
Abstract
Luminescence is characterized by the spontaneous emission of light resulting from either chemical or biological reactions. Because of their high sensitivity, reduced background interference, and applicability to numerous situations, luminescence-based assay strategies play an essential role in early-stage drug discovery. Newer developments in luminescence-based technologies have dramatically affected the ability of researchers to investigate molecular binding events. At the forefront of these developments are the nano bioluminescence resonance energy transfer (NanoBRET) and amplified luminescent proximity homogeneous assay (Alpha) technologies. These technologies have opened up numerous possibilities for analyzing the molecular biophysical properties of complexes in environments such as cell lysates. Moreover, NanoBRET enables the validation and quantitation of the interactions between therapeutic targets and small molecules in live cells, representing an essential benchmark for preclinical drug discovery. Both techniques involve proximity-based luminescence energy transfer, in which excited-state energy is transferred from a donor to an acceptor, where the efficiency of transfer depends on proximity. Both approaches can be applied to high-throughput compound screening in biological samples, with the NanoBRET assay providing opportunities for live-cell screening. Representative applications of both technologies for assessing physical interactions and associated challenges are discussed.
Introduction
Early drug discovery stages often use biophysical methods for target-based screening using purified targets to characterize the disruption of binding interactions by chemical compounds. There are several ways to measure direct interactions between a target and a compound. These include methods based on surface plasmon resonance, thermal shift assay, nuclear magnetic resonance, and calorimetry (isothermal titration or differential scanning). These methods’ weaknesses and limitations are often low throughput, a requirement for purified protein targets, low sensitivity, and high noise. The nano bioluminescence resonance energy transfer (NanoBRET) and amplified luminescent proximity homogeneous assay (Alpha) technologies are two complementary techniques. They have become a mainstay for developing assays to interrogate protein-protein and protein–small molecule interactions in our laboratory. Both methods use luminescence energy transfer between a proximal donor and acceptor to reduce background interference and improve the signal to noise. The approaches use different photon-generating donor and acceptor pairs; NanoBRET uses a luciferase/luciferin and fluorophore pair, whereas Alpha technology uses donor and accepter latex beads to generate and detect singlet oxygen, respectively. The assay principle for detecting inhibitors of molecular interactions is the same in both cases, in which a loss of energy transfer accompanies the disruption of an interaction between two binding components. Both methods are amendable to high-throughput screening (HTS) and exhibit high sensitivity (e.g., up to 100-fold more sensitivity than fluorescence 1 ) with low spectroscopic interference from compound libraries. 2 Moreover, these assays can easily be miniaturized; they are robust and applicable to complex formats, such as cell lysates. Moreover, the application of the NanoBRET assay to quantify molecular interactions in real time within intact cells represents an essential benchmark for preclinical studies. Details on the two methods will be presented in this short review.
NanoBRET
Bioluminescence resonance energy transfer (BRET 3 ) is similar to Förster or fluorescence resonance energy transfer (FRET) and involves resonance energy transfer between a luminescent donor (luciferase) and a fluorescent acceptor ( Fig. 1 ). Because the donor emits light via enzymatic oxidation of a substrate, external excitation is not required. It allows BRET to avoid typical FRET technology issues such as photobleaching, autofluorescence, and high background. Homogeneous time-resolved fluorescence (HTRF), a method that combines standard FRET with time-resolved (TR) measurements, has been developed to overcome some of the challenges facing traditional FRET. HTRF methods have been reviewed elsewhere.4,5 Luminescence generated by a donor luciferase is transferred to an acceptor to induce fluorescence only when two components are within an appropriate distance (<10 nm). 6 The distance required for the resonance energy transfer corresponds to the dimensions of most biological interactions, making BRET technology ideal for investigating biomolecular interactions in a precise manner. 3 Like FRET, BRET measures the ratio of the acceptor’s and donor’s emissions. Thus, the selection of donor, acceptor, and substrate is of considerable importance for assay design. Since its emergence in 1999, several BRET platforms have been developed, and comprehensive summaries of BRET configurations have been reviewed.7,8 Details of methodology, recent variants, protocols for setting up BRET-based screens, and a few examples were also reviewed. 9 The traditional BRET platforms relies on adenosine triphosphate (ATP)–independent luciferase (e.g., RLuc8) as the energy donor, a green fluorescent protein (GFP) variant (e.g., Venus) as the fluorescent acceptor, and the substrate coelenterazine h. The relatively large size of the luciferases potentially places some limitations on the approach. For example, proteins fail to bind to appropriate binding sites of fused receptors,10,11 and G protein–coupled receptors (GPCRs) tagged with RLuc fail to traffic to the plasma membrane. 12
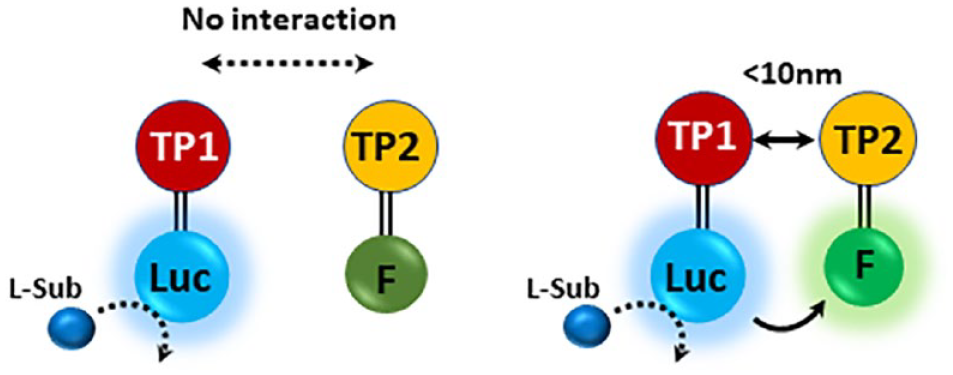
Schematic illustration of bioluminescence resonance energy transfer (BRET) technology. Target protein 1 (TP1) and target protein 2 (TP2) are interacting proteins 1 and 2, tagged with luciferase (Luc) and a fluorescent protein (F), respectively. When the two proteins 1 and 2 are proximal (<10 nm), luminescence produced by luciferase in the presence of luciferin (L-Sub) can be transferred to the fluorescent protein to produce fluorescence.
Characteristics of NanoBRET
NanoBRET, 10 a recent BRET platform, uses NanoLuc (NLuc), a small engineered luciferase (19 kDa). NLuc was developed from the Oplophorus gracilirostris luciferase that catalyzes the oxidation of the imidazopyrazinone substrate, furimazine. Compared with the RLuc, NLuc produces exceptionally bright luminescence light (~150-fold greater) with a slightly blue-shifted (~20 nm) and narrower (~20%) emission spectrum, resulting in high spectral resolution. Its bright luminescence supports high BRET sensitivity and facilitates energy transfer to a broad range of fluorophores and good spectral resolution between donor and acceptor emissions. Furimazine also allows NLuc to be assayed in mammalian cells. Because of its small size and excellent spectral characteristics, NLuc has replaced other luciferases in many BRET platforms. These properties have led to the development of new applications 13 that include assessing gene regulation,14-16 protein stability, 17 and the evaluation of protein-protein interactions (PPIs) in real time at endogenous expression levels in ways that have not been possible before.18,19 NanoBRET technology provides an opportunity to design assays for a broad range of molecular interactions. The key to the approach is to bring a suitable acceptor fluorophore into the proximity of NLuc. The acceptor can either be a small molecule (sometimes called a tracer) or a fluorescent protein, intrinsically fluorescent or rendered fluorescent by appending a fluorophore.
Measuring PPIs Using NanoBRET
NanoBRET has been used to monitor interactions between various protein partners in different cellular compartments with examples including membrane proteins (e.g., GPCRs20,21 and EGFR/GRB222), signaling protein kinases (e.g., KRAS/BRAF 23 ), or transcription regulators (cMyc/MAX 24 ). Typically, luciferases have been introduced through the exogenous expression of a fusion protein. Still, more recently, they have been inserted into endogenous genes using CRISPR/Cas9. 25 For example, NLuc was inserted into the endogenous CXCR4 gene (CXC chemokine receptor 4), which is known to be expressed at low levels (~20 fmol/mg of membrane protein 26 ) in HEK293FT cells. 27 A time-dependent BRET signal was observed in cells transiently expressing a β-arrestin 2–Venus fusion protein (β-arr2/Venus). The authors also demonstrated that the ligand-dependent CXCL12 recruitment of β-arrestin 2 to CXCR4 is dose dependent, and the CXCR4 antagonist (AMD3100) reduced the BRET signal.
The HaloTag28,29 (HT) fusion protein, a self-labeling protein, has been used as an alternative to GFP-based acceptors in BRET experiments ( Fig. 2 ). The HaloTag protein is a 33-kDa protein that can be genetically fused to a protein of interest. Its genetically modified active site is designed to specifically bind an exogenously added chloroalkane ligand under physiological conditions to form an irreversible covalent binding. Because of multiple capabilities using a single genetic construct, HaloTag has emerged as a versatile platform in biomedical research. The pairing of NLuc with a red-shifted HaloTag acceptor exhibits minimal spectral overlap, excellent signal to noise, and dynamic range. 30 It represents a perfect pairing with the potential for studying a wide range of PPIs.
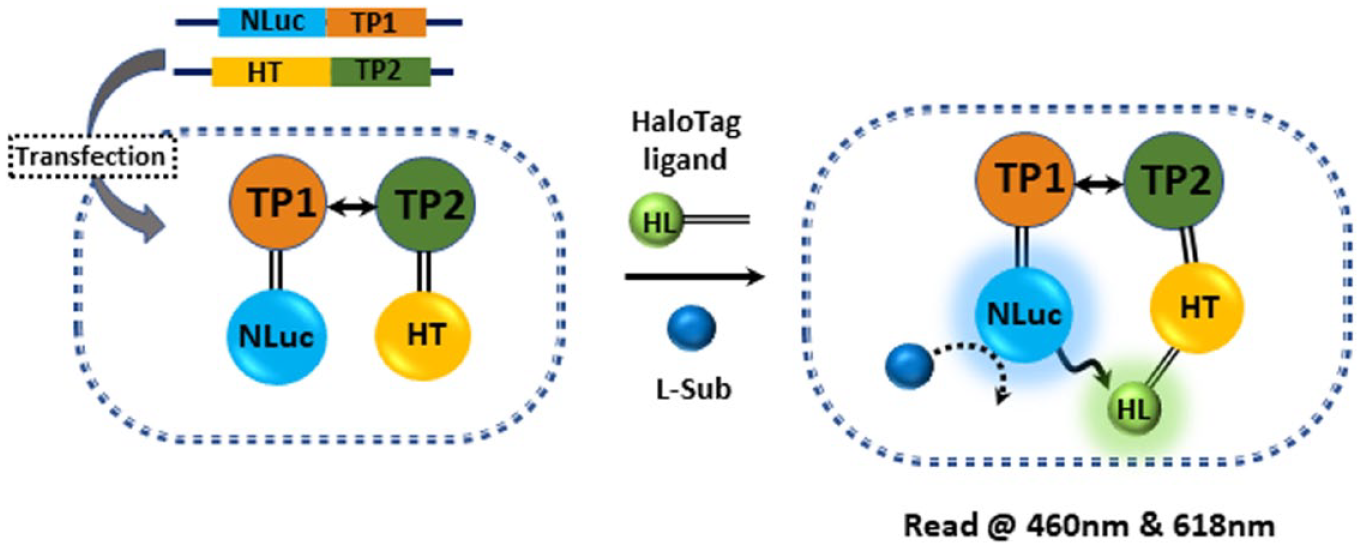
Schematic illustration of NanoBRET with HaloTag (HT) ligand. The addition of NanoBRET HaloTag 618 Ligand (HL) and NanoBRET NanoLuc Substrate (L-Sub) to the cell where target protein 1 fusion (TP1-NLuc, energy donor) and a fluorescently labeled HaloTag-target protein 2 fusion (TP2-HT, energy acceptor) are expressed induces resonance energy transfer from NLuc to HL labeled on a HaloTag.
Applying NanoBRET to High-Throughput Small-Molecule Screening
NanoBRET is adaptable to a high-throughput format and has been used to aid in the development of inhibitors of Bruton’s tyrosine kinase (BTK), 31 bromodomains, 32 the adenosine A3 receptor, 33 and salt-inducible kinases. 34 Some other examples include a study by Pavlinov et al., who used an NLuc/HaloTag strategy to assess Beclin 1–ATG14L interactions. The Beclin 1–ATG14L interaction is a prerequisite for forming the VPS34 initiation complex (VPS15-VPS34-ATG14L-Beclin 1), which is required to activate autophagy. 35 Using this approach, Pavlinov et al. successfully screened a set of 2560 molecules to identify inhibitors blocking the PPI. 36 Sakyiamahet et al. 37 developed a NanoBRET-based protein:ligand interaction platform to identify CXCR4 binders in living cells using TAMRA-Ac-TZ14011, a fluorescent-labeled CXCR4 antagonist, as a BRET acceptor of bioluminescent energy from N-terminally fused NLuc-CXCR4 stably expressed in CHO cells.
Target Engagement Using NanoBRET
A significant challenge in drug discovery is to quantitatively characterize the binding between a candidate inhibitor/modulator and its target in living systems (i.e., target engagement [TE]). This is usually due to the reliance on indirect measurements, such as monitoring the effect on a reporter gene or a cellular process such as phosphorylation. In such cases, the assay’s dose dependency often reflects many compensatory events and requires considerable interpretation. Other factors can also obscure the analysis, such as the compound’s metabolic instability or low cell permeability. The ability to monitor TE in the native state in cells, where the target may be posttranslationally modified or bound to other biomolecules, or small-molecule metabolites, represents a critical benchmark for preclinical drug discovery.
NanoBRET-based TE uses a cell-permeable fluorescent tracer to monitor the binding of compounds to a target protein fused to NLuc. Competition between the tracer and test compound can provide a quantitative analysis of TE. A tracer is typically a known ligand for a target with an appended BRET acceptor dye. The tracer should bind reversibly to a target NLuc-fusion protein to produce resonance energy transfer in the absence of a drug. If a compound can bind and compete with the tracer, its displacement results in reduced energy transfer ( Fig. 3 ). A ratiometric readout of the resulting spectrum (Eq. 1) generates a BRET ratio and offers flexible solutions for measuring binding events.
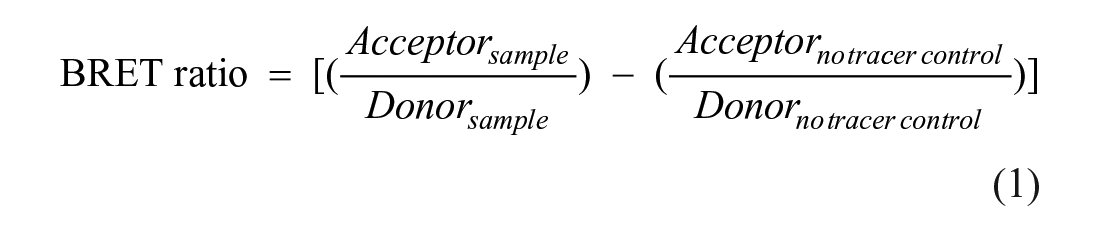
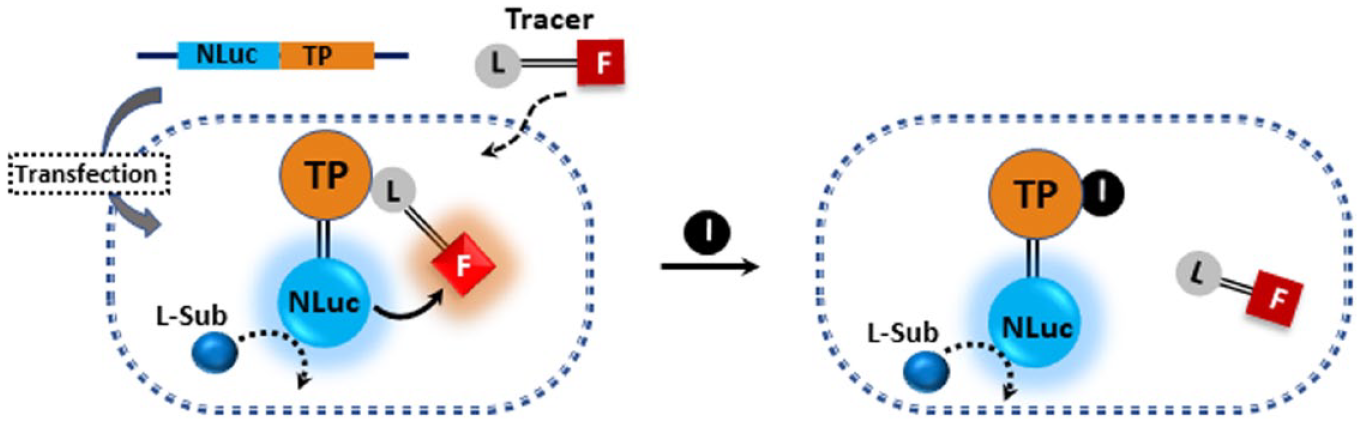
Principle of NanoBRET target engagement. A cell-permeable tracer as a known inhibitor (or a tool molecule, L) conjugated to a fluorescent dye (F) binds to a target protein (TP) to produce energy transfer from NanoLuc luciferase-target protein fusion (NLuc) in cells to a fluorescent dye. The assay includes the following steps: (1) Expression of an NLuc-fused target to produce blue luminescence in the presence of luciferin (L-Sub). (2) Addition of an optimized concentration of red-tagged tracer binds to the target to induce BRET. (3) The addition of a test compound (I) to induce the tracer’s displacement and increase in the blue/red ratio.
Measuring protein kinase inhibitor TE using NanoBRET
The NanoBRET platform represents a valuable tool for guiding medicinal chemistry programs during the development of lead or preclinical candidate compounds, particularly kinase inhibitors in which the intracellular target selectivity is particularly challenging because more than 500 kinases possess a highly conserved active site. Biochemical assays are an essential component of drug discovery. However, they cannot accurately predict TE in living cells for several reasons, such as poor cell permeability, molecular complex formation, or posttranslational modifications. Vasta et al. 38 demonstrated the potential of NanoBRET technology for profiling clinically relevant inhibitors against kinases in live cells. 38 Using six different fluorophore-labeled kinase inhibitors as tracers, they established a significant discrepancy between the observed selectivity and affinity compared with biochemical analysis. However, they showed a good correlation between the cellular TE analysis in HEK293 cells and the published cellular potency values determined by phospho–enzyme-linked immunosorbent assay (ELISA), supporting the better agreement between assays measuring cellular TE and intracellular kinase activity. Profiling crizotinib and dasatinib revealed a narrower selectivity profile than predicted from the biochemical analysis. The NanoBRET assay also showed the effect of subcellular ATP concentration on TE. In addition to evaluating the affinity of a compound for a cellular target, it was also possible to assess its residence time, defined as the inverse rate constant for dissociation. To measure the residence time, the NanoBRET assay started with the test compound’s addition at a saturating concentration to allow binding to equilibrate fully. The assay was then moved to a nonequilibrium condition by washing out the unbound compound, followed by a tracer addition in a dose-dependent manner. Lastly, the employment of a DNA plasmid as a source for each cellular kinase eliminated the burden of protein purification, contributing to the development of a relatively expandable and straightforward platform that could profile a broad range of kinases in a cellular context. Given the availability of an impressive array of well-characterized kinase inhibitor scaffolds, 39 assays for most human and pathogen protein kinases are expected to be developed. Promega offers a range of reagents for conducting NanoBRET TE kinase assays for more than 100 kinases in live cells.
Assessing other TE interactions using NanoBRET
NanoBRET technology has also been employed to monitor responses within the GPCR signaling cascade, 11 including interactions involving G proteins, GPCR kinases, and β-arrestins. 12 In vivo visualization and quantitation of ligand binding to GPCRs was demonstrated by localizing the β2-adrenoreceptor with NLuc in a mouse model of breast cancer using a fluorescent derivative of propranol. 40 In addition, quantitative analysis of intracellular TE has been shown for a panel of histone deacetylase inhibitors 41 and BET family bromodomains. 41 As noted above, the ability to perform real-time measurements in intact cells is an essential element of this approach, augmented by a reliable ratiometric readout that alleviates potential assay interferences such as variation in cell number or expression level between samples. A disadvantage of the approach is the requirement for the exogenous expression of a fusion protein, disrupting the protein’s normal regulation. However, the advent of endogenous tagging 42 promises to open up the approach to endogenous proteins.
Recently, NanoBRET-based TE has been used for real-time monitoring of proteolysis-targeting chimera (PROTAC43–45)–induced enzymatic processes. PROTACs are heterobifunctional small molecules composed of a linker and two ligand moieties: one binds to the target protein, while the other recruits an E3 ligase. PROTACs can remove target proteins by inducing selective intracellular proteolysis via ternary-complex formation, target ubiquitination, and degradation, which has emerged as an alternative targeted therapeutic strategy. Riching et al. 42 reported a live cell platform that used NanoBRET and NanoBit (NanoLuc Binary Technology) to monitor the formation and stability of multiple complexes within a PROTAC pathway. For example, ternary-complex formation induced by a PROTAC can be measured using a HaloTag fusion protein of an E3 ligase conjugated to the NanoBRET 618 fluorescent ligand ( Fig. 4A ). A similar arrangement also allows for ubiquitination to be measured ( Fig. 4B ). A recent example demonstrates the utility of using some of these approaches to investigate the role of covalency in the degradation efficiency of a PROTAC for BTK. 46
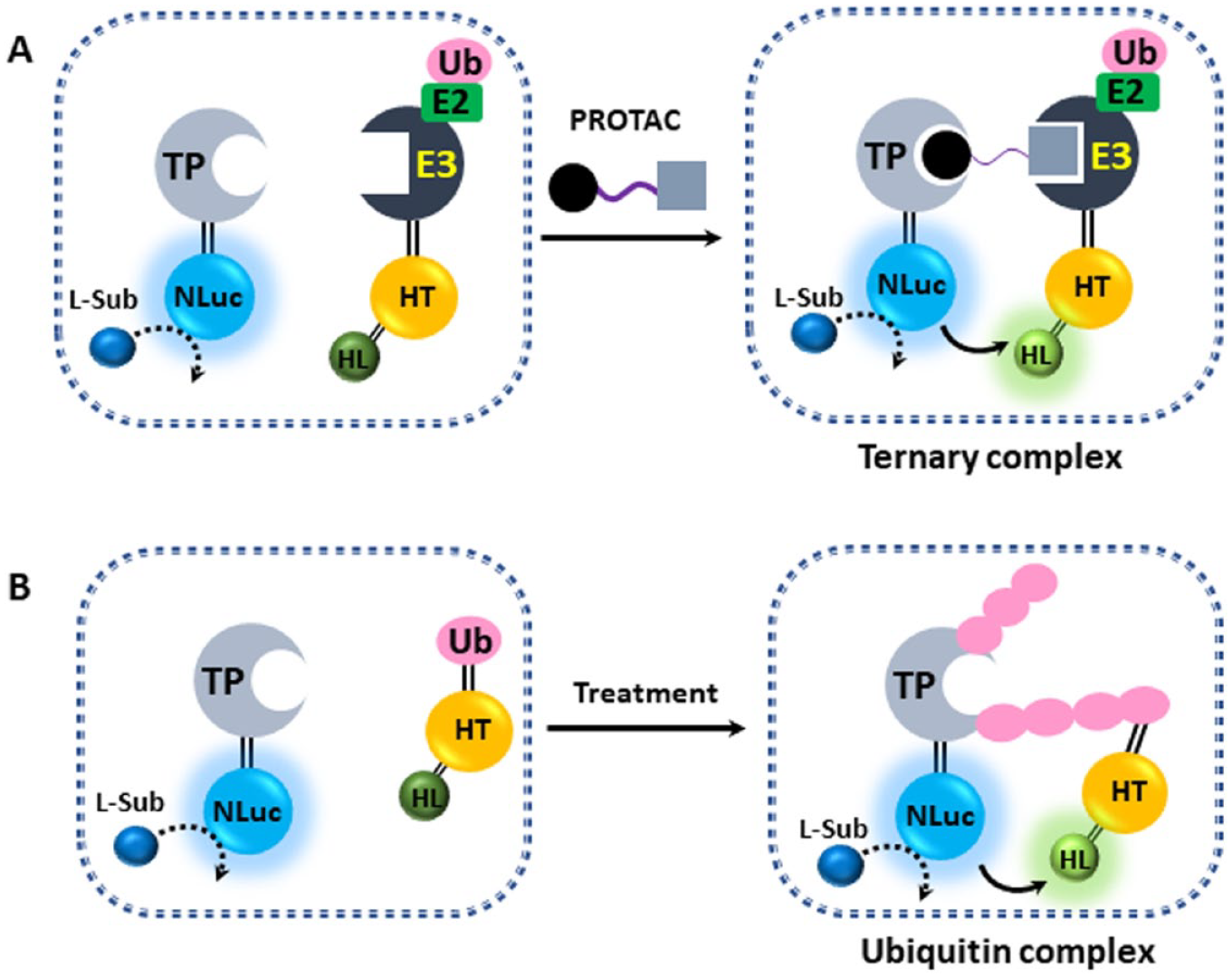
Application of NanoBRET for real-time monitoring of the proteolysis-targeting chimera (PROTAC) degradation pathway. (
Some Considerations When Using NanoBRET
Although NanoBRET-based TE has been extensively developed for kinase profiling, it should be noted that not every protein will tolerate an NLuc fusion. In addition, developing well-characterized ligands for additional protein families will require substantial resources and efforts. For example, labeling known inhibitors with fluorescent dyes and an appropriate linker may induce a dramatic increase in the molecular weight and complexity, particularly for smaller inhibitors, resulting in a shift in their affinity. Baker et al. 47 evaluated the pharmacologic properties of a range of fluorescent ligands against the human adenosine A1 receptor along with the associated linker. They found significant differences in their binding properties, dependent on several factors, including the linker’s position, the linker length, and the fluorophore. While BODIPY 630/650 derivatives were well behaved, it was emphasized that the fluorophore/linker pair and the environmental sensitivity of the resulting fluorescence are essential for the proper utilization of the tracers. Vernall et al. 48 demonstrated the linker’s importance, showing improved selectivity and affinity of the nonselective GPCR adenosine receptor antagonist xanthine amine congener by inserting and optimizing a dipeptide linker between the adenosine receptor pharmacophore and the fluorophore.
Currently, the most versatile fluorescent dye used in NanoBRET tracer design is the BODIPY dye, due to the commercial availability of multiple BODIPY dye variations with a wide excitation/emission spectral ranges and its favorable properties such as high fluorescence and absorption levels, good solubility in a range of solvents, and relative insensitivity to changes in polarity and pH.49,50 Because BODIPY is relatively nonpolar, the tagged ligand’s functional properties are less affected, making BODIPY the most favorable dye for tracer design. Nonetheless, thoughtful consideration of fluorophore dye/linker pair and target environment and choice of the ligand is essential for NanoBRET-based target and ligand binding interrogation.
Alpha Technology (Amplified Luminescence Proximity Homogenous Assay)
Perkin Elmer’s Alpha technology uses the nonenzymatic channeling of singlet oxygen species generated from a photo-excitable latex bead (donor bead) to a second latex acceptor bead in proximity to induce a chemiluminescent signal ( Fig. 5 ). Since Ullman and colleagues’ first development, 51 this technology has empowered the development of robust proximity-based assays to monitor interactions between molecules over a more significant distance than BRET (compare the maximum distances of 200 nm for Alpha technology to ~10 nm for the BRET approaches). Because many singlet oxygen molecules are released per binding event, the signal is greatly amplified, allowing the detection of low-picomolar concentrations of analytes.

Schematic illustration of a representative AlphaScreen/AlphaLISA and its advantages. Biotin-tagged target protein 1 (TP1) binds to streptavidin-coated donor bead, and His-tagged target protein 2 (TP2) binds to Ni-chelated acceptor bead. The interaction between TP1 and TP2 brings the donor and acceptor beads in proximity, allowing singlet oxygen transfer to generate luminescence at 520 to 620 nm (AlphaScreen) or 615 nm (AlphaLISA).
Characteristics of Alpha Technology
Alpha technology comes in two flavors, AlphaScreen and AlphaLISA, with the main difference being the acceptor bead’s emission spectrum. AlphaScreen beads emit over a broad range (520–620 nm), whereas AlphaLISA beads emit at 615 nm. Because of the narrow emission spectrum of AlphaLISA beads, there is much less interference in serum and plasma samples, resulting in improved sensitivity. This sensitivity difference favors the AlphaLISA approach as a good alternative for more complicated ELISA or in-cell Western assays. The donor beads irradiate light at a higher wavelength (680 nm) than the emission wavelength does, which reduces the background significantly, resulting in a high signal-to-background ratio (S/B). Widely used recognition pairs are biotin-streptavidin and pairs based on protein affinity tags, such as 6xHis, Strep, Fc, FLAG, or GST. This flexibility in bead conjugation has enabled its versatile applications, as reviewed by Eglen et al. 52
Measuring PPIs using Alpha technology
The AlphaScreen was the first biophysical technology to detect GPCR heteromerization in pathological human postmortem brains, although other biophysical methods were demonstrated in animal models. 53 Fernandez-Duenas et al. 53 successfully monitored adenosine A2A–dopamine D2 receptor (A2AR/D2R) heteromeric formation in caudate from healthy and Parkinson’s disease subjects with antibodies against A2AR and D2R. Those antibodies were labeled with donor and acceptor beads to induce a singlet oxygen energy transfer dependent on A2AR/D2R complexation. Not only did the assay reveal the existence of GPCR oligomers, but it also demonstrated its potential for HTS with excellent S/B and Z′ of 12.5 and 0.98.
Applying Alpha technology to high-throughput small-molecule screening
The Alpha beads offer excellent stability, superior sensitivity, mix-and-read homogeneous format, and easy automation/miniaturization. Clear advantages are summarized in Figure 5 , indicating why the Alpha technology is one of the primary detection platforms commonly used in HTS applications. There are numerous reports of AlphaScreen/AlphaLISA-based HTS for inhibitor identification that block the interactions between biomolecules, such as protein:protein, 54 protein:peptide, 55 protein:DNA, 56 and protein:RNA. 42 For example, the interaction between the malaria parasite proteins, Apical Membrane Antigen 1 (AMA1) and Rhoptry Neck Protein 2 (RON2), is essential for parasite invasion into red blood cells. A 1536-well plate format assay was designed with a biotinylated RON2L (RON2 peptide) to mimic RON2, which binds streptavidin donor beads, and AMA1 His-tag, which binds nickel chelate acceptor beads. A pilot screen against more than 21,000 small molecules demonstrated strong assay robustness (Z′ = 0.7) and revealed the first small-molecule inhibitor for this interaction. 54 Another example was targeting the heterodimerization of the transcription factors Runt-related transcription factor 1 (RUNX1) and core binding factor β (CBFβ), which is critical for CBF leukemias’ pathogenesis. 57
TE using Alpha technology
The Cellular Thermal Shift Assay (CETSA) from Pelago Bioscience (Solna, Sweden) has emerged as a powerful quantitative label-free TE technique for applications in physiologically relevant environments since its inception in 2013. Although the biophysical principle of CETSA is similar to traditional thermal shift assays, in which specific proteins are denatured over a small temperature range, the readout is not protein unfolding. Instead, CETSA measures the residual protein surviving the heat shock and can be applied to complex systems such as cells and lysates. Critical advantages of CETSA are that it is label free (no requirement of chemical modification of the compound of interest) and readily adaptable to microplates.
Alpha technology has been successfully integrated with CETSA to translate TE events to readable signals using a dual antibody-based proximity system to detect an intact protein. In a representative Alpha-CETSA assay ( Fig. 6 ), a biotinylated anti-analyte antibody binds to streptavidin-coated Alpha donor beads, whereas another anti-analyte antibody is conjugated to streptavidin-coated AlphaLISA acceptor beads. In the presence of the analyte (e.g., a protein of interest), the beads come into proximity. The amount of light emission is directly proportional to the amount of soluble folded protein present in the sample. A thermal melting curve is prepared using known positive and negative controls to identify a specific temperature set point at which compound-stabilized protein target stays in the supernatant. The absence of compound binding leads to protein aggregation/precipitation. The compound-stabilized protein in solution is then detected with two different antibodies that recognize distinct epitopes on the same folded protein. Each antibody is labeled either with acceptor or donor beads, respectively, allowing homogenous Alpha signal generation after the heating step. The assay involves an incubation step in which cells are treated with compound(s) of interest. Heat is then applied to denature the sample proteins, assuming that the ligand-bound protein will have a different melting temperature from the control protein. The remaining soluble target protein in the lysed cells is evaluated using specific antibodies labeled with Alpha beads. PerkinElmer (Waltham, MA) offers an Alpha CETSA TE toolbox assay kit containing anti-mouse immunoglobulin G (IgG) donor and anti-rabbit IgG acceptor beads, which enables the rapid, sensitive, and quantitative detection of a target protein that remains soluble after heat treatment of compound-treated cells without any need for separating soluble and aggregated protein (no washing and no separation steps).
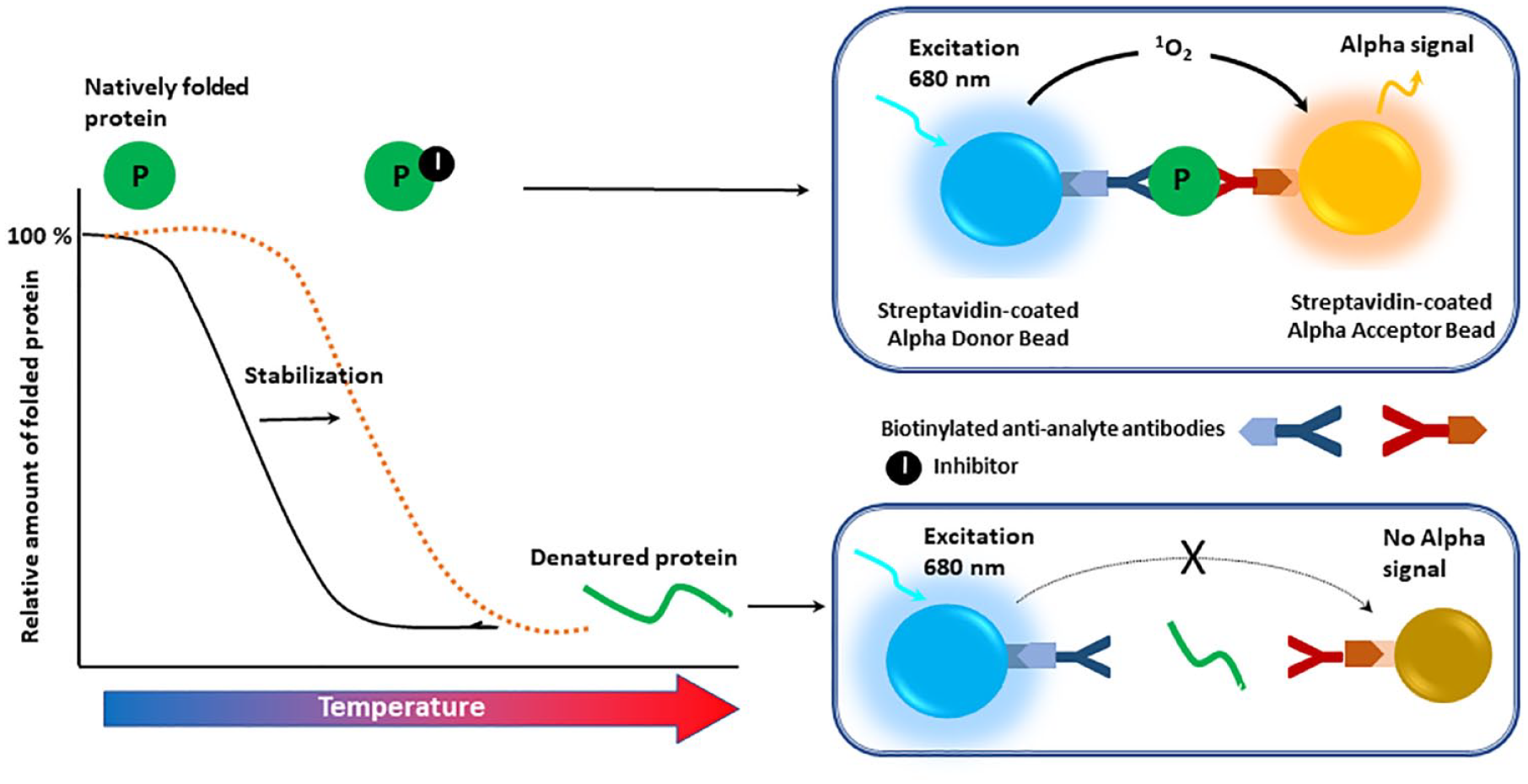
Schematic illustration of a representative Alpha–Cellular Thermal Shift Assay. A biotinylated anti-analyte antibody binds to the streptavidin-coated Alpha donor beads, while another anti-analyte antibody is conjugated to AlphaLISA acceptor beads. In the presence of the analyte (natively folded protein, P), the beads come into proximity. The amount of light emission is directly proportional to the amount of soluble protein present in the sample. Inhibitor binding to the target stabilizes the protein from denaturation, shifting the melting temperature to the right.
High-Throughput Alpha CETSA Assays
Because of the advantages of Alpha technology (no-wash, homogeneous, high sensitivity, a broad dynamic range, and fast time to results), Alpha-HT–CETSA enables the detection and screening of targets of interest in a reliable, high-throughput format using its homogeneous assay platform. Alpha-HT–CETSA in a microtiter plate format was first demonstrated using the human protein kinase p38α (MAPK14) in live HL60 cells. 58 As a pilot study, the readouts from Alpha-CETSA measured directly in solution were compared with a traditional Western blot that involved sample workup for two well-characterized inhibitors in a 96-well format. The authors confirmed a good correlation of both Tm and EC50 values between the two detection methods. Encouraged by these results, Alpha-HT–CETSA was extended to a compound screening against human thymidylate synthase (TS), 59 performed with live K562 cells seeded into 384-well PCR plates. More than 10,000 compounds were robustly screened in a day to identify 65 stabilizing compounds, followed by time-dependent TE and hit validation in parallel using isothermal dose-response fingerprint.
Further HTS applications include screening for direct AR binders in a prostate cancer cell line endogenously expressing AR, 60 a key driver in prostate cancer. Following optimization, an AlphaScreen assay was used to quantify soluble thermostable AR, establishing a robust assay statistic (Z′ = 0.54–0.74). The assay identified known AR antagonists and agonists and provided apparent intracellular Ki values. Another application was demonstrated against two clinically validated oncology targets, BRAF and PARP1 (poly[ADP-ribose] polymerase1). 61 Here, the authors examined a set of 896 kinase inhibitors and found 13 BRAF stabilizers. The authors also looked at a group of 6288 compounds identified from a biochemical fluorescence polarization (FP) assay against PARP1. They found that 81% of the compounds showed >50% stabilization activity, suggesting a strong correlation between FP and Alpha-HT–CETSA. The authors showed that 13 compounds that are not active in Alpha-CETSA are also inactive in a cellular PARylation assay, highlighting the potential for assessing the cell permeability of lead compounds. Due to the synergistic strengths of Alpha and CETSA technologies, to date, all report of HT-CETSA measuring endogenous proteins in cellular lysate used Alpha technology as reviewed by Henderson et al. 62 HT-CETSA assays (many of which are Alpha-HT–CETSA) have been successfully applied to more than 30 targets, including targets localized to the nucleus, the mitochondria, the cytoplasm, and the plasma membrane (https://www.perkinelmer.com/lab-products-and-services/application-support-knowledgebase/alpha-cetsa-knowledgebase.html).
Some Consideration When Using Alpha Technology
Although suitable for developing sensitive and robust assays for HTS, 63 Alpha technology exhibits several limitations. The first is the light sensitivity of the beads, in particular, the donor beads. Short-term exposure to indirect ambient light (e.g., while pipetting) is usually not an issue if precautions are followed to keep bottles and tubes wrapped in foil, plates covered with black lids or aluminum seals, and the avoidance of bright light. Because of the light sensitivity, assays can be read only once, permitting endpoint assays only. To reduce assay costs, many labs have focused on miniaturization rather than the Alpha reagents’ scaling. Veloria et al. 64 demonstrated that reducing alpha beads used for the AlphaScreen assay in conjunction with minor reoptimization provides a robust and cost-effective alternative to traditional AlphaScreen methods without miniaturization or reduction in assay volume. This new platform was suitable for HTS and saved up to 90% on assay detection costs over the manufacturer’s protocol. 64 Lastly, while AlphaScreen is tolerant to spectroscopic interference, compounds can still interfere by quenching the signal directly. Reactive compounds or chelators can also inhibit by quenching or reacting with the singlet oxygen.65,66 Therefore, orthogonal assays to discriminate assay interference should be used. One typical counter screen uses TruHits, marketed by PerkinElmer, to identify false hits that quench singlet oxygen.
For the Alpha-HT-CETSA application, a ligand interaction with the target protein must drive a detectable thermal stability change. If the compound binds to only one domain of a multidomain protein, it can go undetected if it does not globally affect its thermal stability. Therefore, before developing Alpha-CETSA assays, the ability of CETSA to report binding should be accessed from resources such as those available at Pelago Bioscience (https://www.pelagobio.com/), who has access to an extensive thermal stability database. In addition, one of the biggest challenges is loss of antibody recognition upon ligand binding to the target protein, inducing the suppression of antibody recognition and loss of signal.58,59,67 Therefore, careful selection of antibodies using proper controls is required to ensure antibody binding to the protein and produce a robust signal in Alpha-CETSA assay.
Conclusions
Because of the high sensitivity and a range of technologies, luminescence-based technologies are expected to play an essential role in future drug discovery paradigms, from hit identification to lead validation. NanoBRET and Alpha technologies can be used to provide important insights into TE in live cells at an early stage of the drug discovery process. Because both technologies do not require purified enzymes, they can be easily automated and miniaturized for cost-effective and time-efficient high-throughput evaluation of preclinical drug candidates. However, the two technologies also have unique features and limitations, as summarized in Table 1 and described above, and must be used carefully.
Comparison of NanoBRET and Alpha Technologies.
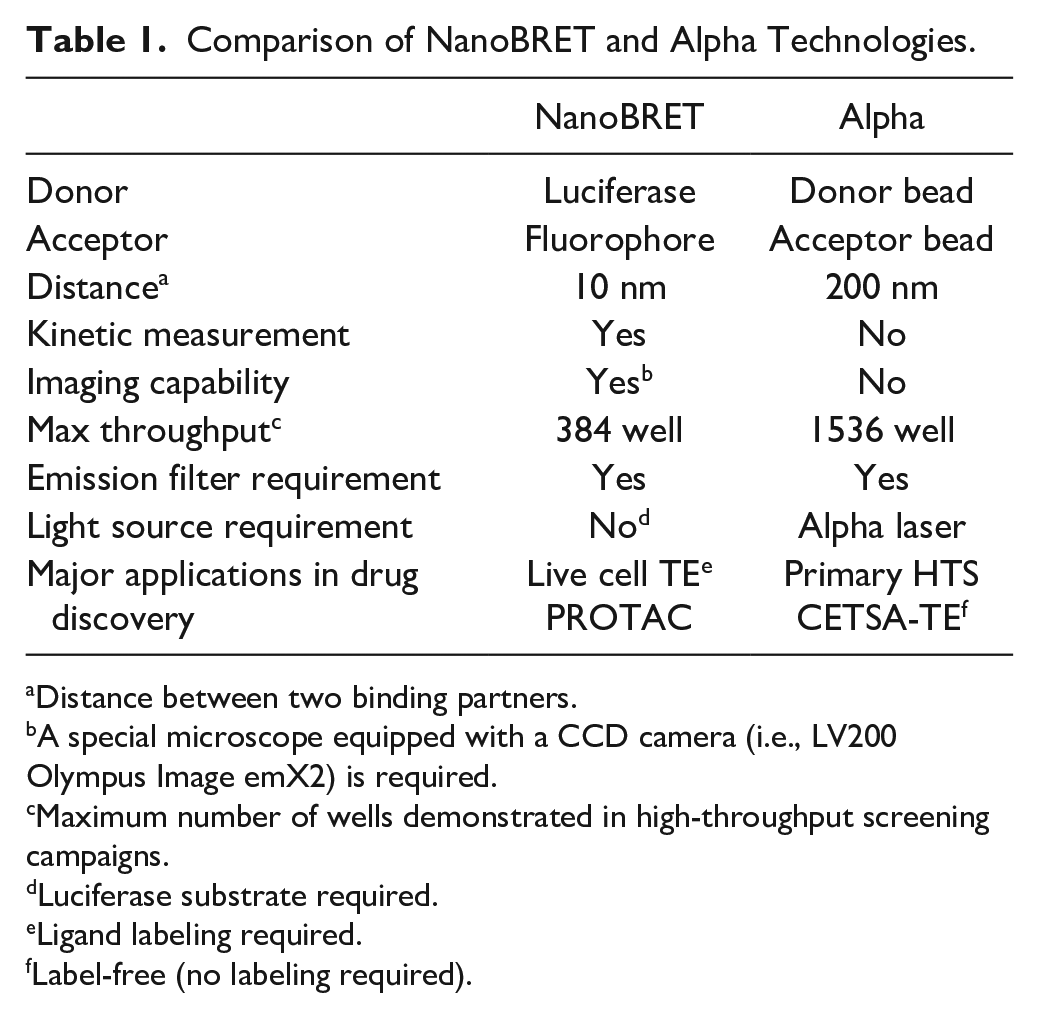
Distance between two binding partners.
A special microscope equipped with a CCD camera (i.e., LV200 Olympus Image emX2) is required.
Maximum number of wells demonstrated in high-throughput screening campaigns.
Luciferase substrate required.
Ligand labeling required.
Label-free (no labeling required).
NanoBRET technologies have been used widely to monitor various ligand-binding, intracellular signaling, receptor-receptor proximity, and receptor trafficking events in live cells. BRET can potentially be used to monitor the proximity of any protein or ligand if appropriate fusion constructs and fluorophore conjugates can be produced. Furthermore, given recent advances in the properties and utilities of a range of luciferase/luciferin systems, the next few years promise to be an exciting time for the application of NanoBRET in conjunction with CRISPR/Cas9 endogenous tagging to examine a range of molecular interactions in cells with high responsiveness and spatial resolution.
Footnotes
Acknowledgements
The authors appreciated the financial support from the Cancer Prevention and Research Institute of Texas (CPRIT, RP160657) and the Welch Foundation (F-1390). The authors also thank scientists at Promega and Perkin Elmer for their kind consultations on various biophysical applications.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: the authors appreciate the financial support from the Cancer Prevention and Research Institute of Texas (CPRIT, RP160657) and the Welch Foundation (F-1390).