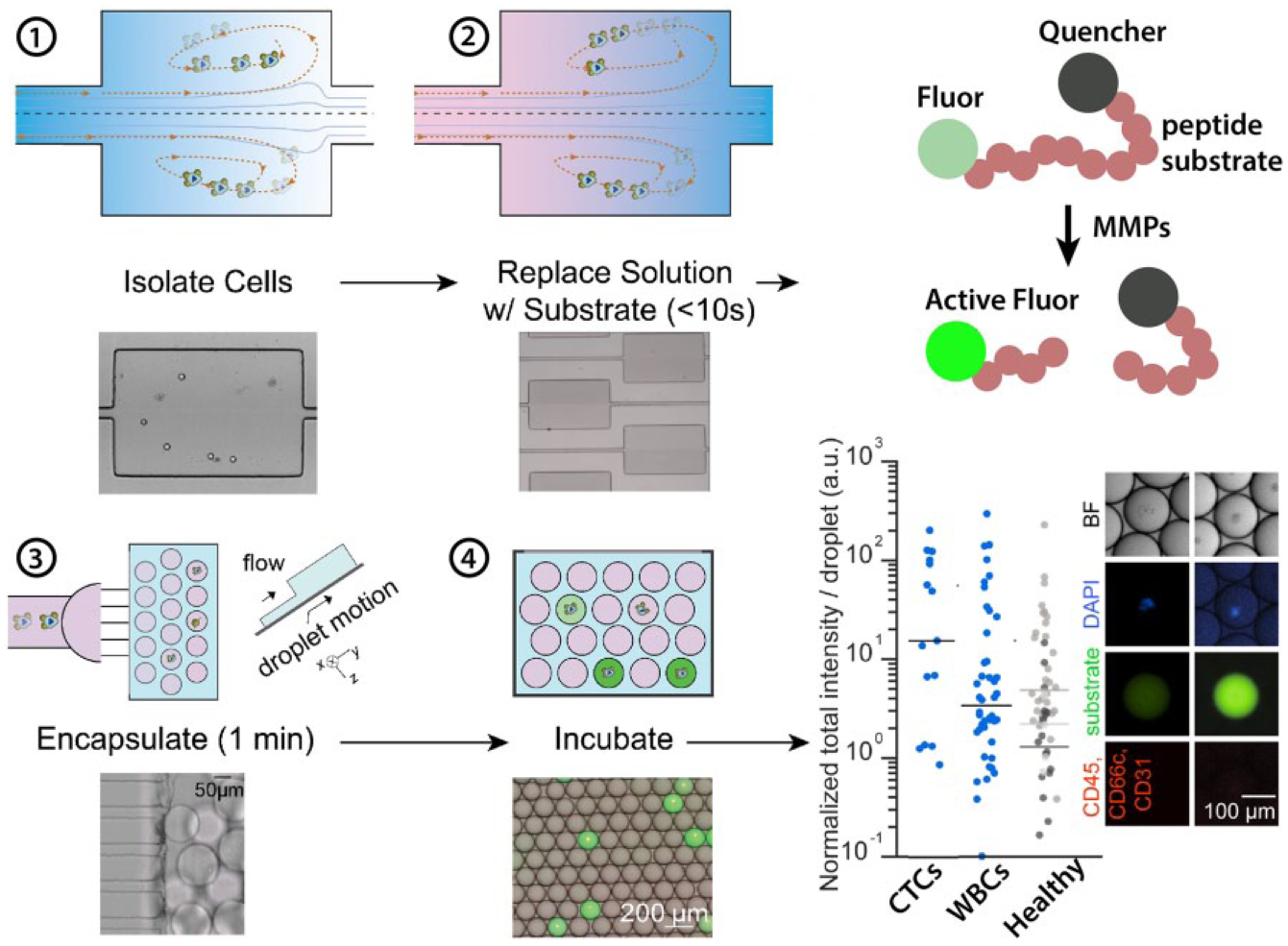
Tracking the secretion of single cells in microdrops. (1) Cells are trapped in microfluidic vortices. (2) While remaining trapped, background solution is replaced with a solution containing a fluorogenic MMP substrate comprising a peptide with fluorophore and quencher. (3) Once exchanged, cells in solution containing the substrate are encapsulated in nanoliter-scale drops using a step emulsification device. (4) Cells are incubated in drops and active MMPs lead to turnover of substrate and accumulation of fluorescent signal, given the small volume of the drop. Circulating tumor cells (CTCs) from metastatic prostate cancer patients have higher MP activity on average than white blood cells (WBCs) from the same patient or from healthy controls. Adapted from Dhar et al. 41 with permission.
Deformability
New technologies developed by us51,52 and others53–55 make use of microfluidic flows to deform and quantitatively measure hundreds to thousands of cells per second. These approaches have not been integrated with downstream sorting capabilities. Because of the issues with latency between data collection and the ability to offload data for analysis, standard high-speed CMOS cameras are not compatible with high-speed sorting pipelines. Two potential approaches to incorporate this functionality include (1) measuring scattered light of a deformed cell to indicate its deformed shape, upon which a variety of active sorting approaches can be triggered, and (2) using time-domain imaging technologies (like STEAM56,57 or FIRE58,59) to translate the spatial information of a deforming cell into rapidly assessed and analyzed time-domain signals from photomultiplier tubes and photodiodes, on which sort decisions can be made. 56 In addition, the deformability and shape of particles or cells affects their migration in a channel flow, which can be used to develop passive, high-throughput sorting approaches by designing microfluidic flow channels to amplify these shape- and deformability-dependent effects.60–62 Cell deformability also leads to modulation in acoustic properties,63–65 potentially enabling acoustic sorting-based selection at significant throughput. Finally, in recent work Pagano et al. 66 performed directed evolution of premalignant lung epithelial cells based on their ability to migrate through small pores in a membrane. Through repeated cycles they found that these could select for a population with heritable traits of reduced size or increased deformability.
Contractility
Force generation is a critical function for a variety of individual cells and tissues; however, quantitative techniques to isolate cells based on force generation at high throughput are lacking. We recently developed an approach to quantify single-cell force generation massively in parallel based on micropatterned fluorescently labeled elastic protein patterns. 67 These patterns are stretched in a manner correlated to applied stress and can be analyzed quickly by automated fluorescence microscopy and image analysis algorithms. As an example of a cell type with therapeutic potential, we applied the platform to quantify the force generated by macrophages during phagocytosis of antibody-opsonized patterns. We could quantify rare outlier cells with significant differences from the average ( Fig. 6 ). Manual cell picking would allow selection; however, is not easily scalable. Automated cell picking can address some of the challenges, 68 but with limited throughput. Combining such a platform with high-speed optics-based cell picking69,70 or microraft technologies 71 could enable high-throughput selection for a directed evolution workflow.
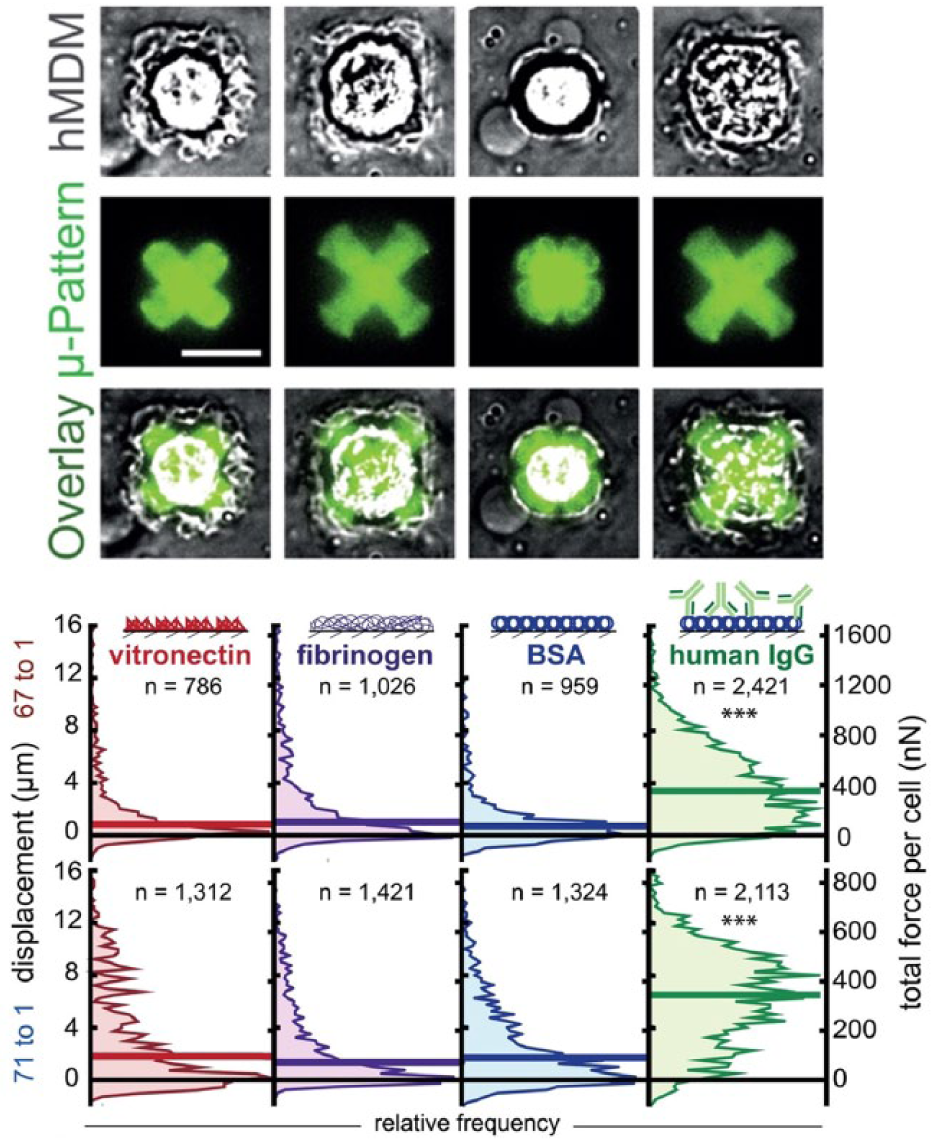
Diversity of force generation in human monocyte-derived macrophages. Images of macrophages contracting protein patterns embedded into an elastomeric substrate (FLECS platform). Opsonin dependence of the phagocytic force was observed. Vitronectin, fibrinogen, bovine serum albumin (BSA), and human IgG were patterned in 50 μm cross-shapes on a stiffer 67:1 base–crosslinker (B:C) (top) and softer 71:1 substrate (bottom). HIgG elicited the most contractile response from the largest fraction of macrophages; however, outlier macrophages were observed for each condition with high force. Adapted with permission from Pushkarsky et al. 67
Adhesion
Interestingly, mesenchymal stem cells (or marrow stromal cells) were initially selected by their differential adhesion to rigid tissue culture polystyrene plates. The concept of cell adhesion-based selection can be generalized with new technologies. By applying known forces to cells adhered to protein-coated substrates or other cells, high-throughput selection based on adhesion is possible. By using microfabrication approaches to create controlled gaps between two protein-coated surfaces, we have been able to characterize and sort cells based on relative adhesion to protein surfaces. 72 Using this system, we found that metastatic breast cancer cells would transfer to collagen IV and collagen I, while normal polarized breast epithelial cells would not transfer to the adjacent ECM-coated surfaces. 72 In other work, shear stress in a microfluidic channel was used to separate out populations of low-adhering pluripotent stem cells compared with other cell populations. 73 A similar approach to controlling shear stress was applied by Plouffe et al. 74 to separate endothelial, smooth muscle, and fibroblast cells based on a combination of adhesion to specific ligands and shear stress in winding microfluidic channels. Perhaps more relevant for evolution related to trafficking to inflamed vasculature, Chang et al. also immobilized E-selectin on the surface of channels and were able to enrich leukocytes based on differential rolling adhesion. 75 Using a different microfluidic design that allows continuous enrichment, Choi et al. also showed high-purity separation of cell lines based on adhesion to P-selectin-coated channels. 76 Analogous approaches could be used to select lymphocytes with properties that enable rolling adhesion and homing to tumor vasculature.
Cell Killing
Although it integrates other functions discussed above (e.g., secretion of lytic agents or applying force to phagocytose target cells), an important function for cell therapies is the ability to target and kill a particular subpopulation of diseased cells. This is critical in the development of anticancer therapies, 77 and recent studies suggest that selective elimination of senescent cells may provide value for addressing aging-related pathologies.78,79 Although immune cells (e.g., T cells, NK cells, macrophages) have an intrinsic ability to perform cell-killing functions, there are important goals to kill quickly, accurately, and repeatedly. There is interest, for example, in a “serial killer” phenotype that avoids exhaustion 80 but has high control of the antigen density dependence of killing to avoid off-target effects. 77
High-throughput selection based on this complex phenotype is challenging, but recent work suggests that the confined volumes of monodisperse drops can provide an advantage. Therapeutic cells of interest can be introduced into droplets with one or more target cells (or off-target cells), and the speed of killing, quantity of killed cells, accuracy, and long-term killing phenotype can be potentially quantified. Recent work from Sarkar et al. showed a proof of concept in which they characterized the cell-killing behavior of natural killer cells and an immortalized natural killer cell line (NK-92), but no sorting was performed. 81
Several technical challenges include controlling the ratios of cells within drops, and high-speed sorting of drops following cell killing to recover the optimal performing phenotype. Controlling droplets to contain only a single therapeutic cell while including multiple target cells is challenging given the normal expectation of Poisson encapsulation statistics in droplets.82,83 The use of inertial ordering phenomena to uniformize concentrations in flow could potentially address this challenge,84,85 while the use of active imaging flow sorters could also improve the accuracy following encapsulation, by selecting only certain ratios to further incubate. High-speed droplet sorters that are compatible with the larger-volume droplets necessary to culture cells during cell killing are also a challenge since larger droplets are prone to breaking in standard dielectrophoretic (DEP) sorters. New sequentially addressed DEP geometries can be gentler in their manipulation of large droplets and an important advance for long-term incubation-based studies in microdrops with eventual high-speed sorting to select rare outliers with enhanced cell-killing behavior. 86
Directed Evolution of Therapeutic Cells
Once the appropriate high-throughput cell selection/sorting tools are available, the process of directed evolution can be initiated ( Fig. 2 ). Although I have highlighted T-cell-based cell therapies, a number of cell types are being considered for cell therapies and could be the target of directed evolution. This includes natural killer cells 87 and macrophages; 88 mesenchymal stem cells, which possess unique homing and secretory functions;6,89 pluripotent stem cell-derived tissue-specific cells, such as beta cells; 90 or effectively any cell type that maintains some proliferative ability without undergoing early senescent processes. The directed evolution cycle consists of the following steps:
Mutagenesis using random mutagens or site-directed approaches91,92 to create a large population of mutants on which to perform selection. A number of approaches to introduce genetic diversity have been explored, including well-studied chemical and physical mutagenesis approaches that lead to nucleotide insertions, deletions, or substitutions. 93 A number of more recent techniques for directed evolution of proteins are reviewed, 94 and CRISPR-based approaches that can target the area of mutagenesis in a tunable window of the genome also appear promising. 95
Selection based on a complex trait using the high-throughput tools discussed above.
Growth, to expand the selected mutant populations and co-select mutations that improve the selected physical trait and do not interfere with cell growth and division.
Mutant fitness can then be quantified based on the quantitative selection/sorting tools used in step 2 and the cycle repeated until a threshold level of fitness is achieved. These mutants can then be analyzed by whole-genome sequencing, RNA-seq, and mass spectrometry to identify genomic and expression-level changes leading to the new phenotype. Quantitative comparisons to wild type and a multitude of other clones derived from the same selection process (e.g., in an analogous process to genome-wide association studies) would allow for understanding conserved regions of genes and networks that are most involved in controlling the selected phenotypes. Starting again with wild-type cells, targeted mutations on these genes can then be performed to determine if the phenotype can be recapitulated in a precision manner, which would be more amenable to cell therapy engineering.
Epigenetic Evolution
In addition, or in parallel to genetic changes, epigenetic changes may also lead to new phenotypes that are relatively stable over many division cycles. For example, engineered MILs from a patient were found to maintain their tropism through expansion, as discussed above. 27 The ability of epigenetic changes to be implemented quickly without modulating the genome and have limited heritability could be viewed as a key advantage for single-cell-directed evolution. Because epigenetic marks are largely inherited during cell division, 96 it may be possible to maintain the same base genome (no mutagenesis) and apply epigenetic “mutagenesis” to achieve a range of epigenetic states to apply these automated selection approaches against. The idea of epigenetic mutagenesis, or the creation of epigenetic diversity that is heritable across cell divisions, has not been widely explored, but could be addressed in multiple fashions: (1) by creating a multitude of cellular environments that signal to cells and modulate epigenetic state, (2) by using specific drugs known to modulate chromatin structure and epigenetic marks (5-aza, chaetocin, DZNep, etc.) at low doses, or (3) by random or site-directed CRISPR-dCas9 that possess a targeting functionality to specific sequences of the genome fused with a chromatin-modifying protein domain. 97 Random modifications can be achieved by incorporating guide RNA sequences targeting genomic sites in a stochastic fashion. Stochastic epigenetic modulation can also be achieved through the administration of small-molecule inhibitors of common epigenome-modifying enzymes, applied at sublethal doses to cells. Examples include methyltransferase inhibitors (zeublarine, MG98, RG108), histone deacetylase inhibitors (vorinostat, trichostatin A, ACY-1215), histone acetyltransferase inhibitors (C646, curcumin), protein methyltransferase inhibitors (UNC0646, UNC321), and histone methylation inhibitors (DZNep). Upon dosing, large-scale changes in epigenetic marks along with increased phenotypic heterogeneity are triggered. 98 If successful, this alternative may be a particularly fruitful direction as epigenetic selection and engineering of cells with a desired trait could be applied directly to a particular patient’s cells to create optimal personalized therapies without genetic modification, which triggers additional safety concerns.
Challenges with Directed Cellular Evolution
The conclusions obtained from a directed evolution process for cells can be confounded by nonlethal mutations that do not contribute to a phenotype and epigenetic changes in cells that are heritable over generations. Random mutagenesis will lead to significant off-target nonlethal mutations along with mutations that give rise to a phenotype, which is not ideal. However, by repeating processes of evolution in parallel to develop several clones with the same phenotype, off-target mutations should appear in a more statistically random manner than the mutations of interest and therefore be computationally identified. In fact, this is how genome-wide associated studies address random variations across populations that may not be associated with a trait. 99 A related issue is that there may be multiple genomic solutions to the same phenotype (i.e., nonuniqueness). This would be actually quite interesting to observe but would interfere with this analysis approach. The issue could be addressed by performing continued evolution of a selected phenotype, such that essential versus nonessential mutations would be observable.
Extrinsic and Intrinsic Heterogeneity Slows the Rate of Genetic Evolution
As discussed above, the breeder’s equation includes a factor, h2, that takes into account that not all phenotypic variation contributing to selection is a result of heritable differences. In organisms, this takes the form of traits influenced by environmental factors or learned behavior. Similarly, cells experience a number of nongenetic factors that affect phenotype. These include cell–cell interactions in culture, cell–substrate/ECM interactions, soluble paracrine factors (collectively extrinsic heterogeneity), and cell cycle and stochastic chemical processes within a cell (intrinsic heterogeneity). 100 Some of these factors can also lead to the establishment of heritable epigenetic marks as discussed above. These effects can dilute the selection effect and should be considered and/or controlled when developing an evolutionary cycle. Environmental effects could potentially be controlled (e.g., with cell micropatterning, 67 cell cycle synchronization, or controlled ECM 101 and soluble factors). Alternatively, by modulating the designs of micropatterns or seeding cells in combinatorial arrays of ECM, one can introduce heterogeneity itself.
Population Reversion
Evolution based on epigenetic changes may not be long lasting, which could mitigate the long-term efficacy of a cell therapy. Evidence for reversion of a selected cell population back to a starting population distribution is seen in flow cytometry experiments. 102 Following selection by flow cytometry of outlier hematopoietic cells with high Sca-1 expression, the progeny of these selected cells slowly reverted to a population with a Sca-1 distribution representative of the average preselected state after about 5 days. 103 Similarly, when selecting for either high or low Nanog-expressing embryonic stem cells the population also reverted to the preselected bimodal condition after about 19 days of culture. 104 The underpinnings of reversion in such experiments may be epigenetic state differences or other stochastic transcriptional and structural differences that arise in a population cultured under specific conditions. 102 These differences might become more fixed when culture conditions are permissive or following multiple rounds of selection. When selecting for T helper cells that secrete varying levels of interferon γ, cells re-introduced in vivo maintained a memory of secretion magnitude and probability in the population for greater than 1 month.38 When performing multiple rounds of selection, a biophysical phenotype was maintained in epithelial cells for more than 8 weeks postselection without mutagenesis. 66 Additional studies are needed to further confirm whether the reversion timescale can be extended for populations selected over multiple cycles without genetic modifications. On the other hand, in some cases (e.g., enhanced cell killing) a transient phenotype may be beneficial, reducing the potential for long-term nonspecific side effects of a therapy.
Conclusions
Nature has used evolution to shape complex functions from molecules to multicellular organisms over billions of years, following rules of natural selection. Over the last millennia humans have adapted evolution to our purposes for the domestication of animals and agriculture, by artificially selecting and allowing the reproduction of organisms with perceived beneficial traits. Within the last decades, taking advantage of automation and enzyme activity quantification approaches, bioengineers have applied directed evolution processes to engineer enzymes, 105 recognized by the Nobel Prize in Chemistry to Frances Arnold in 2018. We have also begun to evolve simple traits at the cellular level. Now that cellular products are reaching the clinic, it is an opportune time to develop new approaches to quantitatively evaluate phenotypes of cells that provide therapeutic benefit and select cells based on these traits with high throughput. In particular, it is becoming apparent that cell function in intricate and dynamic physical and chemical environments within the body requires engineering of complex physical traits that are the result of changes in a number of biochemical circuits, where holistic evolutionary processes are expected to have significant advantages. Automation approaches to sort cells based on functional phenotypes are critical to reduce the timescale of directed cellular evolution and ultimately enable the next generation of functionally engineered cell therapies within the next decades.
Footnotes
Acknowledgements
I would like to acknowledge H Kittur, M Dhar, and I Pushkarsky for assistance with creating figures. I would also like to acknowledge the pioneering work of Frances Arnold which sparked my initial interest in applying evolution as an engineering tool to biology over 10 years ago.
Declaration of Conflicting Interests
The author declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author received no financial support for the research, authorship, and/or publication of this article.