
Abstract
Hypoxic solid tumors induce the stabilization of hypoxia-inducible factor 1 alpha (HIF1α), which stimulates the expression of many glycolytic enzymes and hypoxia-responsive genes. A high rate of glycolysis supports the energetic and material needs for tumors to grow. Fructose-1,6-bisphosphate aldolase A (ALDOA) is an enzyme in the glycolytic pathway that promotes the expression of HIF1α. Therefore, inhibition of ALDOA activity represents a potential therapeutic approach for a range of cancers by blocking two critical cancer survival mechanisms. Here, we present a luminescence-based strategy to determine ALDOA activity. The assay platform was developed by integrating a previously established ALDOA activity assay with a commercial NAD/NADH detection kit, resulting in a significant (>12-fold) improvement in signal/background (S/B) compared with previous assay platforms. A screening campaign using a mixture-based compound library exhibited excellent statistical parameters of Z′ (>0.8) and S/B (~20), confirming its robustness and readiness for high-throughput screening (HTS) application. This assay platform provides a cost-effective method for identifying ALDOA inhibitors using a large-scale HTS campaign.
Introduction
Glycolysis is a critical source of metabolic intermediates for efficient anabolism.1,2 Accumulating evidence has indicated that cancer cells significantly use glycolysis in a hypoxic environment to survive and proliferate. 3 Hypoxia is typical of solid tumors because of decreased blood flow and disorganized tumor vasculature and provides a stressful environment that can favor tumor growth.4–6 In response to hypoxic stimuli, overexpression of the hypoxia-inducible transcription factors-1 and -2 (HIF1/2) promotes angiogenesis and activates cell survival pathways.7,8 Tumor hypoxia induces the expression of the glucose transporter 1 and many glycolytic enzymes. 9 A recent study showed that aldolase A (ALDOA) contributes to the maintenance of the HIF1 activity. 10 ALDOA catalyzes the reversible aldol cleavage of fructose-1,6-bisphosphate (F-1,6-BP) into D-glyceraldehyde-3-phosphate (G3P) and dihydroxyacetone phosphate (DHAP). ALDOA expression occurs in a wide variety of human tumor types,11–14 and high ALDOA levels are associated with poor patient survival. 14 Two biological activities of ALDOA may promote the features of the cancer cell phenotype. First, the glycolytic activity of ALDOA is essential for providing metabolic building blocks for the synthesis of cancer cell biomass.1,2 Second, a glycolysis-HIF1 feed-forward loop maintains the operation of HIF1 in the hypoxic environment of solid tumors. 10 Based on these findings, the inhibition of ALDOA leads to an inherent reduction in cancer cell metabolism and HIF1 activity. Despite its therapeutic significance, the identification of inhibitors of ALDOA activity lacks assays amenable to high-throughput small-molecule screening.
As illustrated in Figure 1A , ALDOA activity is typically assayed by monitoring NADH levels via a coupled enzymatic reaction using glycerol-3-phosphate dehydrogenase (α-GDH). The drawback of traditional methods that rely on direct measurement of either absorbance or intrinsic fluorescence of NADH is low sensitivity, larger reagent volume, and compound interference. Although electrochemical15,16 or amperometric 17 detection methods are challenging to adapt to high-throughput applications. Previously, we reported a fluorescence-based platform 18 incorporating the Elite NADH assay kit to convert NADH to a robust fluorescent signal. The fluorescence-based strategy enabled an ALDOA activity assay with enhanced sensitivity and reduced reagent requirement over the traditional methods. Although its potential for high-throughput compound screening was demonstrated by screening 65,000 compounds, interference from fluorescent compounds is still a factor, and the cost of the assay kit ($379/400 assays) is potentially prohibitory for extended screening campaigns or young investigators with limited funding.
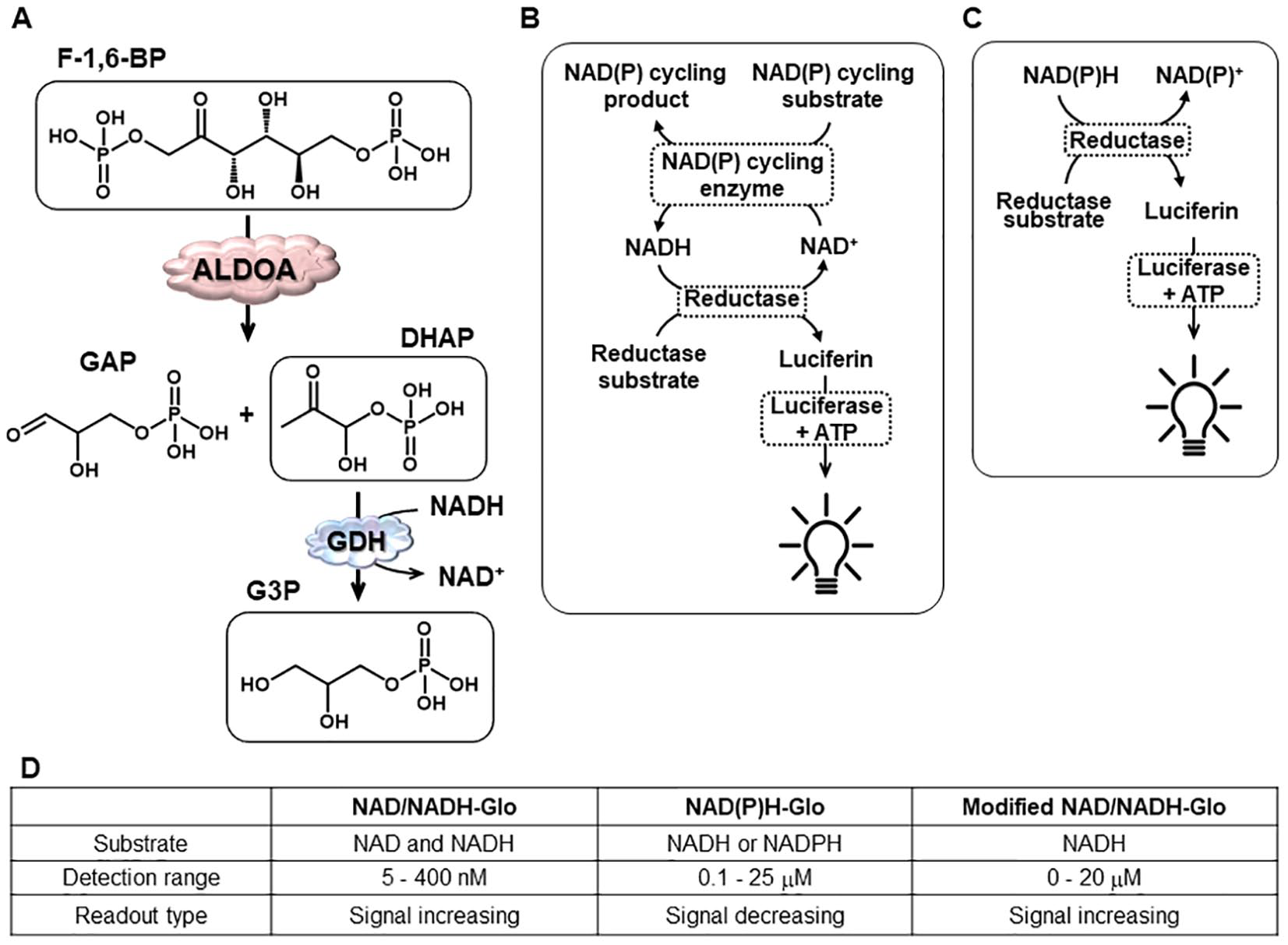
Schematic illustrations of aldolase A activity assay (
Unlike fluorescence, luminescence does not rely on photoexcitation. Thus, the background signal is minimal, allowing a more significant signal/background (S/B) and therefore a higher sensitivity. In addition, a lack of interference by fluorescent substances makes this technology highly desirable for high-throughput screening (HTS) applications, although the signal strength is weaker than fluorescence. Here, we describe a luminescence-based platform to assay ALDOA activity by taking advantage of a commercial NAD/NADH-Glo kit. Some reformatting was necessary to achieve the appropriate dynamic range and to reduce the screening cost. This assay protocol demonstrated robustness in a 384-well plate format with significantly reduced usage of the NAD/NADH-Glo reagents compared with the manufacturer’s original protocol (~74% reduction of detection reagent). A combinatorial library highlighted the potential to survey small molecules for inhibition of ALDOA activity using the assay.
To the best of our knowledge, this represents the first report of a luminescence-based ALDOA assay for high-throughput small-molecule screening and, as a result, further facilitates the development of ALDOA-targeted cancer therapeutics. Following our previous progress on inhibitor screening against ALDOA, we anticipated that both luminescence and fluorescence-based platforms could be used in a complementary manner, factoring in the strengths and weaknesses of each.
Materials and Methods
Reagents
Aldolase enzyme (ALDOA) was prepared as described previously. 18 F-1,6-BP, α-GDH, and β-NADH were purchased from Fisher (Hampton, NH; F0752, G6880, and N8129). Dihydroxyacetone phosphate (DHAP), triethanolamine (TEA), Triton X-100, dithiothreitol (DTT), Trizma base, and bovine serum albumin (BSA) were purchased from Aldrich (St. Louis, MO; 37442, 574797, P1379, D5545, T6791, and A2153). Naphthyl 2,6-bisphosphate (ND1) was synthesized in house, as previously reported. 19 The NAD(P)H-Glo (G9061) and NAD/NADH-Glo (G9071) assay kits were purchased from Promega (Madison, WI) and used following the manufacturer’s protocols for initial evaluation. The Elite NADH assay kit was purchased from eEnzyme (Gaithersburg, MD). White 384-well polystyrene plates were purchased from Fisher (Nunc No. 262360).
ALDOA Activity Assay
The ALDOA activity assay was performed in an assay buffer (1× TEA, pH 7.4, 0.1% BSA, 1.5 mM DTT, and 0.01% Triton X-100) using a 384-well white polystyrene plate at a volume of either 20 µL or 10 µL/well. ALDOA enzyme diluted in an assay buffer was added to an assay plate, followed by compound dispensing (or dimethylformamide [DMF] to mimic a compound). After a 30-min incubation, a mixture containing F-1,6-BP, NADH, and GDH was distributed to initiate the reaction at final concentrations of 20 µM, 20 µM, and 1.67 U/mL, respectively. The ALDOA/GDH-coupled enzymatic reaction was performed at room temperature for 1 h before stopping with a detection reagent, as described below. The initial concentration of ALDOA in the assay was 2.5 nM but was reduced to 1.25 nM during the optimization. The volumes of components were adjusted according to the compounds’ final concentration.
Modified NADH Detection Assay Using NAD/NADH-Glo Kit
An NADH detection procedure was composed of 3 steps: 1) stopping the ALDOA activity, 2) neutralization of the reaction, and 3) incubation with a detection reagent. Initially, 5 µL of 0.5% HCl (v/v) was added to a well of 20 µL of ALDOA activity assay to stop the reaction. After incubation for 10 min at room temperature, 5 µL of 0.5 M Trizma base solution was added and incubated for 10 min to neutralize the acid. A detection reagent was prepared according to the manufacturer’s protocol, except that the amount of NAD cycling enzyme and NAD cycling substrate were reduced by 10- and 2-fold, respectively, to achieve an extended dynamic range. Thirty microliters of detection reagent was dispensed to each well. After a 30-min incubation, relative luminescence units (RLUs) were recorded on an EnVision plate reader (PerkinElmer, Waltham, MA). After the protocol reformation, steps 2 and 3 were combined as follows: 8 µL of the premixed solution of 2.5 M Trizma and the detection reagent at a 1:34 ratio (v/v) was dispensed to each well. An NADH calibration curve was obtained by assaying 20 µL of NADH, dissolved in an assay buffer at various concentrations using the NAD(P)H-Glo kit following the manufacturer’s protocol.
ALDOA Counter Assay
A solution of GDH diluted in an assay buffer was incubated for 0.5 h with compounds followed by the addition of a DHAP and NADH mixture. After 1 h of reaction, the assay was processed according to the modified NADH detection assay described above. The final concentrations of DHAP, GDH, and NADH were 20 µΜ, 1.6 U/mL, and 20 µΜ, respectively.
Assay Validation and Automated Compound Screening
DMF tolerance was tested by varying the concentration of DMF in an ALDOA activity assay. Normalized activity at different levels of DMF was calculated according to the luminescence in the absence of DMF as 100% active. An ND1 dose response was conducted by replacing DMF during ALDOA activity assay with varying concentrations of ND1 dissolved in an assay buffer, the same as the DMF tolerance test. An ND1 dose response was also performed using an Elite NADH assay kit, as previously reported. 18 ALDOA diluted in an assay buffer and DMF or compounds were dispensed using a Janus automated workstation (PerkinElmer). Substrate mixture, HCl solution, and detection reagent mixture were distributed using MicroFlo Select (Biotek, Winooski, VT). A full-plate validation was performed by filling a 384-well plate with negative control (all assay reagents) except for columns 1 and 24, which were filled with the positive control (all assay reagents excluding ALDOA). For example, 10 µL of ALDOA diluted in an assay buffer, 2 µL of 5% DMF solution at 10-fold dilution, and 8 µL of substrate mixture was dispensed in sequence for ALDOA activity assay. After 1 h of ALDOA reaction, the assay was processed according to the modified NADH detection assay protocol described above. The compound screening was conducted as described for full-plate validation by replacing DMF with compounds. To screen compounds at higher concentrations, the volume and/or concentration of ALDOA solution and substrate mixture were revised accordingly.
Compound Library
Both scaffold ranking libraries (SRL) and positional scanning libraries (PSL) were obtained from the Torrey Pines Institute for Molecular Studies (Port St. Lucie, FL). An SRL containing 78 scaffold mixture samples was initially tested. Each of the 78 scaffold mixture samples is composed of individual compounds with the same core scaffold; the core scaffold is varied from sample to sample. The different compounds in each sample are represented in approximately equal molar concentrations, and the total number of individual compounds in a given sample varied from 2000 to 700,000, leading to a total of >30 million compounds. The PSL allows one to identify the individual compounds most likely responsible for driving the assay response for a given scaffold. Each of the individual samples in the PSL has a given R at a fixed position in the same scaffold. The average number of individual compounds in each PSL is 1000 to 2000 compounds per well, leading to a total of ~160,000 compounds per PSL. PSL 1978, 2225, and 882 contained 100 to 200 mixture samples, respectively. Samples were initially dissolved in 5% DMF at 500 µg/mL and dispensed directly to the assay plate.
Data Processing and Analysis
Data from the NADH calibration were fit to a linear regression model in Sigmaplot 13.0 (Systat Software, San Jose, CA). The percentage inhibition of compounds was calculated based on the positive control as 0% inhibition and negative control as 100% inhibition. The data from ND1 dose dependency was by fit to a four-parameter logistic curve using Sigmaplot 13.0. For the assay quality validation, Z′ was calculated according to the equation previously reported. 20
Results and Discussion
Optimization of a Luminescence-Based NADH Detection Platform for ALDOA Inhibitor Screening
Previously, we reported a fluorescent-based ALDOA assay platform by coupling an ALDOA activity assay with the Elite kit, which relies on diaphorase-driven NADH oxidation to convert a nonfluorescent probe to a fluorescence signal. Here, we sought a luminescence-based platform to monitor NADH, albeit using the same coupled assay arrangement ( Fig. 1A ) but instead using a luminescence-based NADH and/or NAD detection kit developed by Promega. 21 Two commercial luminescence-based NAD/NADH detection kits by Promega were considered ( Fig. 1B–D ). 21 The NAD/NADH-Glo assay ( Fig. 1B ) was designed to measure total NAD and NADH, whereas the NAD(P)H-Glo assay ( Fig. 1C ) was designed to measure NADH or NADPH selectively. Because NAD/NADH-Glo cannot discriminate NADH or NAD and the detection is limited to less than 400 nM, which is much lower than the NADH concentration (20 µM) used in our fluorescent ALDOA activity assay, we first explored the feasibility of using the NAD(P)H-Glo kit, which has a higher detection limit of 25 µM. A standard solution of NADH (0–80 µM) was used to assess the assay. The observed luminescence (RLU) was linearly proportional to the concentration of NADH over the range of 0 to 20 µM (r2 = 0.997; Fig. 2A ). Next, the previously developed ALDOA activity assay 18 was integrated with the NAD(P)H-Glo assay kit with some modification. The major modifications were changing the buffer to TEA and the F-1,6-BP concentration to 20 µM, near its Km (19.7 ± 0.6 µM; Suppl. Fig. S1 ), so that all possible types of inhibitors could be identified. The NADH concentration was adjusted to 20 µM in accordance with the detection limit of the NAD(P)H-Glo assay kit. In summary, the ALDOA activity assay employed ALDOA, F-1,6-BP, NADH, and GDH at final concentrations of 2.5 nM, 20 µM, 20 µM, and 1.67 U/mL, respectively, where the ALDOA catalyzed reaction was optimized to convert ~30% of the substrate (F-1,6-BP) to DHAP in 1 h. This is expected to furnish approximately 7 µM of NAD+. Because the NAD(P)H-Glo kit measures NADH, the change in RLU is inversely proportional to the progress of an ALDOA-catalyzed reaction (i.e., “signal decreasing”). The resulting RLU corresponded well to the standard calibration curve for the NADH. Although feasible, the remaining NADH (70%) raised the background substantially, resulting in a low S/B of ~ twofold, which is the typical disadvantage of an assay that results in a decrease in signal. We further investigated how the background signal affects the S/B by varying the concentration of NADH. The signals of both negative and positive controls increased proportionally with NADH ( Fig. 2B ), resulting in a ΔRLU increase up to 10 µM and then plateaued ( Fig. 2C ). Thus, the S/B ratio is inversely proportional to the NADH concentration ( Fig. 2D ), with the highest S/B corresponding to the lowest NADH concentration. This initial examination suggests that an S/B of ~9 could be obtained when 1 µM of NADH is used in a coupled assay, a value that exceeds that obtained using a fluorescence-based platform (S/B of 4). 18
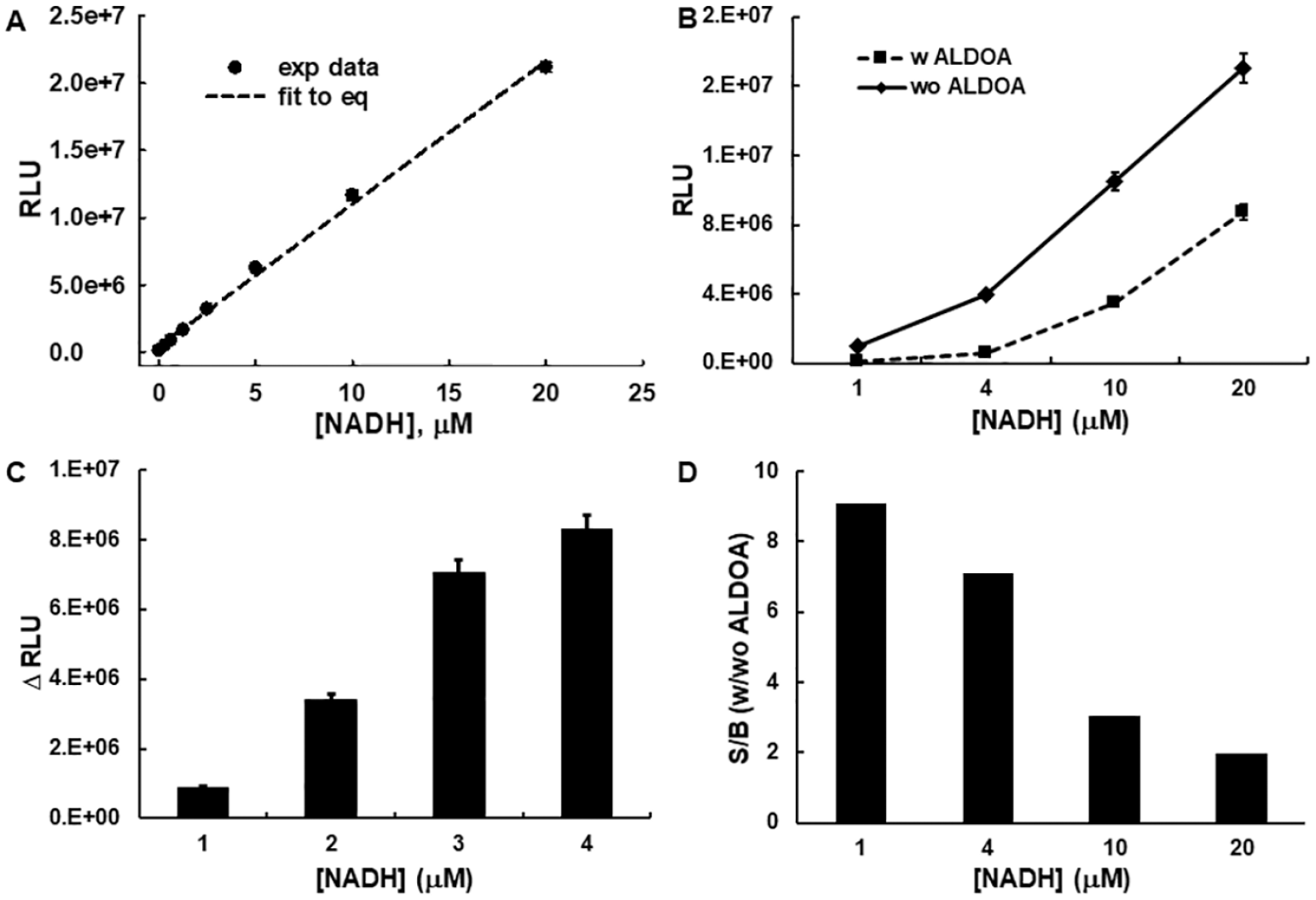
Investigation of NAD(P)H-Glo for the adaptation to aldolase A (ALDOA) activity assay. (
As an alternative approach, we explored the potential of using the NAD/NADH-Glo kit. Although the NAD/NADH-Glo kit was initially designed to detect both NADH and NAD+ with a limit of 400 nM, we found that it could be modified to measure NADH or NAD+ selectively with its dynamic range extended up to 20 µM ( Fig. 1D , denoted as “modified NAD/NADH-Glo”). Specifically, an acidification step (dispensing 5 µL of 0.5% HCl) was added immediately after the ALDOA activity assay to remove any remaining NADH. This was based on the differential stability of NAD and NADH under strongly acidic conditions. 22 The acid treatment can also serve to inactivate an enzyme activity. In an additional step, however, the sample has to be neutralized by dispensing a 0.5 M Trizma solution (5 µL) before adding detection reagents. Ultimately, acid quenching/neutralization allowed selective detection of NADH and the complete quenching of ALDOA activity. A second modification was applied to extend the linearity of the assay up to 20 µM NADH. This was achieved by reducing the amount of the NAD cycling enzyme and substrate by 10-fold and 2-fold, respectively, as instructed by the manufacturer. 23 The initial experiment of monitoring ALDOA activity employing the modified NAD/NADH-Glo protocol showed an ~48-fold S/B when ALDOA (2.5 nM) and NADH (20 µM) were used. This represents a 5-fold improvement over the NAD(P)H-Glo protocol and a 12-fold improvement over our fluorescence-based assay. The assay performance over varying concentrations of the ALDOA enzyme confirmed that the luminescence readout was dose dependent ( Fig. 3A ). Because a greater than 20-fold S/B was achieved at 1.25 nM ALDOA, its concentration was fixed at 1.25 nM for further assay development. The initial assessment confirmed the superiority of this new luminescence-based platform to measure ALDOA activity. Although this luminescence-based NAD/NADH-Glo method has been used in various tissues/cells/enzymatic reactions, it has not, to our knowledge, been used either for assaying ALDOA or in an HTS campaign. Although the integration of commercial detection kits for assay development offers a benefit of saving labor and time, advancing such assays to HTS campaigns is still challenging because of their relatively expensive cost. The cost of the NAD/NADH-Glo kit corresponds to $0.04/µL or ~$1.2/assay if 30 µL of a detection reagent is used, as stated in the manufacturer’s protocol. For an affordable HTS campaign, we therefore assessed the potential of further reformatting this detection method.
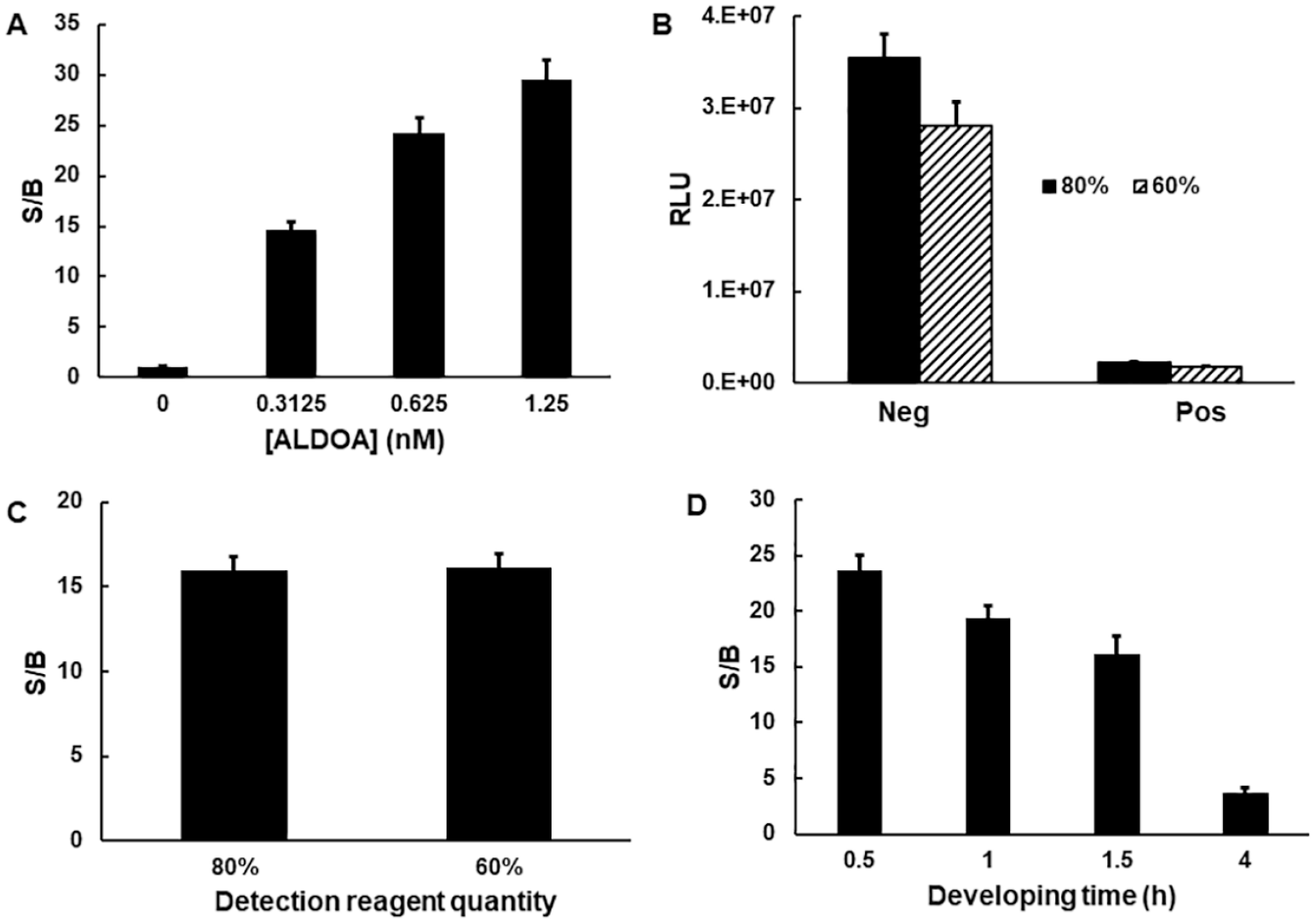
Investigation of NAD/NADH-Glo for the adaptation to aldolase A (ALDOA) activity assay. (
The modified NAD/NADH-Glo protocol requires that the volume of detection reagent be the same as the total volume of the assay plus stop reagents. Our initial protocol employed a detection volume of 30 µL to match the volume of the assay mixture (20 µL) plus stop reagents (acid + base, 10 µL). However, by increasing the concentration of the Trizma solution fivefold, we were able to dispense 1 µL rather than 5 µL, while also reducing the detection reagent by up to 40% (based on 26 µL). As shown in Figure 3B , both the negative and positive controls were equally affected by the amount of detection reagent used, resulting in the S/B ratio being mostly the same in either case ( Fig. 3C ). This result confirmed that the reduction of the detection reagent usage from 30 µL to 16 µL had little effect on assay integrity, thus providing a rationale for reducing detection reagent cost by as much as ~50%. Next, we investigated the effect of the incubation time of the detection reagent on the dynamic range of the assay and reagent stability. An RLU for a negative control increased linearly up to 1.5 h and then dropped slightly, whereas the positive signal continuously increased. As a result, the S/B was highest between 30 min and 1 h (~20-fold) but then gradually decreased ( Fig. 3D ). Based on this result, we chose an incubation time of 30 min, although the detection reagent maintained stability for up to 4 h. Promega also validated the stability of NAD/NADH-Glo detection reagent up to 6 h at room temperature (unpublished result). RLUs are similar in the presence (1.5 mM) or absence of DTT, confirming that DTT does not alter the NAD/NADH-Glo method (data not shown).
Assay Validation and Miniaturization
Evaluation of a small-molecule library dissolved in 5% DMF at a concentration of 500 µg/mL required a DMF tolerance test. As shown in Figure 4A , the normalized activity was consistent up to 1.25% but showed an ~12% decrease at 2.5% of DMF, indicating that testing a concentration of 250 µg/mL at 2.5% DMF is feasible. Under the optimized conditions, a known inhibitor, ND1, 24 was assessed. It showed dose-dependent inhibition with a calculated IC50 and Hill coefficient of 3.3 ± 0.3 µM and 0.88 ± 0.07, respectively ( Fig. 4B ), in agreement with the fluorescence-based assay. 18 Next, a full-plate validation was conducted in two consecutive experiments using an automated workflow, as illustrated in Figure 4C . Averaged Z′ and S/B values of 0.78 and 19.2, respectively, confirmed the robustness and reproducibility of the assay for the HTS.
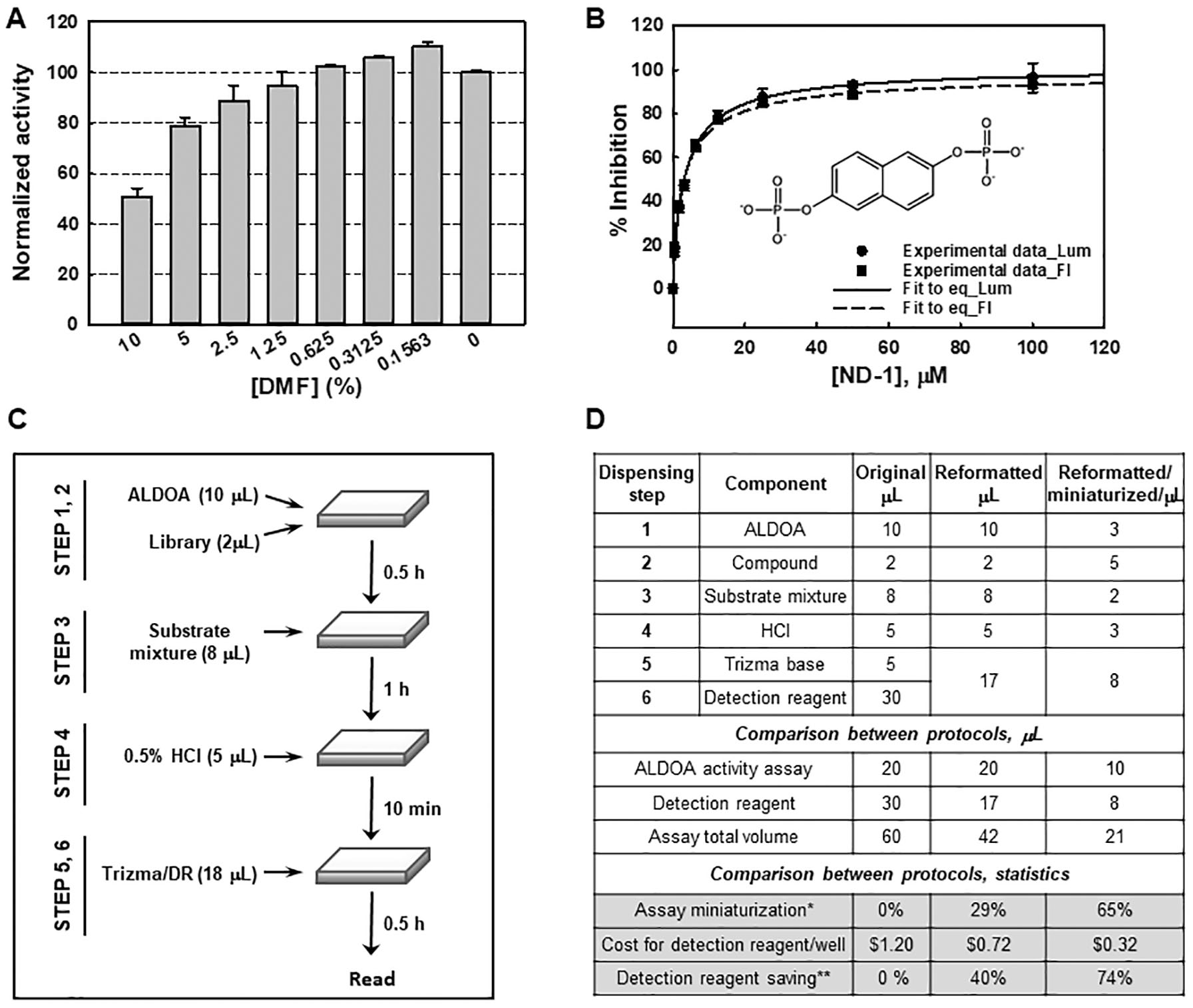
Validation of luminescence-based aldolase A (ALDOA) assay and compound-screening campaign. (
We also determined that the simultaneous dispensing of both the Trizma solution and detection reagent allowed the elimination of a dispensing step. Assay miniaturization was followed by twofold scaling down of the assay volume, resulting in excellent statistics with Z′ and S/B of 0.82 and 19.4, respectively. Thus, we succeeded in miniaturizing and simplifying the luminescence-based ALDOA assay protocol, as summarized in Figure 4D , to afford a cost-effective and sensitive high-throughput compound-screening protocol. This work also shows that the NAD/NADH-Glo platform is easily adaptable, in a cost-saving manner (>70% reduction), to other HTS assays involving either NADH production or consumption.
Compound Screening
Employment of a mixture-based compound-screening approach developed by the Torrey Pines Institute for Molecular Studies allowed for the rapid discovery of ALDOA inhibitors. The formation and use of the Torrey Pines SRL and PSL have been reported previously.25–29 Details of a typical workflow are described in Supplementary Figure S2 . Using the reformatted assay platform, we first tested the SRL at 25, 50, 100, and 250 µg/mL, respectively. The resulting statistics were satisfactory with a Z′ and S/B of 0.83 and 19, respectively, in assaying four 384-well plates. Retesting of 13 samples showing more than 15% inhibition (at the highest concentration of the library, 250 µg/mL) in the same assay and a counter screen was then followed. The counter screen used 20 µM DHAP but no ALDOA to identify false-positive hits due to any assay interference ( Suppl. Fig. S3A ). The quality of the counter screen was excellent as well, with Z′ and S/B of 0.88 and 23.5, respectively. Samples 2225, 1978, and 882 showed reproducible inhibition activity in the ALDOA assay but no activity in the counter screen, with overall inhibition activity ranging between 10% and 40% ( Suppl. Fig. S3B ). Samples 2058, 2165, 2135, and 2437 showed comparable activities in both ALDOA and reference assays, indicating they are false-positive hits due to assay interference. We hypothesized that further investigation of the Torrey Pines’ PSL could allow for the discovery of more potent inhibitors. Sample 2225 contains 38,250 compounds with a scaffold of N-naphthyl-dipeptides. Samples 1978 and 882 contain 38,250 and 72,283 compounds with a scaffold of indole imidazoles and C-6-acyloamino bicyclic guanidine, respectively. PSLs 2225, 1978, and 882 ( Suppl. Fig. S3C ) are composed of 124, 128, and 125 mixture samples, respectively. Screening comprised a total of eight 384-well plates at final concentrations of 100 and 250 µg/mL in duplicate. The statistics were again satisfactory with a Z′ and S/B of 0.88 and ~24, respectively, but none of the samples showed improved potency. Nevertheless, the cost of detection reagent for the screening of a total of 12 plates was ~$1500, a 74% reduction in the cost of ~$5600 using suggested reagent quantities. Such a reduction in cost should significantly extend its application for use in HTS platforms in academic settings.
In conclusion, we have developed a robust, highly sensitive, and cost-effective luminescence-based detection platform to screen ALDOA inhibitors by reformatting a commercially available NADH detection kit. Although a virtual screening 30 strategy could be employed for the identification of ALDOA inhibitors, ultimately a biochemical assay is critical for the progression of a hit to lead. This development involved extending the dynamic range from 400 nM to 20 µM NADH, selectively detecting NADH using an acid quench step, reducing the amount of detection reagent needed by 40%, and miniaturizing the assay by twofold to further cut down on assay cost, leading to an overall reduction in the cost of 74%. Compared with absorbance- or fluorescence-based NADH detection schemes, this luminescence-based method exhibited a considerably enhanced S/B (>12-fold). Furthermore, the IC50 value of a known inhibitor, ND1, is in good agreement with previous reports, which further validates the utility of the luminescence assay. A rapid and cost-effective HTS campaign exhibited excellent assay statistics of Z′ (>0.8), and S/B (~20) across 12 assay plates confirmed the robustness of this platform for more substantial scale HTS campaigns. As none of the compounds tested warranted moving on to the next step, the screening of a larger and more diverse compound library will be followed in the near future. In addition, this reformatted NADH-detection platform can be readily adaptable to the analysis of other NADH-dependent enzymes in an affordable manner.
Supplemental Material
ALDOA_figures_final_Supp_Rev_Final – Supplemental material for A Robust and Cost-Effective Luminescent-Based High-Throughput Assay for Fructose-1,6-Bisphosphate Aldolase A
Supplemental material, ALDOA_figures_final_Supp_Rev_Final for A Robust and Cost-Effective Luminescent-Based High-Throughput Assay for Fructose-1,6-Bisphosphate Aldolase A by Eun Jeong Cho, Ashwini K. Devkota, Gabriel Stancu, Ramakrishna Edupunganti, Ginamarie Debevec, Marc Giulianotti, Richard Houghten, Garth Powis and Kevin N. Dalby in SLAS Discovery
Footnotes
Acknowledgements
The authors thank Dr. Donna Leippe at Promega Inc. for her helpful suggestions throughout the assay development.
Supplemental material is available online with this article.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: CPRIT (RP110532-P1 and RP160657), NIH R01 CA216424, and the Florida Drug Discovery Acceleration Program (the State of Florida, Department of Health) supported this research.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.