
Abstract
Testing of drug effects and cytotoxicity by using cultured cells has been widely performed as an alternative to animal testing. However, the estimation of pharmacokinetics by conventional cell-based assay methods is difficult because of the inability to evaluate multiorgan effects. An important challenge in the field is to mimic the organ-to-organ network in the human body by using a microfluidic network connecting small-scale tissues based on recently emerging MicroTAS (Micro Total Analysis Systems) technology for prediction of pharmacokinetics. Here, we describe an on-chip small intestine–liver coupled model for pharmacokinetic studies. To construct an in vitro pharmacokinetic model that appropriately models in vivo conditions, physiological parameters such as the structure of internal circulation, volume ratios of each organ, and blood flow ratio of the portal vein to the hepatic artery were mimicked using microfluidic networks. To demonstrate interactions between organs in vitro in pharmacokinetic studies, Caco-2, HepG2, and A549 cell cultures were used as organ models of the small intestine, liver, and lung, respectively, and connected to each other through a microporous membrane and microchannels to prepare a simple model of a physiological organ-to-organ network. The on-chip organ model assay using three types of substrate—epirubicine (EPI), irinotecan (CPT-11), and cyclophosphamide (CPA)—were conducted to model the effects of orally administered or biologically active anticancer drugs. The result suggested that the device can replicate physiological phenomena such as activity of the anticancer drugs on the target cells. This microfluidic device can thus be used as an in vitro organ model to predict the pharmacokinetics of drugs in the human body and may thus provide not only an alternative to animal testing but also a method of obtaining parameters for in silico models of physiologically based pharmacokinetics.
Introduction
Prediction of the human response to drugs or chemicals is critically important for medical evaluations, drug discovery, and the study of life science but remains difficult because of the complexity of the human body. To predict the pharmacokinetics of drugs in the human body, animal tests are widely conducted as an effective method to evaluate in vivo disposition; however, animal tests are characterized by various drawbacks such as species differences, high cost, and ethical issues. 1
To solve these problems, screening of drug effects and cytotoxicity by using cultured cells in vitro has been widely performed as an alternative to animal testing. 2 However, conventional cell-based assay methods cannot be used to evaluate multiorgan effects because it is difficult to maintain the homeostatic properties of cells in vitro and to coculture different types of cells on dishes and well plates. Therefore, absolute prediction of the pharmacokinetics of substances in the human body by using conventional in vitro methods is difficult.
An important challenge in the field is to establish organ or tissue models of the human body by preparing miniaturized tissue using recently emerging microfluidic technology for cell-based assays.3–5 Thus far, various microfluidic models of the liver,6–8 lung,9,10 kidney,11,12 gut,13,14 and other organs15–17 have been proposed using microfluidic systems. Reconstruction of organ function and characteristic structures in vitro has been demonstrated using microfluidics and microstructures. Furthermore, approaches that mimic organ-to-organ networks in the human body based on microfluidic networks connecting small-scale tissues have been proposed for the prediction of pharmacokinetics.18–21 Sung et al. 20 proposed micro cell culture analogs (microCCA) that can coculture two or more cell types in microfluidic devices to predict response to drugs and chemicals for establishment of a physiologically based pharmacokinetic (PBPK) model in silico. However, the physiological blood pathway has not been still structurally mimicked in the microfluidic device. To construct the PBPK model that appropriately models physiological conditions for prediction of reactions of the whole body, it is necessary to mimic in vivo parameters such as the blood circulation system and organ volume relation because the parameters strongly relate distribution function and metabolism function in vivo. 22
We previously proposed a microfluidic device with embedded stirrer-based micropumps and a two-compartment culture chamber to mimic in vivo conditions and evaluate the function of intestinal absorption. 13 In pharmacological studies, the metabolic function of the liver and the absorption function of the small intestine are both important because they highly influence the bioavailability and bioactivity of ingested substances. In the present study, we developed a device to model small intestine and liver based on physiological blood circulation and organ volume for pharmacokinetic studies. To investigate interactions between organs in a physiological manner, we cultured small intestine model cell, liver model cell, and lung model cell separately, but the cultures were connected to each other through a microporous membrane or microchannels in the device to prepare a simple model of physiological organ-to-organ networks for estimation of the efficacy of orally administered or biologically activate substrates. To achieve conditions that are more physiological than those reported in the literature, we reproduced in vivo parameters such as the structure of the internal circulation, volume ratio of each organ, and blood flow ratio of the portal vein to the hepatic artery on the device. We successfully reproduced chemical reaction phenomena by using the device. This work is a first trial to reproduce both blood circulation and organs volume relation in vitro.
Materials and Methods
On-Chip Organ Model
A model of a human in vivo circulatory system, which consists of organs and complex networks of arteries and veins, is shown in Figure 1a . An on-chip organ model was built based on the in vivo model, as shown in Figure 1b . To reconstruct physiological drug kinetics, the small intestine and liver were modeled in the on-chip organ model, because these organs considerably contribute to drug kinetics based on their absorption and metabolic functions in the human body. The small intestine model was composed of two compartments (i.e., the lumen and portal vein) to mimic physiological small intestinal function. The heart was also modeled through the circulation of fluids in the on-chip organ model. In the present study, the lung was adopted as a target organ for anticancer drugs because lung cancer is highly prevalent worldwide. The lung model was located downstream of the model heart in the on-chip organ model because all blood in the body flows from the heart to the lung before entering the main circulation. The portal vein and hepatic artery channels were connected to each of the model organs to control flow conditions and thereby mimic physiological blood circulation.
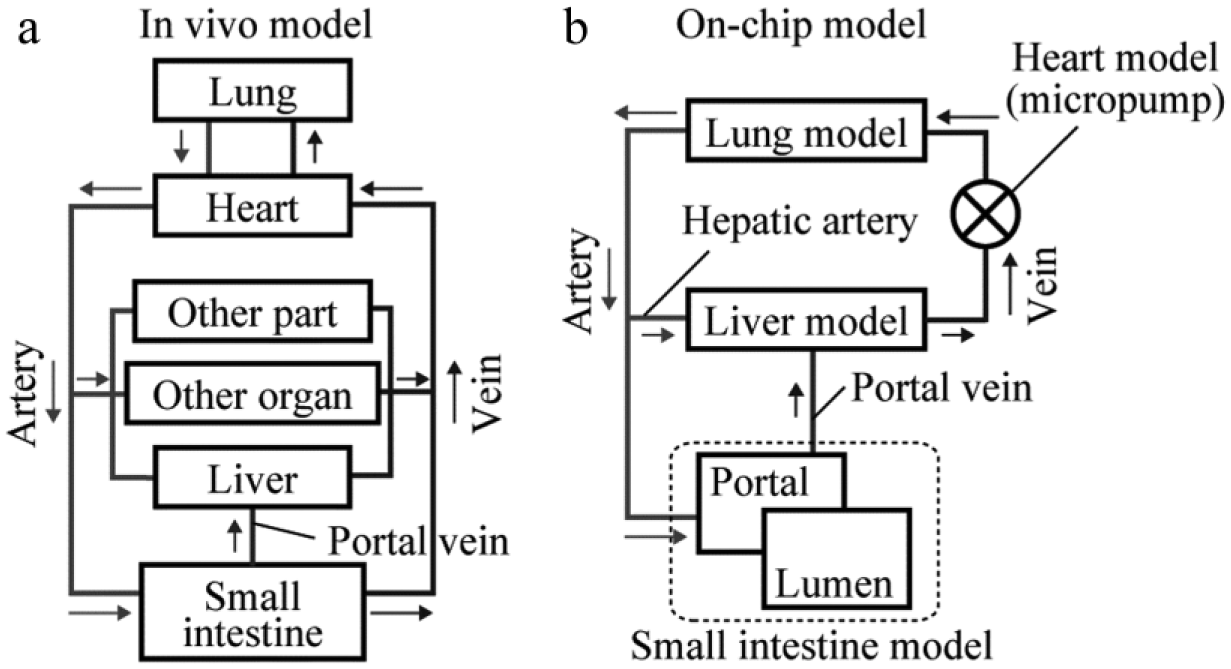
Schematic representation of pharmacokinetic models. (
Microfluidic Device
The microfluidic device was designed to meet the requirements of the on-chip organ model described above ( Fig. 2a ). The device consisted of a small intestine chamber, a liver chamber, a lung chamber, and stirrer-based micropumps connected to microchannels that corresponded to arteries and veins. The small intestine chamber was separated into upper and lower compartments corresponding to the cell polarity (denoted as the lumen side and the portal vein side, respectively) by a microporous membrane. 23 The device was assembled using two layers of polydimethylsiloxane (PDMS) chips that had microchannels designed to mimic the physiological blood flow and organ volume ratio ( Fig. 2b ). The blood flow ratio of the portal vein to the hepatic artery and the organ volume ratio of the small intestine to the liver in the human body are approximately 3:1 and 2:1, respectively.24,25 The parameters for the microchannels on the PDMS chips were adjusted and calculated to balance the pressure drop between the microchannels and thus maintain the flow ratio consistent with the physiological condition; that is, the blood flow ratio of the portal vein to the hepatic artery was 3:1. The dimensions of the small intestine chamber and liver chamber were 200 mm2 and 100 mm2, respectively, to maintain the ratio of the respective cells at 2:1.
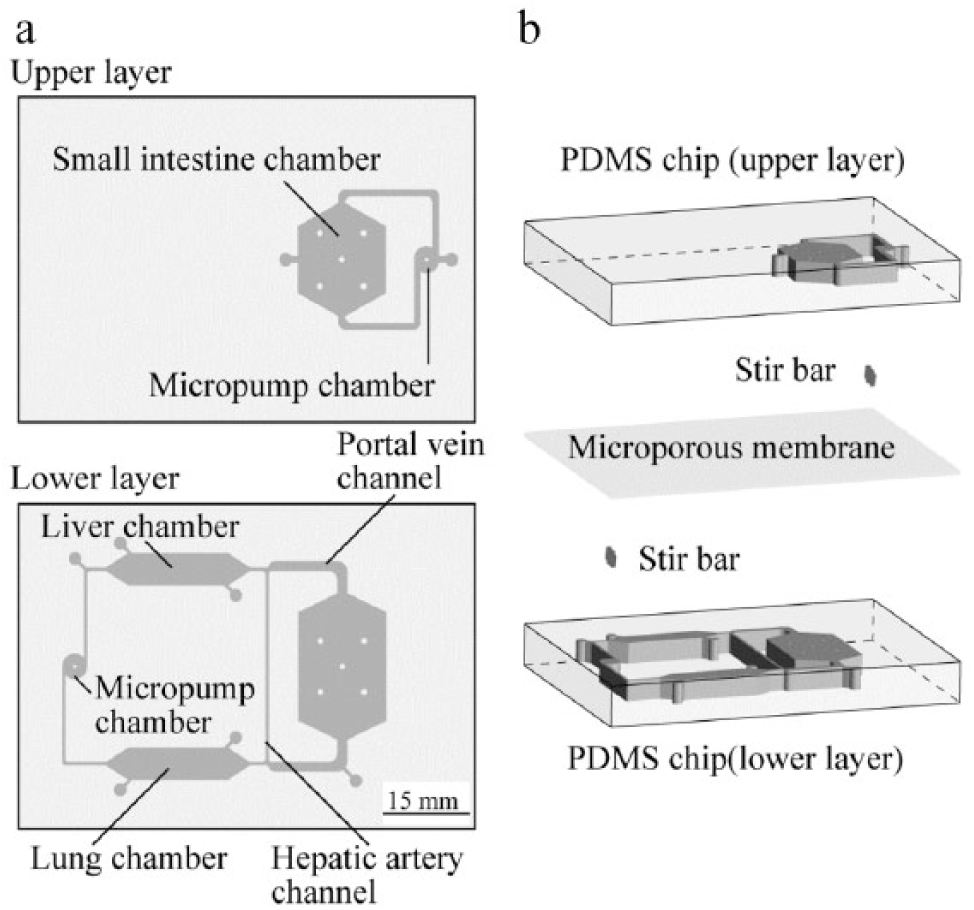
Schematic representation of the microfluidic device. (
The integrated stirrer-based micropump developed in our previous study to model the heart circulates culture medium to maintain uniform distribution of chemicals throughout the device. 13 The circulating medium is important for reducing local concentrations of metabolic waste and maintaining local nutrient levels, as well as for maintaining an even drug distribution for pharmacokinetic studies. Stir bars are enclosed in the micropump chambers and driven using an external magnetic field generated by a motor-driven permanent magnet controlled by a magnetic stirrer controller. Pulsative and unidirectional continuous flow is generated as the rotational motion of the stir bar pushes the liquid from the micropump chamber inlet to the outlet.
The device made by PDMS was fabricated by the conventional microfabrication method as reported in our previous work. 13 For plasma bonding, a polyethylene terephthalate (PET) microporous membrane was coated with aminosilane (KBE-903; Shin-Etsu Chemical, Tokyo, Japan) by dipping the membrane in an aminosilane coupling agent and then drying the agent, because the PET membrane cannot be bonded to PDMS by using the common O2 plasma method. A photograph of the device is shown in Figure 3 .

Photograph of the microfluidic device. Microchannels on the upper and lower layers of the device were visualized by black and gray colors, respectively. The three chambers and micropumps represent the small intestine, liver, lung, and heart.
Device Characterization
The flow ratio in the microchannels was evaluated by particle tracking velocimetry (PTV). A suspension of fluorescent beads (diameter of 1.0 µm, 0.05% v/v; Polysciences, Warrington, PA) in Dulbecco’s phosphate-buffered saline (PBS; Sigma, St. Louis, MO) was used for measurement of the flow velocity. After all the microchannels were filled with the bead suspension, the stir bar in the channel of the lower layer was rotated to generate flow at each rotation frequency. The flow velocity was measured at the portal vein and hepatic artery channels. Motion of the fluorescent beads was observed using a fluorescence microscope (PowerIX71; Olympus, Tokyo, Japan) equipped with a CCD digital camera (DP72; Olympus). The flow ratio of the channels was then calculated from the measured data.
Cell Culture
The small intestine model cell line Caco-2 and the lung model cell line A549 were obtained from Riken BioResource Center (BRC, Tsukuba, Japan), and the liver model cell line HepG2 was obtained from American Type Culture Collection (ATCC, Manassas, VA). All cell lines were cultured in a 100-mm dish in Dulbecco’s modified Eagle medium (DMEM; Sigma) containing 10% fetal bovine serum (FBS; Sigma), 0.1 mM MEM Non-Essential Amino Acids Solution (Life Technologies, Carlsbad, CA), and 1% antibiotic-antimycotic solution (Wako, Richmond, VA) and maintained at 37 °C in a humidified atmosphere containing 5% CO2.
Before the cells were injected into the device, the device was sterilized by filling it with 70% ethanol for 1 h, followed by rinsing with PBS. Thereafter, the PDMS microchannels and the membrane were coated with collagen (Cellmatrix Type I; Nitta Gelatin, Morrisville, NC) by filling the device with 10% collagen solution for 30 min. After trypsinization in the dishes, each cell line was injected into each chamber and the device was kept in an incubator overnight. After the cells attached onto the surface, circulation of the culture medium by the micropump was initiated. During experiments, the culture medium in the device was exchanged every 2 days to supply nutrients for the cells.
Pharmacokinetic Study
The pharmacokinetic study was conducted using three anticancer drugs: 10 µM epirubicine (EPI; Wako), 50 µM irinotecan (CPT-11; Tokyo Chemical Industry, Tokyo, Japan), and 1 mM cyclophosphamide (CPA; Wako). EPI and CPA were injected into the channel in the upper layer corresponding to the lumen side, whereas CPT-11 was injected into the channel in the lower layer corresponding to the blood vessel side. For the CPT-11 test, Caco-2 cells were not cultured in the device. The exposure time was 48 h for each drug. In this experiment, HepG2 cells and A549 cells were adopted as targets of EPI, and CPT-11 and CPA, respectively, because effect and mechanism of each drug are different, as described later.26–28
Cell condition was observed with calcein-AM (Dojindo, Rockville, MD) and propidium iodide (PI; Dojindo) stains. Only calcein-AM, which is converted to green-fluorescing calcein after acetoxymethlester hydrolysis by esterases of living cells, was used for quantification of cell viability. Images were captured by a microscope equipped with a CCD camera and analyzed using image-processing software (ImageJ; National Institutes of Health, Bethesda, MD) to measure fluorescence intensity. As a control for each experimental condition, experiments using culture medium without the drugs were conducted at the same time. The viability of cells in the experiments was calculated by dividing test values by the control values for normalization.
Statistical Analysis
In each experiment, at least three replicated tests were run. Unpaired Student t test was performed for statistical evaluation, and p < 0.05 was defined as statistically significant.
Results and Discussion
Characterization of Flow Conditions
The flow velocity in the microchannels was evaluated by capturing the motion of fluorescent beads in the microchannels in movies. The flow velocities measured at the center of the portal vein channel and the hepatic artery channel for each rotation frequency of the magnetic motor are shown in Figure 4 . Flow velocities between 0 and 0.15 mm/s–1 correlated linearly with the rotational frequency.
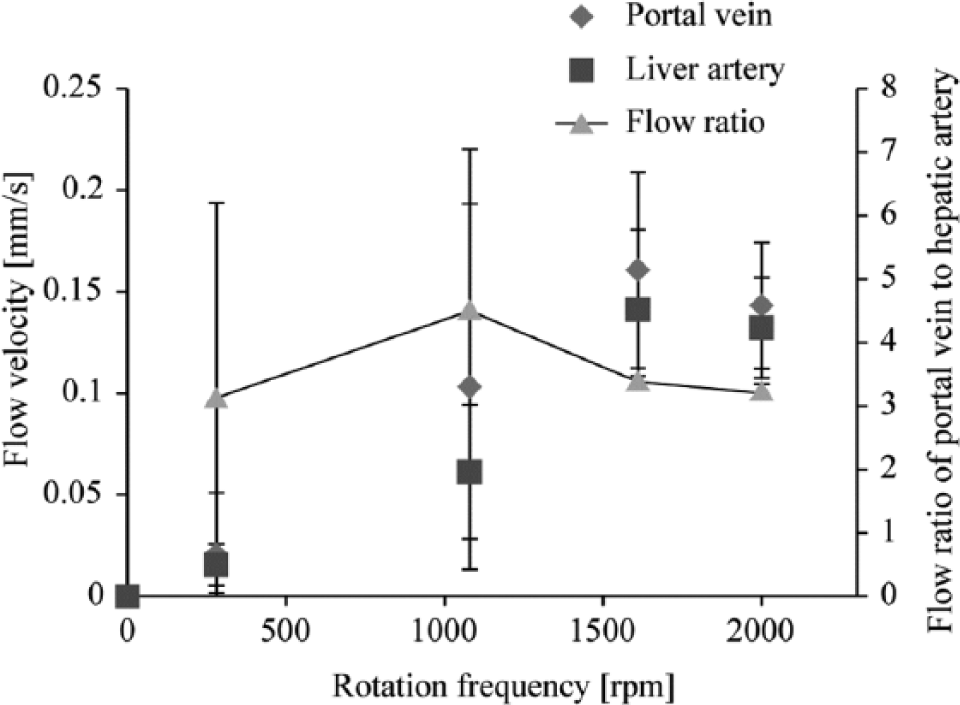
Flow velocity and flow ratio of the portal vein channel to the hepatic artery channel measured using particle tracking velocimetry (PTV) versus rotation frequency of the magnetic motor. The flow velocity shows a linear correlation to the rotational frequency at both the portal vein channel and the hepatic artery channel. The flow ratio of the microchannels is almost 3:1 to maintain the physiological flow ratio of the portal vein to the hepatic artery.
The flow ratio of the portal vein channel to the hepatic artery channel was determined from the measured flow velocity, as shown in Figure 4 . The blood flow ratio of the portal vein to the hepatic artery in the human body is 3:1, and this ratio was maintained in the device by choosing each rotational frequency to balance the pressure drop between microchannels. The data suggest that the flow ratio in the microchannels is controllable by calculating the pressure drop when using stir-based micropumps.
The shear stress in the culture chamber was determined from the measured flow velocity because cultured cells in the device may be inactivated and peeled off by excessive hydrodynamic force generated by the flow stream. The threshold hydrodynamic shear stress for cell attachment has been estimated roughly as 0.2 to 0.5 Pa, 29 whereas the measured maximum hydrodynamic shear stress in the culture chambers was 4.4 × 10–4 Pa at a rotational frequency of 1600 rpm. These values demonstrate the appropriate performance of the stir-based micropump to circulate the culture medium in the device. Although cells exposed to high shear forces show reduced liver-specific function, 30 the present study shows that cells cultured under perfusion conditions were not influenced by shear stress.
In our previous study, when the rotational frequency of the magnetic motor was less than 500 rpm, the flow was strongly pulsatile and unstable. 13 Therefore, the rotation frequency should be higher than 500 rpm. In addition, the blood flow rate and tissue volume of the human liver are approximately 23.2 mL/s–1 and 1500 mL, respectively. 31 To mimic subequal range of the physiological flow rate, we adopted a rotation frequency of 1600 rpm to generate a flow rate of 0.16 µL/s–1 in the 50-µL liver chamber.
On-Chip Coculture of Three Cell Lines
We investigated the performance for coculture of the three cell lines—Caco-2, HepG2, and A549. The results of cell coculture in the device are shown in Figure 5 . The three types of cells formed and kept confluent monolayers in the each chamber on day 3. Conventionally, it is difficult to coculture Caco-2 and HepG2 for a few days or more by using a commercially available membrane culture insert because of oxygen and nutrient deficiency. 32 However, in our coculture test, the device was able to coculture the three types of cells for at least more than 3 days, possibly because of the high oxygen permeability of PDMS and the perfusion culture environment using the microfluidic technique. The environment contributes to supply oxygen and nutrient, as well as to remove waste matters around cells.33,34 This coculture ability of the microfluidics would be one of the big advantages when the device is applied to cell-based assay research.

Photographs of cells cocultured in the microfluidic device for 72 h. The photograph shows images in the each culture chamber: (
Small Intestine Model Assay
To evaluate small intestinal function in the device, we conducted a drug assay using EPI. In this experiment, culture medium including 10 µM EPI was injected into the upper compartment corresponding to the lumen side of the human body. To evaluate the barrier function to substrates, devices cultured with and without a Caco-2 cell monolayer on the microporous membrane located in the upper compartment were prepared. EPI is known to have anticancer effects in cancers of the liver, stomach, breast, and bladder but not the lung. 26 At 48 h after drug injection, the viability of HepG2 cells cultured in the liver chamber as cancer cells targeted by EPI was measured by fluorescein staining using calcein-AM as described above. Figure 6a shows the viability of HepG2 cells with or without the presence of Caco-2 cells as a monolayer on the membrane in the small intestine chamber. When the Caco-2 cell monolayer was present, the viability of HepG2 cells against EPI increased by approximately 35% (p < 0.05). The intestinal wall of the human body has not only an absorption function but also a barrier function to prevent hazardous chemicals from entering the human body. The present findings may reflect that the Caco-2 cell monolayer has a function that blocks hazardous chemicals administered orally, including anticancer drugs, from inducing toxicity in the body.
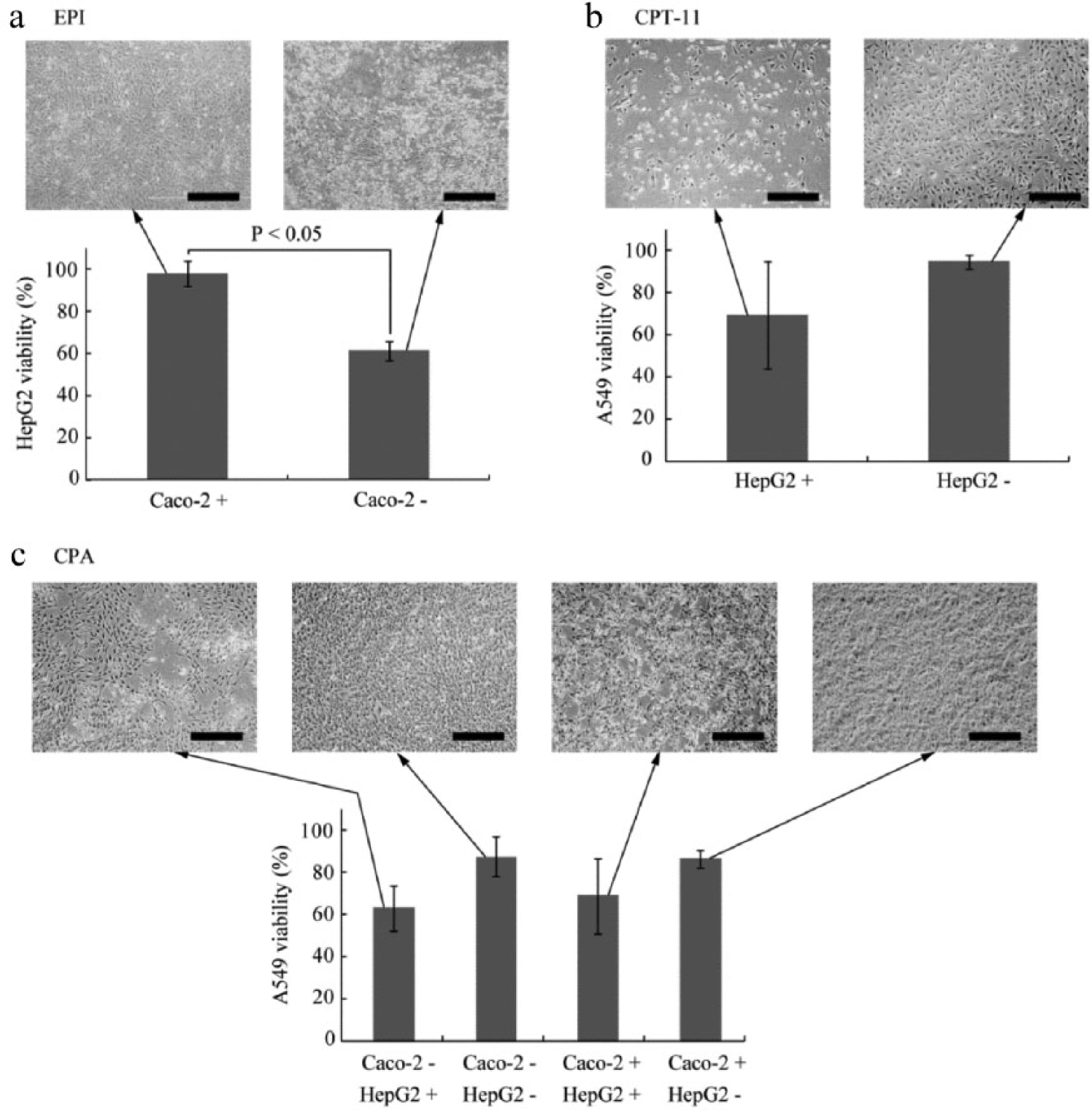
Viability and morphology of target cells treated with (
Liver Model Assay
To investigate liver function using the device, we conducted a drug assay using CPT-11. In this experiment, culture medium including 50 µM CPT-11 was injected into the lower compartment, because CPT-11 is normally administered intravenously at the bedside. To evaluate the metabolism of CPT-11 of only HepG2 cells, we cultured the devices with and without HepG2 cells in the liver chamber, without Caco-2 cells in the small intestine chamber, because Caco-2 cells also have metabolism activity of the same level as HepG2 cells. 35 At 48 h after drug injection, the viability of A549 cells cultured in the lung chamber to model cancer was measured. Figure 6b shows the viability of A549 cells with or without the presence of HepG2 cells, which were used to mimic the metabolic function of the human body. When HepG2 cells were present, the viability of A549 cells against CPT-11 decreased by approximately 25%; notably, the A549 cells showed almost 100% viability when HepG2 cells were not present. CPT-11 is a prodrug that exerts its anticancer effect after hydrolysis by the liver. 27 The findings of the present study suggest that HepG2 cells metabolize prodrugs to provide anticancer effects against cancer cells from other target organs. Although HepG2 cells are also a cancer cell line, the chemical produced by the metabolism of CPT-11 has almost no anticancer effect in liver cancer. 36 Thus, CPT-11 is widely used to treat lung cancer, stomach cancer, and breast cancer but not liver cancer. Our findings therefore show that liver metabolism can be modeled using the presented on-chip organ model.
Combination Assay of Small Intestine and Liver Model
A drug assay using CPA was conducted to evaluate the mutual interaction between small intestine function and liver function. In this experiment, culture medium including 1 mM CPA was injected into the upper compartment corresponding to the lumen side. To evaluate both intestinal barrier function and liver metabolism function, we cultured the device with and without Caco-2 cells on the microporous membrane located in the upper compartment and with and without HepG2 cells in the liver chamber located in the lower compartment. At 48 h after drug injection, the viability of A549 cells cultured in the lung chamber as a cancer model was measured.
Figure 6c shows the viability of A549 cells with or without the presence of Caco-2 cells and with or without the presence of HepG2 cells. CPA exhibited anticancer activity in the presence of HepG2 cells, which implies that it was metabolized by HepG2 cells. In the absence of HepG2 cells, the viability of A549 cells in the presence of the Caco-2 monolayer (68%) was slightly higher than that in the absence of the Caco-2 monolayer (62%). The viability of the A549 cells was over 85% when HepG2 cells were present, regardless of whether Caco-2 cells were present.
CPA is a prodrug that is bioactivated by cytochrome P450 (CYP) 2B6 monooxygenases to provide the anticancer chemical 4-hydroxycyclophosphamide (4OHCP). 28 In clinical therapy, it is typically given orally, and its bioavailability, which is one of the principal pharmacokinetic properties of drugs, has been reported to be approximately 85% to 100%. 37 The ratio of mortality in the presence of the Caco-2 monolayer (32%) divided by mortality in the absence of the Caco-2 monolayer (38%), when HepG2 cells are present, is approximately 84%. Bioavailability of drugs in vivo relates strongly to effects corresponding to cellular mortalities in the cell-based test; therefore, the mortality ratio obtained in the experiment and the bioavailability can be compared. The mortality ratio is similar to the value of its bioavailability reported in the literature. 37 Because bioavailability affects efficacy of drugs directly, it is important to predict a bioavailability corresponding to absorption abilities of an intestine correctly.
Although there is no significant difference in the data of CPT-11 and CPA, a lot of obviously damaged cells were observed on the bright-field images when the viabilities were lower, as shown in Figure 6 . In this study, the living cells were observed by the cell-staining method using calcein-AM, which is one of the most common methods for measuring cellular viability. However, it might be difficult to observe living cells correctly with the device. To obtain more precise results, reactive oxygen species (ROS) or glutathione (GSH) levels of cells have to be measured.
Caco-2 cells have not only an absorption function but also a metabolism function, because the CYP family, which metabolizes drug chemicals, is expressed on Caco-2 cells. 38 It is not a problem in this case because the metabolism in vivo is affected by the liver and colon. Therefore, this example might be a preferable demonstration of showing interactions of organs using the device. The result shown in Figure 6c might include these kinds of interaction.
In the PBPK model, ADME (absorption, distribution, metabolism, and exclusion) processes play an important role in predicting the efficiency of chemicals in vivo. Blood flow pathway and rate and organ volume balance structurally mimicked on the proposed device are responsible primarily for the distribution function. It is well known that chemicals are distributed easily to organs with a high perfusion rate, such as liver, heart, and kidney. On the other hand, it is difficult to distribute chemicals to organs with a low perfusion rate such as muscle, fat, and peripheral organs. In addition, the rates of absorption and metabolism processes depend heavily on organ volume and dimensions. This means that replicating physiological parameters such as blood flow pathway and volume ratio of each organ is quite valuable for the PBPK model. Thus, the concept proposed in this study might become operative, especially when the exclusion process and other organs are integrated into the microfluidic device.
On the other hand, the device still has limitations, such as number of organs, the physiological ratio of cell number and liquid volume, and cellular activities, to replicate the human body perfectly. It is possible that the activity necessary to metabolize prodrugs is lower in HepG2 cells than in physiological hepatocytes, even though HepG2 cells have been widely used for in vitro screening. 39 We had planned on using HepG2 cells to study the feasibility of the device. In this study, the model chemicals were carefully chosen because it was known that HepG2 cells can activate them.21,40 However, to apply the device to a much wider range of chemicals, it is better to use primary human hepatocytes to model liver function in the device. Recently, human induced pluripotent stem cell–derived hepatocytes and other organ cells have become available for cell-based assays. 41 When the human cells with high activities will be easily available and the number of organs will increased on the device, to mimic structurally physiological parameters proposed in this study will be important.
In this study, we proposed a novel concept of a microfluidic device as a multiorgan model that could mimic physiological circulation and organ volume ratio for in vitro investigation of pharmacokinetics. The device was able to coculture Caco-2 cells, HepG2 cells, and A549 cells for more than 3 days, and it revealed the importance of small intestine and liver function when testing anticancer drugs. This is the first demonstration of such a top-down approach to biological modeling using a microfluidic device with a structurally mimicking physiological parameter with on-chip perfusion functionality.
In future studies, we will integrate multiple functions of important organs, such as the kidney, stomach, and pancreas, to replicate the ADME of drug chemicals. To realize higher metabolism function on the device, human primary hepatocytes or human induced pluripotent stem cell–derived hepatocytes should be cultured instead of HepG2 cells. Sequential measurements of cellular activity are required for the pharmacokinetics, although only the data measured at the end point of the experiment were shown in this study. To satisfy this requirement, it is effective to integrate microsensors, which are able to measure cellular conditions, into the device. 42 Parallelization and automation of the devices could be possible with microfabrication technology. The enhanced physiological relevance of this system will promote detailed study of complex biological systems and may be used not only as an alternative to animal testing but also to obtain parameters for in silico modeling. The automated system might be a powerful assay platform in the fields of medical research, drug discovery, and life science.
Footnotes
Acknowledgements
We thank Mr. S. Segawa and Ms. H. Nakamura for assistance in experiments.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by JSPS KAKENHI Grant-in-Aid for Young Scientists (B) and JST CREST.