
Abstract
The aberrant regulation of protein expression and function can drastically alter cellular physiology and lead to numerous pathophysiological conditions such as cancer, inflammatory diseases, and neurodegeneration. The steady-state expression levels of endogenous proteins are controlled by a balance of de novo synthesis rates and degradation rates. Moreover, the levels of activated proteins in signaling cascades can be further modulated by a variety of posttranslational modifications and protein–protein interactions. The field of targeted protein degradation is an emerging area for drug discovery in which small molecules are used to recruit E3 ubiquitin ligases to catalyze the ubiquitination and subsequent degradation of disease-causing target proteins by the proteasome in both a dose- and time-dependent manner. Traditional approaches for quantifying protein level changes in cells, such as Western blots, are typically low throughput with limited quantification, making it hard to drive the rapid development of therapeutics that induce selective, rapid, and sustained protein degradation. In the last decade, a number of techniques and technologies have emerged that have helped to accelerate targeted protein degradation drug discovery efforts, including the use of fluorescent protein fusions and reporter tags, flow cytometry, time-resolved fluorescence energy transfer (TR-FRET), and split luciferase systems. Here we discuss the advantages and disadvantages associated with these technologies and their application to the development and optimization of degraders as therapeutics.
Keywords
Introduction
Normal cell function, cell cycle progression, and cell growth are processes that rely on tightly regulated intracellular signaling pathways and a wide variety of posttranslational protein modifications to upregulate and downregulate these processes in response to extracellular stimuli. Events that are disruptive to both protein homeostasis and function in a manner that causes dysregulation of signaling pathways can lead to pathophysiological conditions such as neurodegeneration, inflammation, and numerous types of cancer.1–7 Genetic lesions in the genes coding for key regulatory proteins may give rise to mutations that modulate enzymatic activity by introducing a gain or loss of function in these proteins. Likewise, genetic lesions in epigenetic and chromatin remodeling proteins may lead to altered gene expression patterns and subsequent changes in protein expression levels. Lastly, the aberrant folding of proteins and formation of large aggregates that disrupt cellular function can cause diseases and disorders known as proteopathies.8,9
Understanding the cellular pharmacology of dysregulated proteins is the key to therapeutic intervention. Historically, the focus of small-molecule-based drug discovery efforts has been to design and optimize ligands with drug-like properties that can modulate the function of disease-causing proteins, through either direct inhibition of enzymatic activity, by inducing a conformational change that perturbs normal function or drug binding affinity, 10 or disrupting protein–protein interactions. 11 Many of the small-molecule drugs found on the market today fall within this paradigm. While many are successful in treating important disease conditions, these approaches have limitations, and there are still many areas of unmet patient need that require novel therapeutic approaches. Rather than focus on controlling protein function, which requires high binding affinity and the constant presence of the drug for efficacy, the field of targeted protein degradation instead aims to destroy disease-causing proteins through substoichiometric catalysis using the innate cellular machinery. This approach has demonstrated differentiated pharmacology, high potency, and selectivity relative to traditional inhibitors.12–26
Cellular protein levels are established by a balance between endogenous protein degradation via lysosomal or proteasomal pathways27–29 and de novo resynthesis, a balance that fluctuates in response to changes in the cellular environment. 30 Targeted protein degraders are small molecules that hijack the natural cellular processes involved in protein recycling and homeostasis.20,31 Degraders can take the form of a bifunctional degradation activating compound (one example, branded BiDAC), also commonly referred to as PROTACs. BiDACs are complex small molecules composed of three regions: (1) a ligand to the protein of interest, (2) a chemical linker region, and (3) a ligand that recruits an E3 ligase. Conversely, they can be monovalent degradation activating compounds (one example, branded MonoDAC), also often referred to as molecular glues.31,32 MonoDACs bind to an E3 ligase and cause an alteration in the protein surface that promotes a cooperative protein–protein interaction with a target protein. Both MonoDACs and BiDACs induce protein degradation by mediating the formation of a ternary complex of an E3 ligase and target protein, thereby inducing polyubiquitination and the subsequent destruction of disease-causing proteins via the ubiquitin proteasome system (UPS).
Targeted protein degradation controls the amount of a harmful cellular protein rather than modulation or inhibition of its function. This novel therapeutic approach also offers many opportunities to differentiate from inhibitor-based therapies. First, by catalyzing the destruction of their target protein, degrader molecules may also disrupt scaffolding functions of proteins otherwise not addressed by small-molecule inhibitors, allowing for drug properties that are unlike other classes of drugs.12,26 Second, degraders may offer higher potency and differentiated cellular pharmacology. Unlike inhibitors, which must remain bound to a single-target protein, degrader molecules can catalyze the ubiquitination and destruction of multiple copies of the same protein since the degraders themselves are not degraded in the proteasome.18,33 This is supported by the finding that covalent degraders have not shown the same level of differentiation from their inhibitor-based counterparts. 34 However, it is important to note that the general effectiveness of covalent-based degraders remains fairly controversial to date.35,36 We anticipate that this catalytic property of degraders allows for lower therapeutic doses and an improved therapeutic window by minimizing off-target toxicity driven by interactions with homologous proteins. Additionally, degraders may offer a broader targetable proteome since they can be based on small-molecule binders and not necessarily an inhibitor of the target protein. Lastly, BiDACs in particular may offer avenues to achieve selectivity where none exists for a traditional inhibitor molecule. Targeted protein degradation is a complex process that provides the opportunity to modulate selectivity at different steps, including through distinctive ternary complex formation with highly homologous isoenzymes and/or differences in catalytic competence of ternary complexes once formed. By enabling interacting proteins to sample multiple orientations when forming ternary complexes, it has been shown that nonselective inhibitors that bind two proteins with highly conserved binding sites can be rendered selective when modified into BiDACs and degrade only one of the proteins if other distal regions of the proteins are less homologous. 37
Optimization of BiDAC potency is complex and challenging as it requires refinement of all three regions of the molecule to strike the right balance in protein binding affinity and linker properties (length, rigidity, vectors from each end ligand) for optimal ternary complex formation, target ubiquitination, and degradation. These parameters need to be maintained and monitored while also optimizing in vivo pharmacokinetic properties. Given this complexity, degrader hit identification and optimization are largely empirical and high-capacity approaches that rapidly assess target degradability are needed to guide medicinal chemistry efforts. Thus, there is an urgent need for high-throughput methods of monitoring targeted protein degradation that are efficient, robust, sensitive, quantifiable, and reproducible. This perspective discusses many of the technologies now currently available to directly measure the degradation of target proteins induced by BiDACs and empower efficient drug discovery in this rapidly emerging therapeutic field.
Western Blot and Capillary Electrophoresis Immunoassays
Much of the early work in the targeted protein degradation field has traditionally relied on Western blots to visualize changes in protein levels in lysates from degrader treated cells. Using this approach, the loss of target protein can be monitored in both a concentration- and a time-dependent manner and can also be used to confirm that the observed protein loss is E3 ligase-dependent and proteasome-dependent when degraders are co-treated with appropriate compounds.38,39 In a typical experiment, cells are treated with test compounds and incubated for a desired time period for sufficient protein degradation. In order to ensure that sufficient total cellular protein (10–100 µg) is loaded onto the gel for subsequent Western blot detection, 40 either degrader compounds must be added to cells in suspension prior to plating the cells in 6- or 12-well plates or compounds can be added to adherent cells plated prior to the experiment. Following the addition of the degrader compound, the plates are covered and incubated at 37 °C with 5% CO2 throughout the experiment. To visualize the degradation of cellular proteins, treated cells are lysed, and proteins are denatured and separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) gel electrophoresis. We have found that a six-well plate of cells typically yields 200 µL of 1–2 µg/µL protein for gel loading. In the case of a traditional Western blot, separated protein bands are then transferred to a nitrocellulose membrane, washed several times, and then incubated with antibodies specific for the protein of interest. The membranes are typically also probed with an antibody to a reference protein (loading control) that is not expected to be degraded. This is followed by the addition of a secondary antibody that recognizes the primary antibody and carries a detectable tag, fluorescence tracer, or enzyme that catalyzes a light-generating detectable signal. The availability of well-characterized and highly specific primary antibodies against the target protein is critical to minimize the nonspecific binding and background signal, thus allowing the generation of high-quality Western blots. There is flexibility in the approach due to a wide variety of commercially available labeled secondary antibodies and instrumentation for signal detection. Finally, target protein degradation is qualitatively determined by normalizing protein bands to a reference protein (loading control) and to cells treated with vehicle alone.
In the field of targeted protein degradation, Western blots have remained the traditional method for visualizing concentration-dependent protein degradation in cells. However, Western blots are extremely resource-intensive and require multiple wash steps and incubation periods to produce a high-quality blot. Quantification of the bands of interest can also take from several hours to days, depending on the software used. Although informative for visualizing protein degradation, Western blots are not ideal for driving fast-paced drug discovery and optimization efforts, which have come to rely more heavily on the high-throughput plate-based assay methods discussed in the following sections. However, recent technological advances have been able to reduce the time needed to visualize targeted protein degradation by Western blot. Multiple detection platforms have emerged (i.e., Wes, Jess, Sally Sue, or Peggy Sue), which utilize the Simple Western system of automating the protein separation and immunodetection steps. This improvement significantly reduces the overall time and effort and means that quantitative results can be obtained in less than 3 h. The Simple Western system is a capillary electrophoresis immunoassay (CEI)-based methodology that can identify and digitally quantitate the relative abundance of a protein of interest ( Fig. 1 , left panel). Researchers prepare protein lysates from treated cells and add them, along with all necessary antibodies and substrates, into a plate, which is loaded in the instrument with single-use capillaries. The instrument then separates proteins by size or charge, immobilizes separated proteins onto the capillary wall, applies primary and secondary antibodies, and develops the signal corresponding to the protein of interest.41,42
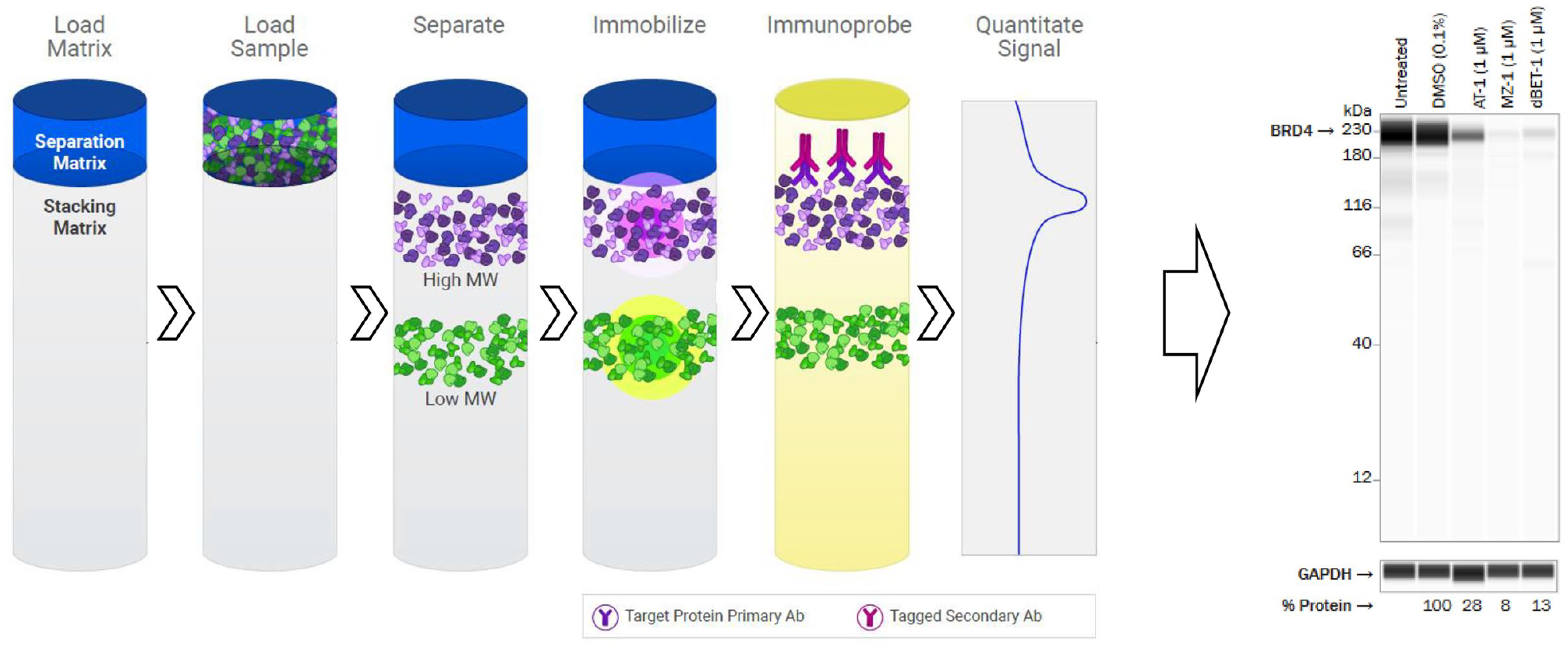
Simple Western technology. The Simple Western automated procedure uses a 24- to 96-well assay plate containing a stacking matrix, a separation matrix, and protein lysates from treated and untreated cells. The instrument loads the stacking and separation matrices, followed by protein samples, which are separated according to size or charge. Proteins are immobilized to the capillary walls, and gel matrices are extracted before primary and secondary antibodies are added to detect proteins of interest. (Image adapted from ProteinSimple marketing materials with permission; https://www.proteinsimple.com/simple_western_overview.html.) The chemiluminescence or fluorescence signal is then processed and presented as a digitalized Western blot representation to visualize targeted protein degradation. In this example, degradation of BRD4 by dBET-1 is demonstrated. (Image adapted from Tocris marketing materials with permission; https://www.tocris.com/tpd.)
Both the Western blot and the Simple Western systems are amenable to the detection of endogenous or ectopically expressed, unmodified, untagged full-length target proteins. Provided the antibody epitopes are not perturbed (a requirement for all immunodetection-based methods), both methodologies also allow the detection of modified, tagged protein or protein domains. Additionally, both techniques are broadly applicable across a full target portfolio of target proteins, provided that validated antibodies against those targets have been characterized. However, the Simple Western systems provide several additional advantages, including the reliable capture and quantitation of high-molecular-weight proteins, as in the case of BRD4 ( Fig. 1 , right panel). By shortening the time needed to obtain results, Simple Western platforms can also increase throughput over Western blots while also improving upon the visualization and quantitation of protein loss. Simple Western techniques have picogram-level sensitivity and a dynamic range of detection of more than 3 orders of magnitude, 43 require significantly less sample than a traditional Western blot, and can run up to 24 samples per instrument in less than 3 hs or 96 samples in approximately 11 h, depending on instrumentation. In our hands, the small sample size requirement of <3 µL at a typical range of 0.01–1 µg/µL (of which only 40 nL is loaded) makes it feasible to collect treated cells from 96-well plates, which is particularly useful when cell or degrader availability is limited. The Jess system adds the option to use fluorescence infrared (IR) and near IR (NIR), as well as protein normalization to the chemiluminescence capabilities of the Wes. This improvement allows multiplexing of proteins with similar size if secondary antibodies are available from different species or primary antibodies are fluorescently labeled. Total protein normalization eliminates known complications of using housekeeping genes to normalize loading.44,45 Lastly, in addition to the quantitation being automated and standardized, these Simple Western systems eliminate run-to-run variability of Western blots by automating loading, transfer, and standardized incubation and wash times.
Although there are notable recent improvements over Western blots, CEI-based technologies remain a relatively low-throughput way of optimizing degraders in a drug discovery setting, and analyses are still subject to the same limitations of traditional Western blots. Western blot and Simple Western technology are not amenable for large numbers of samples. Due to limited throughput, Western blots and CEI-based approaches will be difficult to use for compound library screening, multiple dose points, and time-course measurements of protein degradation. For Simple Western, the polyubiquitinated target proteins are more readily transferred than on Western blots for high-molecular-weight proteins. However, it is also important to note that as proteins become modified with poly-ubiquitin chains, the molecular weight of the target protein can rapidly increase. Therefore, loss of detected signal does not necessarily reflect the degradation of the target alone, which complicates analyses. It is also possible that ubiquitination could block primary antibody recognition, also causing an apparent loss of detectable signal. Most importantly, it is important to point out that the success of these approaches and any of the higher-throughput assay technologies described below relies on the prior rigorous validation of primary antibodies against the target protein of interest. Understanding the degree of nonspecific signal associated with a chosen antibody is crucial to setting the most accurate normalization levels for 0% remaining protein following the treatment of cells with BiDACs.
High-Throughput Flow Cytometry and In-Cell Western
High-throughput flow cytometry (HTFC) and In-Cell Western (ICW) approaches improve on the throughput offered by traditional Western blots for detecting the degradation of endogenously expressed, unmodified, and untagged full-length proteins. Flow cytometry is a widely used laser-based technology that can analyze the expression of cell surface and intracellular proteins by measuring the fluorescence intensity produced by a fluorescent-labeled antibody bound tightly and specifically to the protein of interest. As with Western blots, recent technological advances have also revolutionized flow cytometry as a medium- to high-throughput screening method to aid drug discovery efforts. 46 Traditional flow cytometers require appreciably higher quantities and volumes (500 µL to 3 mL) of labeled cells and corresponding antibodies per sample, thereby increasing the time and cost per data point. However, more compact flow cytometers are now available (i.e., Guava easyCyte by Luminex (Austin, TX), iQue3 by Sartorius (Göttingen, Germany)) that can measure different fluorescent intensities of cellular ligands or proteins in 96-well or higher-throughput formats to increase throughput and reduce cost by requiring much lower volumes of sample and detection antibodies. Relative to traditional Western blots, which require repeated incubation and wash cycles with primary and secondary antibodies, HTFC provides an additional advantage by cutting down the total time needed to go from sample prep through signal detection. HTFC is compatible with extracellular or intracellular protein domains and, while suitable for use with either suspension or adherent cell lines, it is more amenable to suspension cells since these do not require the additional steps necessary to enzymatically or mechanically dissociate adherent cells from the growth surface and each other prior to flow cytometry. Likewise, there are fewer processing steps when the protein of interest is extracellular, thereby bypassing the requirement of permeabilizing cells to antibodies against intracellular targets.
In a typical HTFC experiment, test degraders are added to a 96-well (or higher-throughput) plate containing cells expressing the target of interest and incubated for a time period needed to observe maximal protein degradation. During the incubation period, assay plates are covered and maintained at 37 °C with 5% CO2 for the duration of the assay. The cells are then fixed, permeabilized, and sufficiently washed, and then fluorescent-labeled antibody is added and incubated at 4 °C for 1 h. In the final step, the samples are washed one final time before being analyzed with an appropriate flow cytometer in the case of HTFC.
Another analogous approach that allows protein loss to be determined in intact, albeit fixed, cells is by an ICW approach (
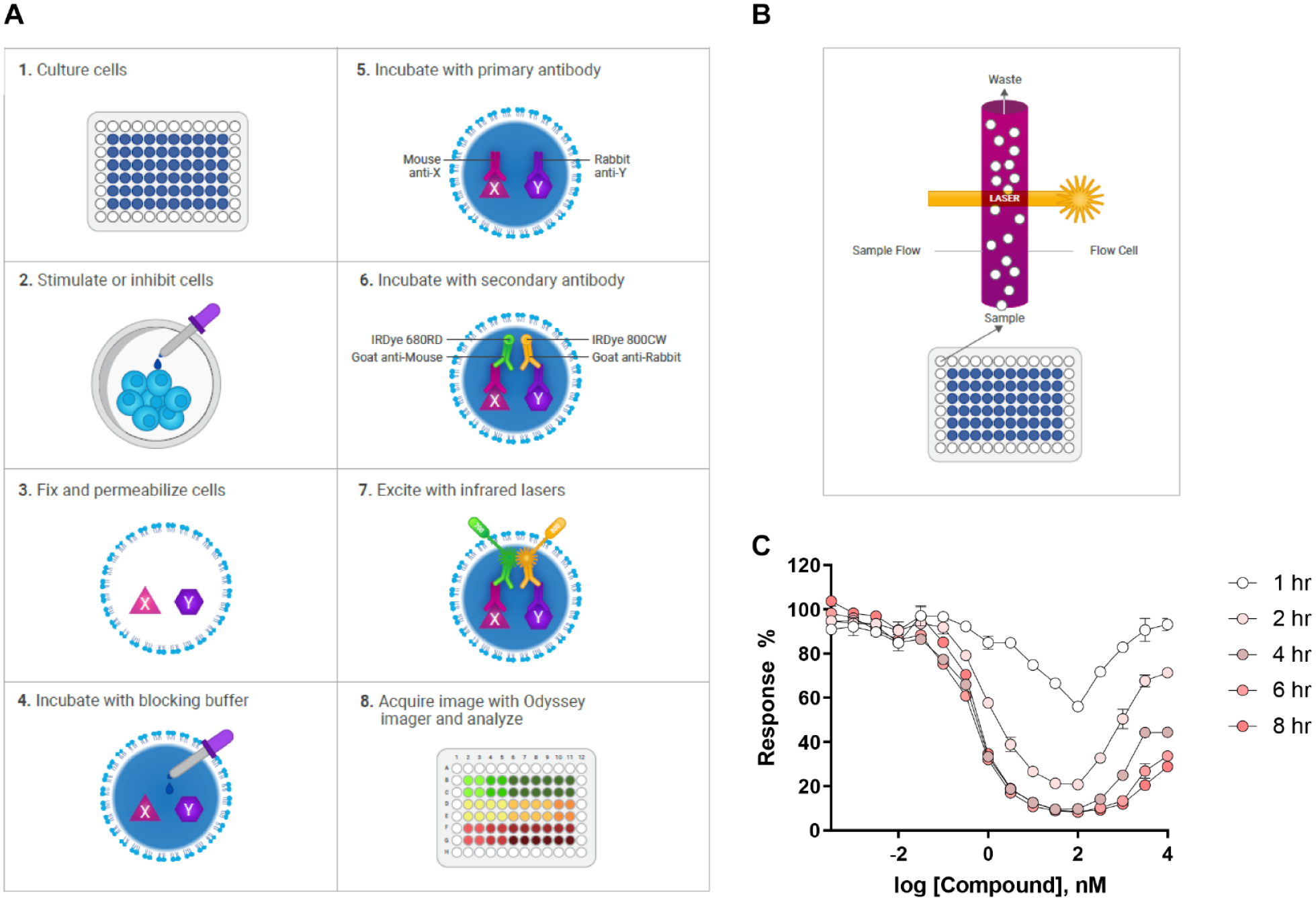
Schematic of ICW and HTFC approaches. (
As in the case of the Western blot and Simple Western approaches, the quality of ICW and HTFC data also relies on the availability of high-quality, well-validated primary antibodies and fluorescent-tagged secondary antibodies. This is even more important for ICW and HTFC since specific and nonspecific signals cannot be differentiated based on association with a protein of the correct molecular weight. The use of monoclonal primary antibodies is preferred to minimize nonspecific binding and background signal. When choosing antibodies, it is important to note that Western blot antibodies recognize fully denatured proteins and will not likely bind to the same proteins in their native, folded state. Additionally, the protocols for HTFC and ICW methodologies largely preserve native protein structure, but epitope masking on the protein surface may occur due to cross-linking. Therefore, all antibodies selected for HTFC and ICW assay development should be immunohistochemistry (IHC) compatible for the greatest chance of success. As with Western blot, once an antibody is identified, the detection of the protein of interest can be readily transferred to any cell line, allowing rapid comparison of larger numbers of cell lines with the same technology, albeit with a much higher throughput in the case of HTFC and ICW.
Despite these advantages, it is important to note that the formation of ternary complexes between the target and the E3 ligase prior to target protein ubiquitination and degradation may complicate data analysis when using these methods. The formation of ternary complexes combined with cross-linking during fixation may also mask the primary antibody epitope on the surface of the protein, thereby artificially lowering the detected signal, which could be falsely interpreted as degradation of the target. However, there are ways to implement cell engineering methods, such as CRISPR knock-in or lentiviral infection, to coexpress a common small tag (i.e., HA) at the N- or C-terminus of the target protein. In this experimental setup, the detection step would then employ a fluorescent-labeled antibody against the tag, rather than the target protein directly. The utilization of affinity tags may also be useful when there is no validated primary antibody available for a target of interest. While new technologies such as the iQue3 have certainly made HTFC a much more high-throughput method in terms of flow cytometry, the general throughput of HTFC for drug discovery remains impacted by sample prep time that is more time- and resource-intensive. To power rapid and highly efficient targeted protein degrader drug discovery, 384-well (or higher-throughput) homogenous plate-based assays with fewer sample processing steps and shorter data turnaround times could be more transformative in terms of efficiency.
AlphaLISA SureFire Technology
AlphaScreen (Amplified Luminescent Proximity Homogeneous Assay Screen) is a versatile bead-based proximity assay technology developed to measure a wide variety of analytes, including tagged or endogenous cellular proteins. 49 It is a quantitative and high-throughput alternative to the methods described above and substantially decreases the hands-on and total assay times needed for signal readout, which is crucial to speeding up the pace of drug discovery in the targeted protein degradation space. The technology offers flexibility in detection since the type of bead can be designed to recognize a variety of different engineered protein tags or each bead can be coated with specific antibodies (AlphaLISA) against different epitopes on the target protein ( Fig. 3A ). When bound to the same protein, Alpha donor and acceptor beads come into proximity and are excited by near far-red light (680 nm), and a chemical reaction ensues in the “donor” bead that causes ambient O2 to enter an excited singlet state. On its own, the creation of singlet O2 is not enough to generate a signal, but when “acceptor” beads are in close proximity, the transfer of singlet O2 triggers a chemiluminescent response, which in turn produces an amplified fluorescent signal. In the case of AlphaLISA SureFire, acceptor beads contain europium, which has improved spectral characteristics compared with precursor AlphaScreen beads that use rubrene.
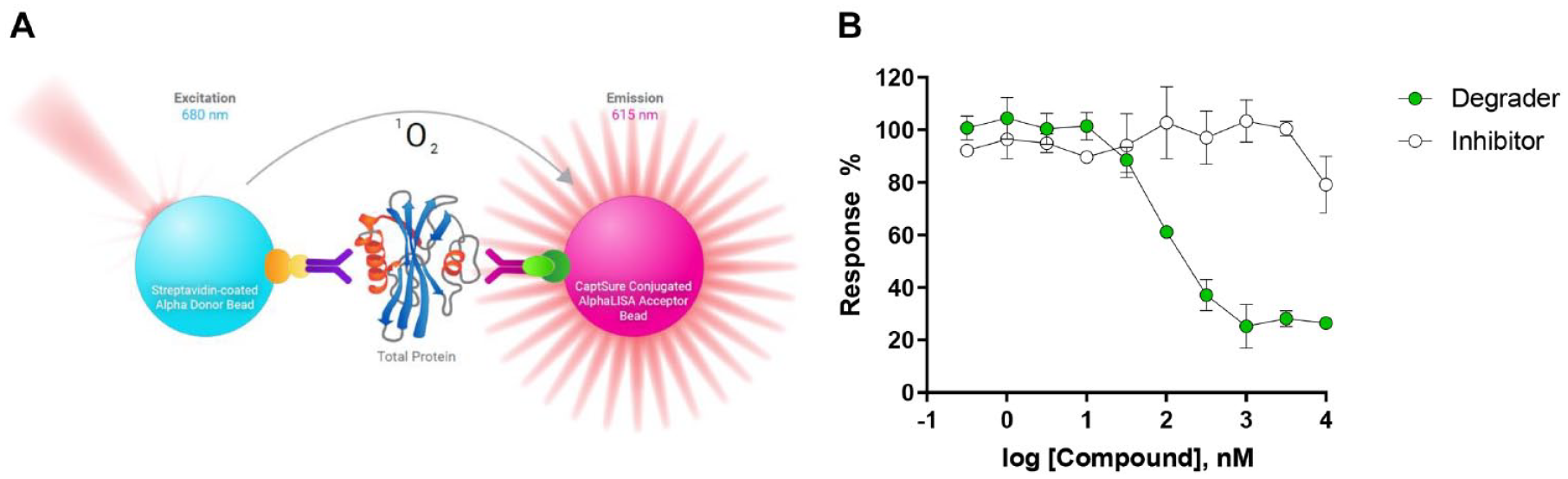
Schematic of the AlphaLISA SureFire Ultra Assay principle. (
There are two commonly used methods for running these assays, depending on the specific needs and cell type. In a two-plate high-throughput screening (HTS) assay format, test degraders are first added to the 384-well plate, mixed with an appropriate number of cells expressing the target of interest, and incubated for a desired time period. The plates are covered during incubation at 37 °C with 5% CO2 during the assay and then lysed with lytic buffer for a previously optimized period of time to ensure complete cell lysis. In a subsequent step, a small amount of lysate is transferred to an Alpha HTS plate, and bead detection reagents are then added to the sample (either simultaneously or sequentially) and incubated before the signal is measured in an appropriate plate reader. The length of the detection step incubation will depend on the quality of interactions between the beads and protein, where binding to an affinity tag may be significantly shorter than when capturing endogenous proteins using antibody-coated beads, which depends more on the quality of the validated antibodies. The fluorescence signal can be normalized to 0% response (high signal: cells treated with DMSO) and 100% response (low signal: no Alpha beads) controls to determine the degree of targeted protein degradation ( Fig. 3B ). Alternatively, the assay can be further streamlined and performed in a single 384-well plate. By reducing the number of transfers, the one-plate method may be more reproducible over time. It is important to point out that AlphaLISA SureFire technology only allows for endpoint assays, requiring cell lysis to observe the signal from the detected proteins of interest. It is also possible to visualize how the degradation of the target protein evolves over time by running the assay in multiple HTS plates and lysing each plate at different time points. Overlaying these curves in a single plot provides a nice visualization of how DC50, Emax, and the degradation hook effect change with time.
One of the biggest advantages of this technology is that the approach lends itself to faster optimization, automation, and miniaturization, making it amenable to routine and high-throughput screening of compounds. The signal is intrinsically amplified and leads to femtogram-level sensitivity and a wide dynamic range of 4–5 log units. Alpha assay platforms can be used in both biochemical and cell-based assays and can allow for the detection of analytes ranging in size from small molecules to large viral particles, with a maximum distance of 200 nm between the donor and acceptor beads. The superb sensitivity and dynamic range of the Alpha technology make it the most amenable technology for use in detecting intracellular, secreted, or membrane-bound proteins or other biomarkers either individually or in multiplex assays where multiple targets are detected simultaneously in the same samples. For cell-based assays designed to measure targeted protein degradation, this level of signal in a 384-well format can dramatically cut down on plate reader readout time (0.5–5 s per well). This sensitivity permits the detection of low abundance proteins in 20 µL volumes of sample, even those endogenously expressed and derived from limited material (e.g., primary cells). Compared with Western blots, which may require up to 1 × 106 cells per sample, AlphaLISA SureFire can be used to measure protein degradation with 5000 cells per condition, or lower if the target protein is expressed at high levels. A further enhancement is enabled by the AlphaLISA SureFire Ultra version of this technology, which exploits proprietary CaptSure immobilization technology to ensure high-fidelity coating of acceptor beads with a validated antibody against the target protein of interest. This lowers background signal over previous iterations of the bead-based technology, further enhancing the sensitivity and dynamic range and enabling the detection of targets in cell media containing phenol red, other complex matrices, and cell or tumor tissue lysates to broaden its applicability to drug discovery, preclinical studies, and translational research.
Using the appropriate antibodies, it may be possible to differentiate between target engagement and subsequent degradation by using antibodies against posttranslational modifications on a target protein. For example, the relative impacts of inhibition and degradation for a BiDAC compound targeting a kinase that has autophosphorylation activity can be assessed directly using antibodies that recognize the phosphoprotein specifically, especially when compared with measurements of the total protein (phosphorylated and unphosphorylated). Getting a readout on both events may improve our understanding of how degraders may or may not differentiate from their inhibitor precursors in driving desired phenotypic responses to target protein modulation. However, the requirement of two antibodies per readout may make it challenging to apply this technology to less well-characterized proteins. For these proteins, beads that address affinity tags might be more amenable if antibodies to native targets are not available for assay development.
Despite all these advantages, it is worth noting a few limitations of this technology. Alpha beads are expensive and are highly sensitive to ambient light, necessitating a darkened area for adding assay reagents and the taking of due considerations to eliminate prolonged exposures to light during the final incubation period prior to readout. Additionally, the temperature of the plate reader can affect the rate of generation and diffusion of singlet O2 by as much as 10% per degree Celsius. Therefore, to minimize run-to-run variability, assay plates and plate readers should be maintained in a well-regulated environment devoid of temperature fluctuations. Lastly, transition metals (Fe2+, Fe3+, Cu2+, Ni2+, Zn2+, Al2+) are strong singlet oxygen quenchers when present in appreciable amounts. It is also widely known that metals such as Zn+2 are sometimes ubiquitous and can leach into compound samples during synthesis and workup procedures and be disruptive to HTS campaigns. 50 To ensure that responses are due to compound activity and loss of the protein target rather than signal quenching due to heavy metal ions, caution should be taken to validate all results in a counterscreen employing a chelation agent such as EDTA. (Note: According to the manufacturer, AlphaLISA technology can tolerate up to 100 mM EDTA in samples.)
Time-Resolved Fluorescence Energy Transfer-Based Assays
Time-resolved fluorescence energy transfer (TR-FRET) assays such as HTRF or Lance (Perkin Elmer, Waltham, MA) are an alternative high-throughput approach available to monitor changes in cellular protein levels 51 and aid efficient and rapid drug discovery efforts in the targeted protein degradation field. Similar to the AlphaLISA SureFire technology, TR-FRET is a straightforward homogeneous mix-and-read sandwich-based immunoassay approach that does not require multiple wash steps prior to signal detection. Rather than utilizing a bead-based detection technology, degrader-induced loss of target protein is determined through quantification of a ratiometric FRET signal between two fluorophore-tagged antibodies that bind to two different epitopes on the same protein, one labeled with a donor and one labeled with an acceptor fluorophore ( Fig. 4A ). After incubation of cells with degrader molecules, the cells are lysed, and detection antibodies are added to the lysates. Like Alpha-based assays, HTRF and Lance enable highly reproducible and highly sensitive measurements (detection limits down to 10 pg/mL), allowing cell densities in the range of 2000–1000 cells per well in assay volumes as low as 10 µL in low-volume 384-well plates. The hands-on and total assay time are also comparable to AlphaLISA protocols.
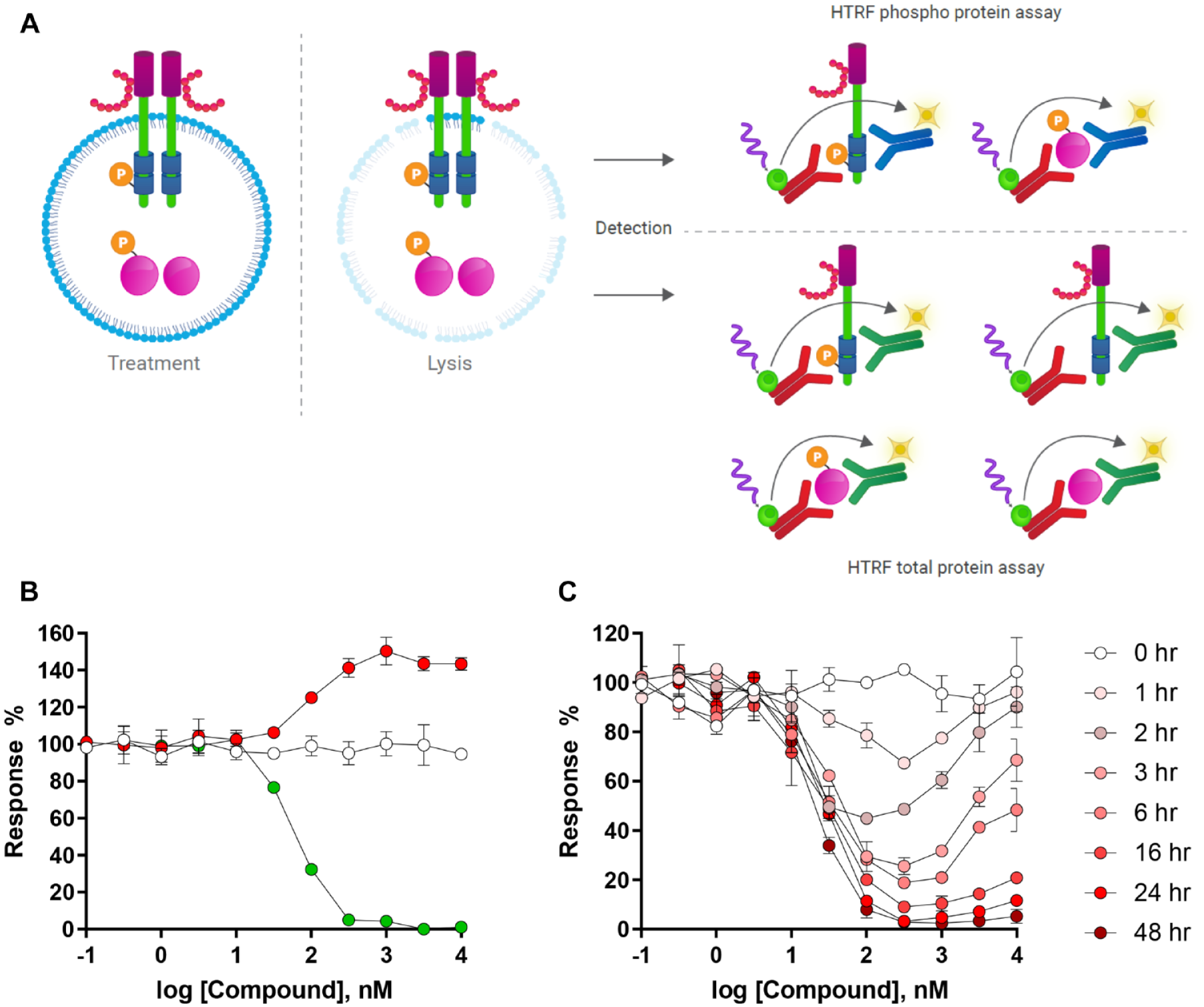
Schematic of the HTRF TR-FRET assay principle. (
In a typical experiment, test degraders are added to the 384-well plate, mixed with an appropriate number of cells expressing the target of interest and incubated for a desired time period. The plates are covered during incubation at 37 °C with 5% CO2 during the assay. After the incubation period with degrader compounds, lytic buffer is added to cells for at least 30 min and detection antibodies are added to the plate and allowed sufficient time to recognize and bind to the remaining target and are incubated for at least 4 h to ensure an optimal signal-to-background ratio. This final incubation step is required to ensure that each detection antibody has ample time to bind to two different epitopes on the protein target. Plates are then scanned in an appropriate plate reader and FRET signal is normalized to 0% response (high signal: cells treated with DMSO) and 100% response (low signal: no cells) controls to determine the degree of targeted protein degradation. The directionality of the fluorescence response is directly correlated with changes in protein level, where an upward response reflects compound-induced stabilization of the protein and a decrease in signal indicates compound-driven loss of protein ( Fig. 4B ). A sigmoidal fit of the resulting data allows the potency (DC50) and efficacy (Emax) to be determined at any given time point. Similar to the AlphaLISA technology, TR-FRET-based assays are endpoint assays, requiring cell lysis to observe the signal from the detected proteins of interest. To build an understanding of the kinetics of target degradation, TR-FRET assays must be set up in parallel using multiple HTS plates so that cells can be lysed, and detection antibodies added after each time point ( Fig. 4C ). Overlaying these curves in a single plot provides information regarding the shift in DC50, Emax, and widening of the degradation hook effect observed with time.
As with many of the previously described approaches for measuring protein degradation in cells, TR-FRET-based assays such as HTRF rely heavily on the availability of validated antibodies to detect and measure endogenous or ectopically expressed, unmodified, untagged full-length target proteins. For the detection of each biomarker, it is critical that both antibodies bind to the same protein using different epitopes to enable the FRET signal while also keeping background signal down to a minimum. The assays can be multiplexed, measuring more than one biomarker per sample, but none of the antibody epitopes can overlap in that case. If there is any overlap, multiple biomarkers must be detected using duplicate assay plates, each receiving detection antibodies against an individual biomarker. As with the Alpha technology, the approach can be used to measure inhibition of posttranslational events on the target protein, allowing for quantitation of pathway inhibition as well as total protein levels. Moreover, selection of mutant-specific antibodies might allow assays to be developed to specifically monitor the targeting and degradation of oncogenic mutant proteins in heterozygously expressing cells without the confounding effects of detecting the wild-type version of the same protein. This would be a powerful approach for optimizing degraders that are selective for the oncogenic driver mutants while minimizing degradation of the wild-type version of the same protein in normal tissues. The requirement of two antibodies per readout may make it challenging to apply this technology to less well-characterized proteins, but similar to the AlphaLISA SureFire technology, there are many options for assay development with this technology. The availability of validated antibodies for less well-characterized proteins may be a challenge; HTRF assays can be adapted to detect endogenously or ectopically expressed engineered tagged proteins by instead employing validated antibodies against one or more affinity tags to enable the FRET signal. Additionally, it is always possible to redevelop or reoptimize existing assays to improve assay quality using any new validated antibodies that become available, provided that it is custom labeled with the correct fluorophore.
While the flexibility of the assay build is an attractive feature of this type of assay technology, the final assay design requires a pair of validated fluorescent-labeled detection antibodies, and these reagents can be expensive to purchase throughout the drug discovery process. The cost per data point for TR-FRET assays is probably the largest disadvantage of this technology when used for prolonged drug discovery efforts, although costs can be defrayed significantly by custom-labeling antibodies in bulk and avoiding costly purchases of ready-made assay kits. Despite higher cost, the flexibility, adaptability, and transferability of TR-FRET-based assays offer some advantages. Once an HTRF assay is established against a target protein in one cell line, it is usually readily transferable to other cell lines expressing the same protein. Further, TR-FRET-based technologies such as HTRF implement the use of long-lifetime fluorescent lanthanide chelates on the acceptor antibody, rather than more commonly used fluorophores, and allows the emission measurement to be taken at some delayed time after the excitation of the donor fluorophore. Additionally, the donor and acceptor fluorophore employed by the HTRF technology also induce a large Stokes shift. The combination of these factors reduces background signal and improves the overall signal-to-background ratio, increases dynamic range, and extends signal stability without the fear of significant quenching from ambient light. Additionally, HTRF assays can be further miniaturized to 10 µL volumes. Even at these volumes, these assays are amenable to 24 h incubation times with compounds, provided that the assay plates are covered for the duration of the assay. Any edge effects that may occur due to sample evaporation with time are usually offset by the ratiometric fluorescence readout from each sample well, which helps to factor out volumetric differences across wells, thereby improving reproducibility and overall data quality.
Nano-Glo HiBiT Technology
The most recent HTS technology to enter as a powerful drug development tool in the targeted protein degradation space is the Nano-Glo HiBiT Lytic Detection System (Promega, Inc., Madison, WI). The HiBiT technology is based on NanoLuc Binary Technology (NanoBiT), in which NanoLuc luciferase is broken down into two complementary components, an 11-amino-acid HiBiT tag and a 17.6 kD polypeptide named LgBiT. 52 Briefly, the 11-amino-acid HiBiT peptide tag can be introduced into an endogenous copy of the gene encoding the protein of interest using CRISPR (endogenous system) or engineered into a recombinant DNA expression construct that can be introduced into cells by plasmid transfection or lentiviral infection (ectopic system). In the presence of the complementary LgBiT protein, the NanoLuc luciferase is reconstituted and can actively utilize a substrate to catalyze the generation of a bright luminescence signal that is directly proportional to the amount of HiBiT-tagged protein of interest in the cell lysate ( Fig. 5A ). The linear range of detection covers protein concentrations that range over several orders of magnitude, and the resulting luminescent signal is stable for hours and thus suitable to be used in HTS campaigns that drive drug discovery efforts for protein degraders.
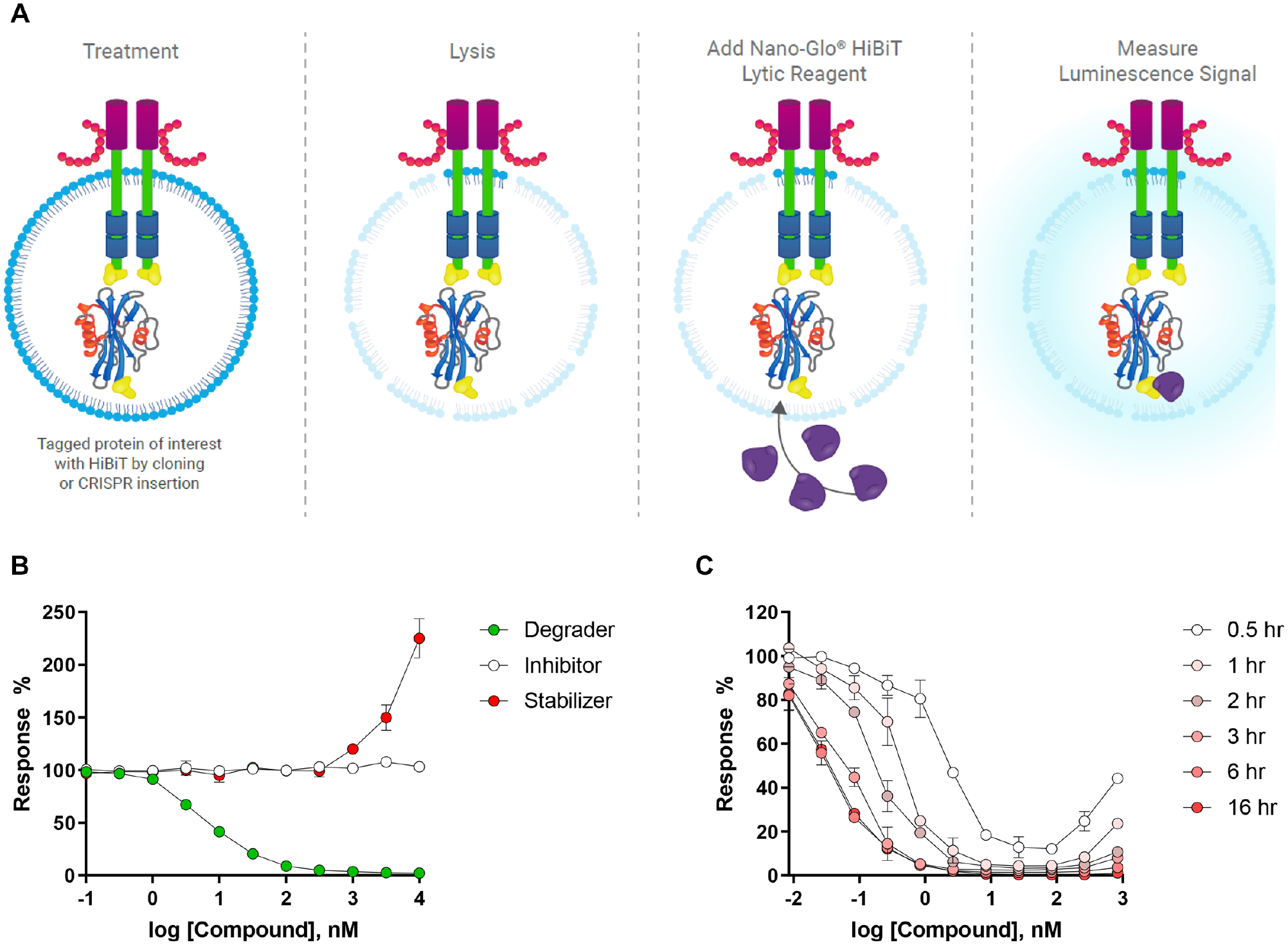
Schematic of the Nano-Glo HiBiT Lytic Detection System. (
Factoring in the time needed for CRISPR/Cas9 knock-in of the HiBiT tag into the protein sequence of interest and to generate a cell line stably expressing HiBiT-tagged target protein, the overall assay development time requires a minimum of 3–4 weeks. Provided that guide RNAs are available for each locus and transfection protocols for the chosen cell line are known, the HiBiT tag can be knocked in at either the N- or C-terminus. However, it is also important to note that the availability of suitable guide sequences may limit tag placement for some protein targets. After engineered cells are allowed to grow out in culture, the intensity of HiBiT signal from the expressed target protein is measured at different cell densities. Poor signal intensity from a large pool of cells could signal inefficient transfection and/or knock-in efficiency and may require the process to be repeated a second time to boost the overall signal. Another alternative to boost HiBiT signal is to try to identify single clones where the transfection was highly efficient, resulting in robust luminescence signal of the HiBiT-tagged protein relative to an equivalent amount of cells from the mixed pool, most of which may not contain the HiBiT tag. If a single clone is required for a high-quality assay with robust HiBiT signal, identifying a clone will add an additional 2–3 weeks to the assay development timeline. This could be considered a disadvantage of this approach over the Alpha SureFire or HTRF technology, which do not require CRISPR modification of the target protein. However, once a stably expressing clone or population of cells has been generated, which produces sufficient signal, the final steps of assay development move quickly and enable straightforward homogeneous mix-and-read assays that do not require multiple wash steps before signal detection.
In a typical HiBiT endpoint assay, test degraders are added to the 384-well plate, mixed with an appropriate number of cells expressing the HiBiT-tagged target protein, and incubated for a desired time period. The plates are kept covered during incubation at 37 °C with 5% CO2 during the assay. After the incubation period with compounds, the cells are lysed with Nano-Glo HiBiT Lytic Detection Reagent (composed of Lysis Buffer, LgBiT protein, and furimazine substrate) for only 10 min with gentle agitation, which is among the fastest turnaround time from lysis to signal readout of all the previously described assay technologies. The plates are then scanned in an appropriate plate reader and luminescent signal can be normalized to 0% response (high signal: cells treated with DMSO) and 100% response (low signal: no cells, only growth media) controls to determine the degree of targeted protein degradation. The directionality of the luminescence response is directly correlated with changes in protein level, where an upward response reflects compound-induced stabilization of the protein, and a decrease in signal indicates compound-driven loss of protein ( Fig. 5B ). A sigmoidal fit of the resulting data allows the degradation potency (DC50) and efficacy (Emax) to be determined at any given time point. Similar to the other previously described 384-well-compatible assays, the kinetics of target degradation can be studied by setting up several copies of assay plates and lysing the cells and adding LgBiT detection reagent at increasing intervals of time to track the degradation over time. Overlaying these curves in a single plot provides information regarding the shift in DC50, Emax, and widening of the degradation hook effect observed with time ( Fig. 5C ).
Alternatively, the HiBiT technology can be adapted to a single-plate method for collecting real-time kinetics information. In this setup, the LgBiT protein can be introduced ectopically by lentiviral infection into cells already expressing the HiBiT-tagged target protein. Both HiBiT and LgBiT tags are expressed simultaneously in the cells, allowing the reconstituted luminescent NanoBiT enzyme to be present throughout the experiment, and signal can be measured over time by supplementing cells with the Nano-Glo Endurazine and Vivazine Live Cell Substrates (https://www.promega.com/products/luciferase-assays/reporter-assays/nano-glo-extended-live-cell-substrates). Endurazine and Vivazine allow a slow rate of ester hydrolysis by cellular esterases, which leads to the steady release of furimazine substrate throughout the experiment. Once formed, furimazine serves as a substrate for NanoLuc and NanoBiT luciferases. Since this is a single nonlytic reagent addition step, this approach is more cost-effective in terms of assay plate and HiBiT reagent costs. This nonlytic single-plate approach allows for real-time measurement of targeted protein degradation for periods lasting from minutes to hours to days. However, although it saves on material costs, it should be noted that real-time degradation on the scale of hours to days will require either a microplate reader with a microplate cell incubator attachment or a robotic system that can transfer the microplate between the incubator and a standard plate reader at the desired time points.
Lastly, it is also important to mention that coexpression of LgBiT protein and the HiBiT-tagged target protein in the single-plate method will result in the full reconstituted 13 kD NanoLuc luciferase being present at the N- or C-terminal end of the target protein. This could modulate the ternary complex formation that would normally occur between a BiDAC, target protein, and E3 ligase. 18 The small HiBiT tag (11-amino-acid peptide, ~1.3 kDa) minimizes interference or modulation of the ternary complex formation between the target protein and the E3 ligase and other components of the UPS and represents an improvement over larger full-length reporter gene tags. Traditional reporters such as HaloTag (33 kD), YFP/GFP/RFP (27 kD), and NanoLuc (19 kD) are large in size and could disrupt protein–protein interactions made with the native target and interfere with polyubiquitination and/or degradation of the target protein. Moreover, these large protein reporters may themselves contain several lysine residues, which could become ubiquitinated upon formation of the ternary complex. Ubiquitination of the reporter gene tag itself would also result in degradation of the target protein as a single construct, but it would not be driven through ubiquitina-tion of the target itself and could be misleading to drug discovery efforts. This may also occur with the 11-amino-acid HiBiT tag, but with significantly lower frequency than would occur with a large reporter gene tag enriched with lysine side chains.
Concluding Remarks
The optimization of protein degraders is a complex undertaking that requires refinement around all three regions of the molecule, including the binding ligands to both the target and E3 ligase, as well as the linker region of the molecule. While the targeted protein degradation field has long used Western blots as the traditional method for measuring the activity of degradation activating compounds, there is an urgent need for high-throughput assay methods to inform and drive medicinal chemistry efforts and rapid drug discovery. We have reviewed several different methods that have been employed in this field, ranging from low to high throughput (summarized in Fig. 6 ). The high-throughput assay technologies are all robust, sensitive, reproducible, and quantifiable, but each comes with associated limitations, as discussed. Where possible, we have offered potential workarounds to overcome some of these limitations.
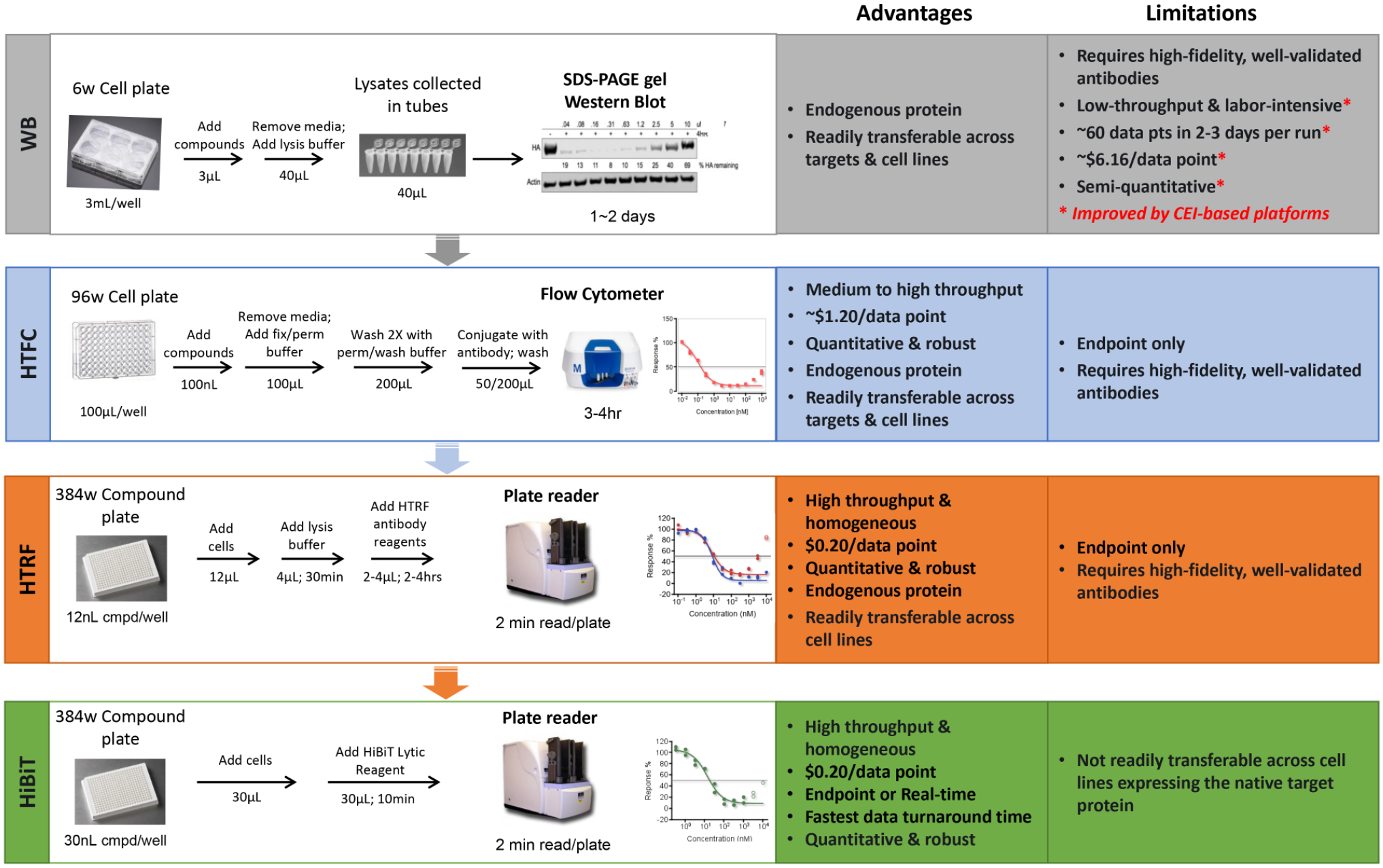
Recent advancements in assay technologies to drive rapid degrader drug discovery.
The decision of which assay should be used to drive a drug discovery program is one of the most important decisions to be made early on, and a number of factors should be considered, including cost, time, and feasibility. It is important to note that the overall strategy will also impact assay selection. Some targets may benefit from an approach where data-heavy HTFC or ICW-type approaches may be informative for the optimization of BiDACs that drive a particular phenotypic result. On the other hand, some target proteins may warrant a more pragmatic tiered screening funnel with faster data turnaround times using some of the homogenous fluorescence-based assays discussed in this perspective. A tiered assay approach would rely more heavily on a primary protein degradation assay but would also incorporate additional technologies and platforms to more fully characterize BiDACs as they become more optimized throughout the drug discovery process. In our experience, this tiered approach allows for maximized efficiency in data collection to drive earliest BiDAC discovery and optimization efforts. Regardless of the strategy implemented, it goes without saying that all assays and reagents used to detect the target proteins (i.e., antibodies) must be thoroughly validated before compound screening commences.
If it is straightforward to use CRISPR to knock in the HiBiT tag to the N- and/or C-terminus of the target protein, HiBiT is the preferred method for a workhorse screening assay used to drive the rapid optimization of degradation potency, Emax, and the catalytic efficiency of degraders because it is amenable to both endpoint and continuous read assay formats, thereby offering more datatype/readout flexibility at a cost comparable to TR-FRET. On the other hand, TR-FRET is a suitable alternative, provided that highly specific well-validated antibodies are available against the target of interest, and there are already many assay-ready kits that are commercially available for a number of highly valued drug targets. TR-FRET does not offer the flexibility of both endpoint and continuous read assay formats, nor can it match the wide dynamic range of HiBiT, but it is certainly robust enough to support high-throughput drug discovery efforts. However, TR-FRET does offer the attractive option for follow-up screens in multiple different cell lines expressing the same target. Since the cost of both technologies is comparable, the target itself will likely dictate which of these two technologies is most suitable for degrader development. Finally, it is important to note that any one of these technologies improves upon the throughput offered by traditional Western blots and is suitable for research aimed at improving our understanding of the downstream pharmacology and consequences of targeted protein degradation. However, we recommend that these workhorse screening assays be complemented by other technologies and platforms to fully characterize BiDACs, including confirming the degradation of endogenous protein by tradition Western blot to ensure that results align with a HiBiT-tagged protein or using viability assays such as CellTiter-Glo to determine if there is any cytotoxic effect of BiDACs that may be misinterpreted as degradation in a primary screening assay. Regardless of the preferred strategy, as research in the targeted degradation field intensifies over the coming years, a shift to more high-throughput technologies and automation to support these types of platforms would be a worthy investment to accelerate drug discovery efforts.
Footnotes
Acknowledgements
The terms MonoDAC and BiDAC are trademarks of C4 Therapeutics, Inc.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.