
Abstract
There is a growing need in drug discovery and basic research to measure multiple second-messenger components of cell signaling pathways in real time and in relevant tissues and cell types. Many G-protein–coupled receptors activate the heterotrimeric protein, Gq, which in turn activates phospholipase C (PLC). PLC cleaves phosphatidylinositol 4,5-bisphosphate (PIP2) to produce two second messengers: diacylglycerol (DAG), which remains in the plasma membrane, and inositol triphosphate (IP3), which diffuses through the cytosol to release stores of intracellular calcium ions (Ca2+). Our goal was to create a series of multiplex sensors that would make it possible to simultaneously measure two different components of the Gq pathway in living cells. Here we describe new fluorescent sensors for DAG and PIP2 that produce robust changes in green or red fluorescence and can be combined with one another, or with existing Ca2+ sensors, in a live-cell assay. These assays can detect multiple components of Gq signaling, simultaneously in real time, on standard fluorescent plate readers or live-cell imaging systems.
Keywords
Introduction
Cell signaling involves the concerted activity of multiple second-messenger pathways. Figure 1 is a simplistic diagram of just Gq signaling, and it illustrates how many different proteins and second messengers are involved in parallel pathways. It is the balance of these different signaling components, coordinated in both space and time, that ultimately dictates the response of the cell. Although this is well understood in theory, the practice of measuring signaling is often reduced to two time points—before and after drug—and to a single second messenger. When kinetic measurements of signaling are possible, a new level of precision and insight guides new experiments and optimized assays. In the cases that it has been possible to image multiple components of a signaling pathway in the same cells,1–4 the interplay between the different components has provided new insights into the biological system and the downstream consequences of a drug’s actions.
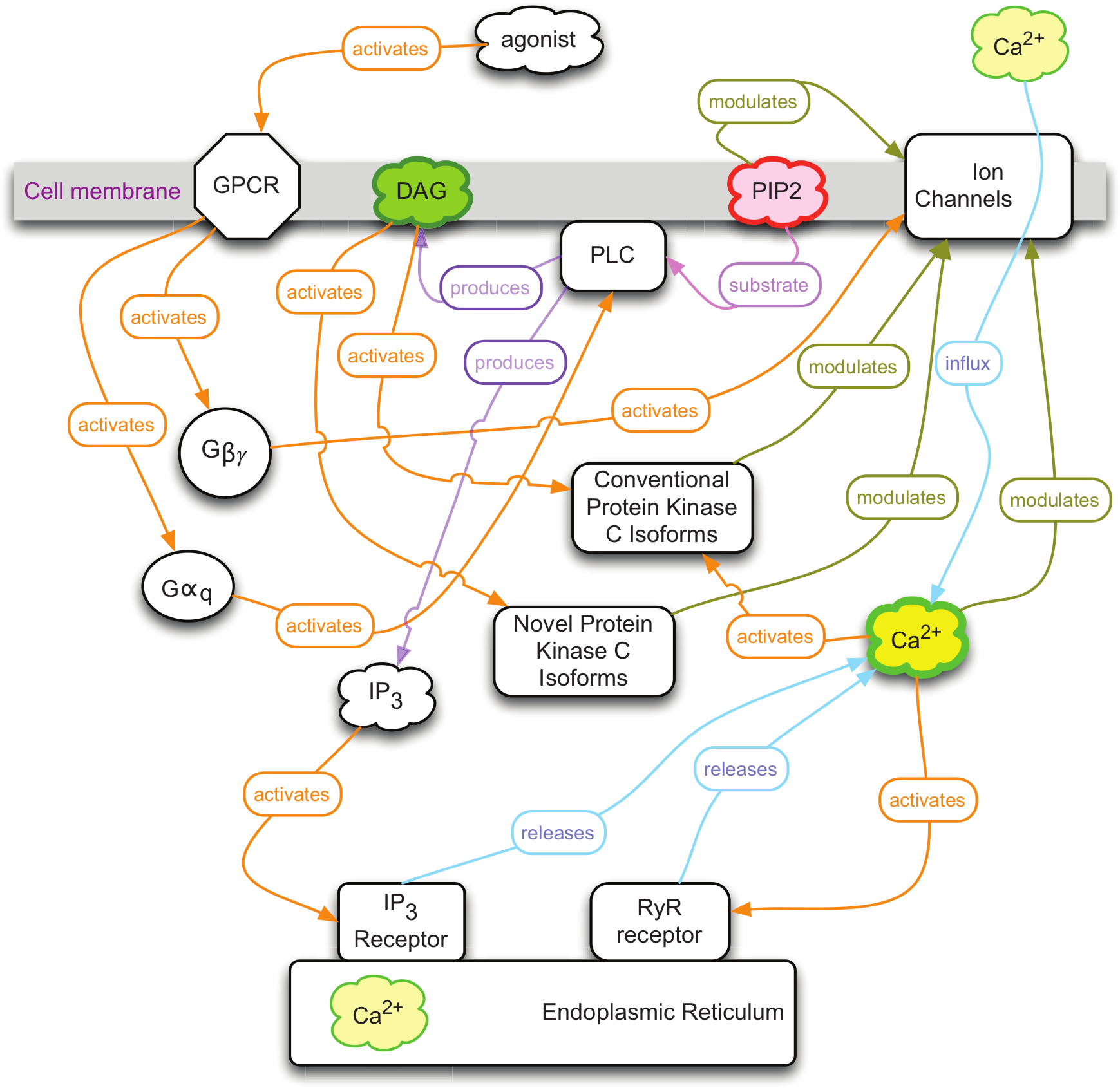
A simple diagram of G-protein–coupled receptor (GPCR) signaling through Gq. The activated receptor catalyses the activation of the heterotrimeric Gq protein. The Gqα activates phospholipase C (PLC), which cleaves phosphatidylinositol 4,5-bisphosphate (PIP2) to produce both diacylglycerol (DAG) and inositol 1,4,5-trisphosphate (IP3). The IP3 triggers the release of Ca2+ from intracellular stores, and the combination of raised intracellular Ca2+ and DAG activates conventional protein kinase C enzymes with a multitude of downstream targets. The goal of this work was to create independent sensors for DAG and PIP2 that could be multiplexed with one another, as well as existing Ca2+ sensors, to detect the concerted pattern of Gq signaling activity. No one sensor in isolation is capable of unambiguously detecting Gq signaling since other pathways can produce elevated levels of DAG, PIP2, or Ca2+.
Multiplex sensors capable of simultaneously detecting different signaling components are particularly important to advancing our search for drugs that interact with G-protein–coupled receptors (GPCRs). This importance can be appreciated in the context of either the traditional view of GPCR signaling or in the framework of the more recent agonist-bias signaling at particular GPCRs.
Traditional models of GPCR signaling involve activated G-protein α and βγ subunits as the crucial first components of signaling, which then act upon effectors. Even in this relatively simple model, there are multiple effectors and multiple second messengers, all acting in concert. Most, if not all, of these second messengers can be influenced by other signaling pathways. Even in this model, multiplex sensors are necessary to decode the pathway or pathways involved in the response to the activation of a particular GPCR. For example, Gq signaling produces a rise in intracellular Ca2+, but many other pathways do as well. To unambiguously identify Gq signaling, it is necessary to measure other components as well.
In the more recent model of agonist-biased signaling, an even more pressing case can be made for multiplex sensor systems.5–9 A wide variety of evidence, including recent structural studies, has culminated in a multistate model of GPCR activation in which different agonists stabilize a particular receptor in a conformation that activates a unique pattern of intracellular signaling. In simple terms, different agonists can produce different levels of Gα, Gβγ, and arrestin signaling. This selectivity is particularly important in the scenario where a receptor can access both an important activity, such as analgesia, via one pathway and unintended consequences, side effects, via another. 10 Multiplex sensors that can simultaneously measure multiple pathways will be critical to assessing the biological relevance of a particular drug. 7
What are the design criteria for optimal multiplex sensors? First, they need to work in living cells and provide kinetic data for each signaling pathway. This means they need to provide strong signals that can be sampled at the Nyquist frequency, which for cellular signaling events (200 ms to 5 s) can be up to 10 Hz. Second, each sensor needs to consume as little of the visible spectrum as possible so that there is minimal crosstalk with other sensors. Finally, each sensor has to specifically detect the second messenger at physiologically relevant concentrations.
Fluorescent protein-based sensors meet many of the design criteria: they work in living cells, they produce strong signals that can be sampled repeatedly and quickly, and the protein domains they carry have evolved to specifically detect a particular second messenger. 1 However, early sensors based on Förster resonance energy transfer (FRET) between two different fluorescent proteins rarely produce the sort of robust signals necessary for automated detection. Furthermore, the broad absorption bands of the donor and acceptor fluorophores consume most of the visible spectrum.11,12
More recently, a new generation of fluorescent protein sensors has been developed that only uses one fluorescent protein, produces large changes in fluorescence, and has the potential for multiplexing. Many of these new sensors carry a single, circularly permuted fluorescent protein that converts analyte binding into changes in fluorescence intensity. The green fluorescent G-CaMP Ca2+ sensors,13–15 the red R-GECO Ca2+ sensors, 16 the green ElectricPk voltage sensor, 17 the green cGMP sensor, 18 and our own prototype sensors for diacylglycerol (DAG) use this approach. 19
Many drugs act at GPCRs on the cell surface. Some of these receptors couple to the heterotrimeric protein, Gq, which activates phospholipase C (PLC). PLC in turn cleaves phosphatidylinositol 4,5-bisphosphate (PIP2) to produce two second messengers: DAG, which remains in the plasma membrane, and inositol triphosphate (IP3), which diffuses through the cytosol to release stores of intracellular calcium ions (Ca2+). This coordinated increase, in both DAG and cytosolic Ca2+, triggers the activation of conventional isoforms of protein kinase C (cPKC), which then phosphorylate many different targets. To explore the potential of multiplex sensors for the Gq pathway, particularly in the context of drug discovery and laboratory automation, we improved our green fluorescent DAG sensors; created a new, robust red fluorescent PIP2 sensor; and multiplexed these with one another or with existing green and red Ca2+ sensors.
Materials and Methods
PCR-amplified fragments of the PKCδ coding region and a circularly permuted green fluorescent protein (GFP) from G-GECO1 16 were combined and cloned into a modified version of the mammalian expression vector pcDNA3.1 using the In-Fusion Cloning system (Clontech Laboratories, Mountain View, CA). The pcDNA3.1 vector was obtained from Life Technologies (Grand Island, NY). Sixty-four unique candidate diacylglycerol sensors resulted from combining fragments of PKCδ with circular permuted enhanced GFP (cpEGFP). Thirty-two constructs contained the cpEGFP insert in the full-length PKCδ, and an additional 32 constructs contained enhanced GFP in a truncated PKCδ where the C2 domain was deleted.
HEK293 cells 20 were cultured in Eagle’s minimal essential medium (EMEM) supplemented with 10% fetal bovine serum and penicillin-streptomycin at 37 °C in 5% CO2. The cells and EMEM were purchased from ATCC (Manassas, VA). Prior to cell seeding, 96-well glass-bottom plates were coated with poly-D-lysine. Cells were seeded on the plates, transfected using Lipofectamine 2000 Transfection Reagent according to the manufacturer’s protocol, and incubated for 24 to 48 h at 37 °C in 5% CO2. Then, 60 ng sensor DNA was co-transfected with 40 ng human M1 muscarinic acetylcholine receptor per well. Penicillin-streptomycin liquid and Lipofectamine 2000 were obtained from Life Technologies. Poly-D-lysine was purchased from Fisher Scientific (Pittsburg, PA).
EMEM culture medium was replaced with 1× Dulbecco’s phosphate-buffered saline (DPBS) prior to screening transfected cells for fluorescence. A Zeiss Axiovert S100TV inverted microscope equipped with computer-controlled excitation/emission filter wheels, shutters, and a Qimaging Retiga Exi CCD camera (Surrey, Canada) was used to image cells at 25 °C using the 10× objective lens (N.A. 0.3). A combination of 480 ± 20–nm excitation and 535 ± 25–nm emission filters was used resolve the green fluorescence from the DAG sensors, and 572 ± 20–nm and 630 ± 30–nm filters were used to collect the R-GECO signal. Cells were analyzed for increases or decreases in fluorescence intensity upon addition of carbachol, phorbol 12,13-dibutyrate (PdBU), DMSO, or ionomycin. To analyze the image stacks, background fluorescence was defined as a region of the image that contained no cells. The average value of this region was subtracted frame by frame from the measurements of the mean pixel values of the fluorescent cells. Fluorescence intensity data were plotted and analyzed with IGOR (Wavemetrics, Oswego, OR).
For transient expression and screening in an automated fluorescence plate reader, HEK293T cells were cultured in Corning Co-Star polystyrene 96-well plates (Corning, Tewksbury, MA) coated with poly-D-lysine. Then, 70 µL of a 25-µg/mL poly-D-lysine solution was added to each well for 1 h, and then the well was rinsed 1× with sterile DH2O and dried (coating concentration ~5 µg/cm2). HEK293T cells were plated at 35,000 cells/well in 100 µL growth medium per well without antibiotics so that the cells would be 90% to 95% confluent at the time of transfection (approximately 24 h later). For each transfection (i.e., one well in a 96-well plate), 160 ng plasmid DNA (120 ng sensor + 40 ng receptor) was diluted in 25 µL Opti-MEM, 0.48 µL Lipofectamine 2000 was diluted in 25 µL Opti-MEM, and these were then mixed and added to the cells. Cells were incubated in this mixture for 4 to 6 h, and then the mixture was replaced with fresh medium. Prior to scanning a plate on the Biotek Synergy Mx, EMEM culture medium was replaced with 250 µL of 1× DPBS per well. Plates were read at 25 °C, using monochromators set to 488/20 nm excitation and 530/20 nm emission to resolve the green fluorescence from the DAG sensor.
Results
To reliably detect the activation of the PLC pathway, we created a series of genetically encoded, fluorescent DAG sensors. Sixty-four candidates were produced that fused a circularly permuted green fluorescent protein to PKCδ, which is a novel isoform that only responds to DAG because only the C1 domain is functional.21,22 Two robust prototype sensors, previously described, were recovered from this initial effort. 19 In both of these sensors, the circularly permuted green fluorescent protein and the linker are positioned between the pseudosubstrate and the C1 domains of PKCδ. Surprisingly, one sensor increases fluorescence as a result of activation (Upward DAG), whereas an insertion only 6 amino acids away produces a sensor that decreases fluorescence as a result of activation (Downward DAG). To our knowledge, this is the first example of small change in the position of the fluorescent protein producing an inversion of the signal produced by the sensor. However, it is well known that minor adjustments in the linkers interconnecting the circularly permuted fluorescent protein and the analyte-sensing domains can have a large impact on the amplitude of the fluorescence change.13,14 To optimize these prototype sensors, we created an additional 156 variants of the original Upward and Downward DAG sensors. This produced the Upward DAG2 sensor, which produces a much larger response than the initial Upward DAG sensor, while it continues to display very similar response kinetics. These sensors can be readily coexpressed with the red fluorescent R-GECO1 Ca2+ sensors to simultaneously measure both second messengers in living cells ( Fig. 2A ). Both the onset of the Ca2+ response and the return to baseline are considerably quicker than the DAG response, which is consistent with previous measurements.23,24
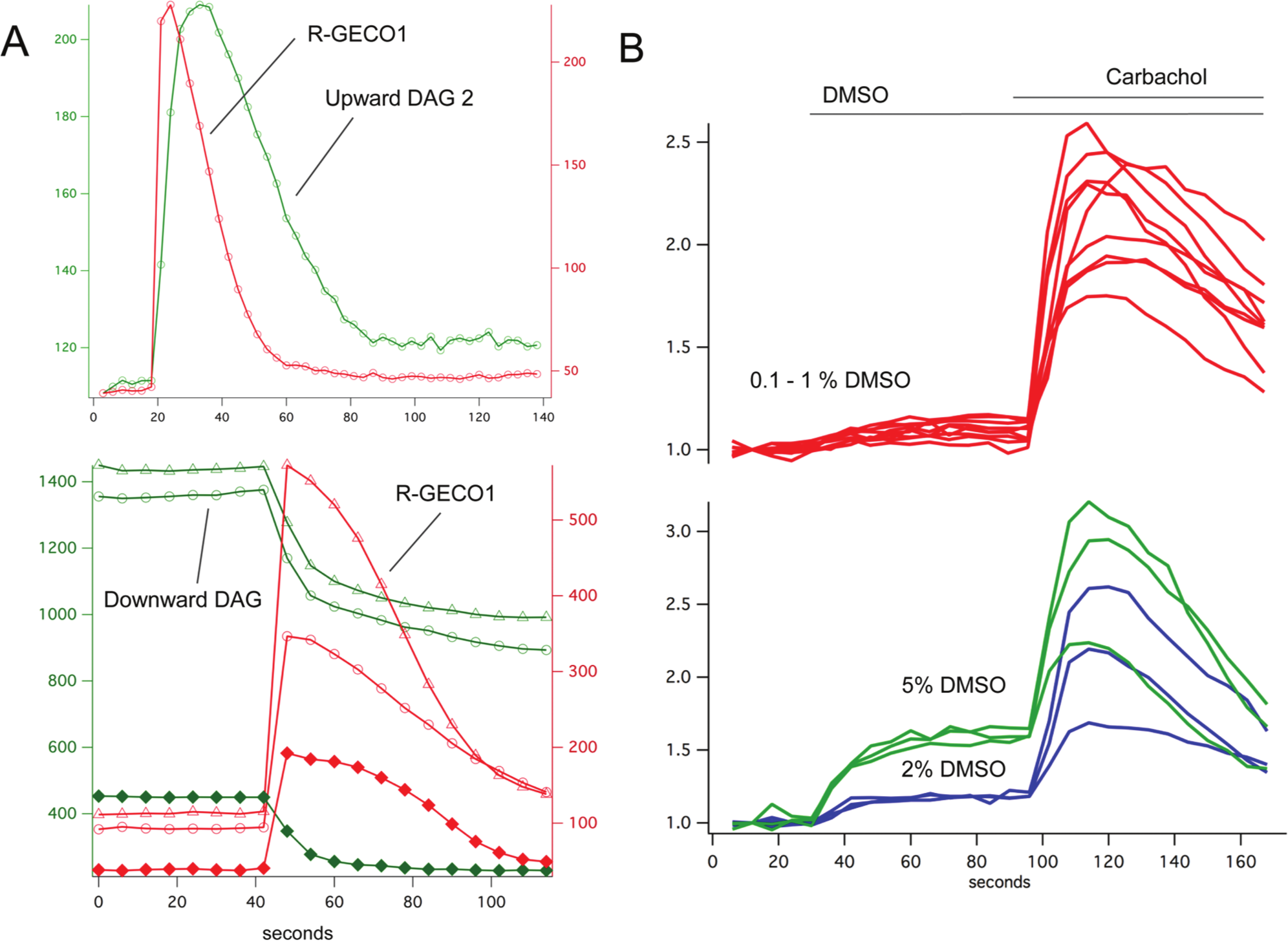
The green fluorescent sensors Upward DAG2 and Downward DAG can be coexpressed with the red fluorescent R-GECO1 to simultaneously measure Ca2+ and diacylglycerol (DAG) signaling in living cells (
Many compound libraries are carried by DMSO, a vehicle that can cause artifacts in live-cell assays. To test the effects of DMSO on the DAG assay, DMSO of different concentrations was added to the culture, followed later by carbachol to evoke the maximal sensor response ( Fig. 2B ). At moderate final concentrations of 0.1% to 1%, the DMSO produced no effect, whereas at higher concentrations, artifactual, DMSO-triggered changes in fluorescence did occur.
PLC hydrolyzes PIP2 to produce both DAG and IP3. To independently check the fidelity and kinetics of the DAG sensors, we created a red fluorescent PIP2 sensor by fusing the pleckstrin homology (PH) domain of PLCδ to two different components of the recently described dimerization-dependent red fluorescent proteins. 25 Previous work has shown that the translocation of the PLCδ PH domain can be used to measure PIP2 turnover, 26 and if the PH domains carry FRET pairs of fluorescent proteins, a small change in FRET occurs when PLC is activated. 27 To create a more robust sensor that does not involve FRET and that produces a larger signal with a single fluorescent protein, we fused the PH domain to each member of the ddRFP pair. One pair of constructs produced a particularly strong red fluorescent signal at the membrane that rapidly disappeared with M1 receptor activation ( Fig. 3A ). This red fluorescent PIP2 sensor, PIP2r, was coexpressed with the green fluorescent G-GECO1 Ca2+ sensor, 16 and stimulation of the M1 receptor produced a rapid, simultaneous rise in Ca2+ and fall in PIP2 levels. Changes in Ca2+ can have profound effects on many cellular processes. To explore the relationship between intracellular Ca2+ levels and the signals produced by our DAG and PIP2 sensors, we first raised Ca2+ levels by adding the ionophore ionomycin and then activated the DAG sensors with PdBU and the PIP2r sensors with M1 receptor activation. Raising intracellular Ca2+ had no apparent effect on the DAG and PIP2 levels ( Fig. 3C , D ).
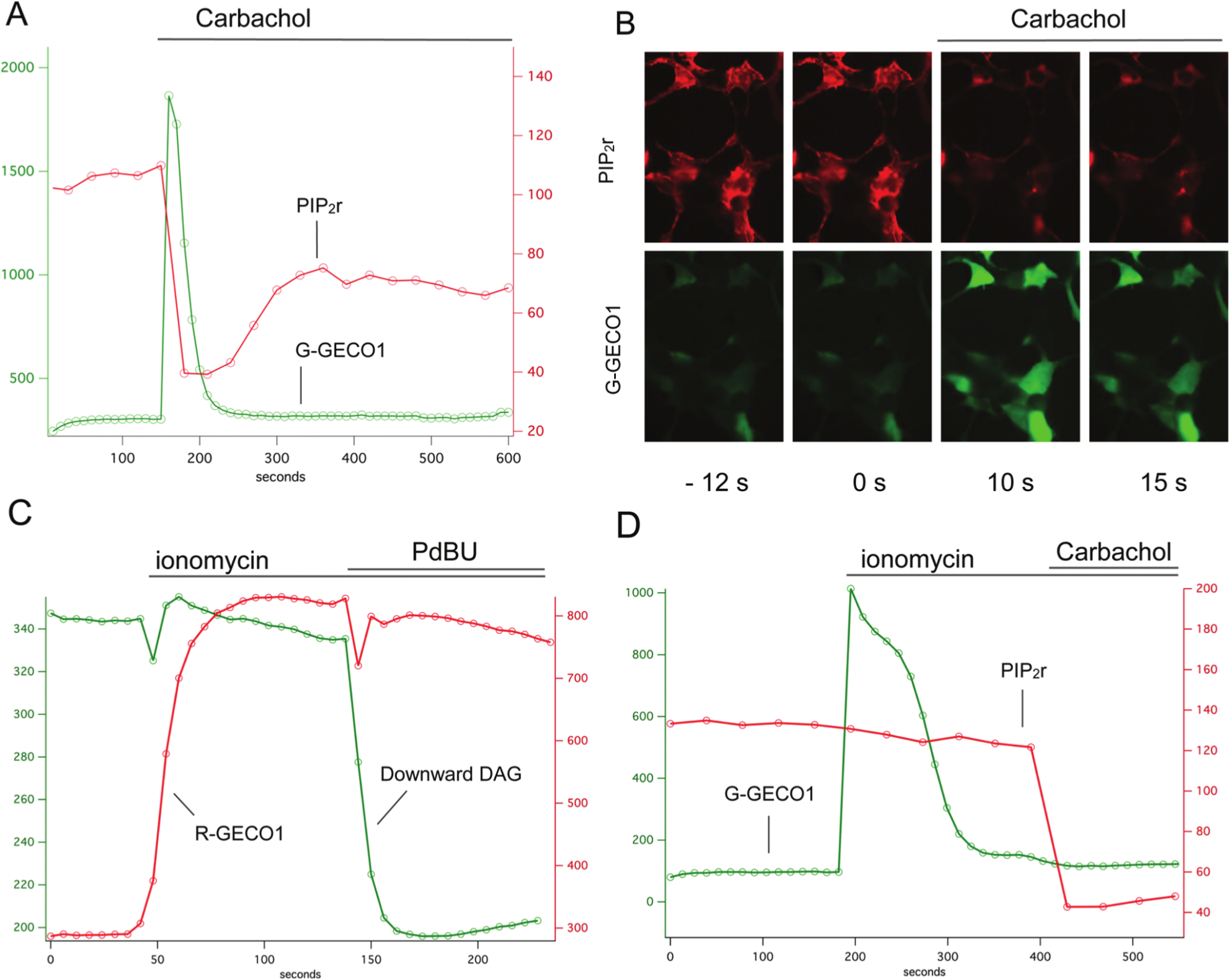
Multiplexing diacylglycerol (DAG), phosphatidylinositol 4,5-bisphosphate (PIP2), and Ca2+ sensors. The red fluorescent PIP2 sensor (PIP2r) was coexpressed with the green G-GECO1 Ca2+ sensor and the M1 receptor. Carbachol addition triggered a simultaneous increase in green fluorescence and decrease in red fluorescence (
Coexpression of the Upward DAG2 or Downward DAG2 sensor with the PIP2r sensor provides a new view of both the substrate and product of PLC ( Fig. 4 ). To our knowledge, this is the first time that genetically encoded biosensors have been used to simultaneously measure substrate and product. M1 receptor activation produced a remarkable change in the intensity and distribution of both probes. As expected, the PIP2r sensor rapidly leaves the membrane and loses fluorescence, whereas the Upward DAG sensor translocates to the membrane and increases fluorescence ( Fig. 4A ). The onset of the response of the Upward and Downward DAG2 sensors, as well as PIP2r, is kinetically quite similar. However, the return to baseline is considerably slower for the Downward DAG2 and PIP2r sensors ( Fig. 4B ). Because this return to baseline varies depending on our sampling rate, our interpretation is that the apparent return to baseline for Upward DAG2 is artificially accelerated by photobleaching and similarly prolonged in the cases of Downward DAG2 and PIP2r.
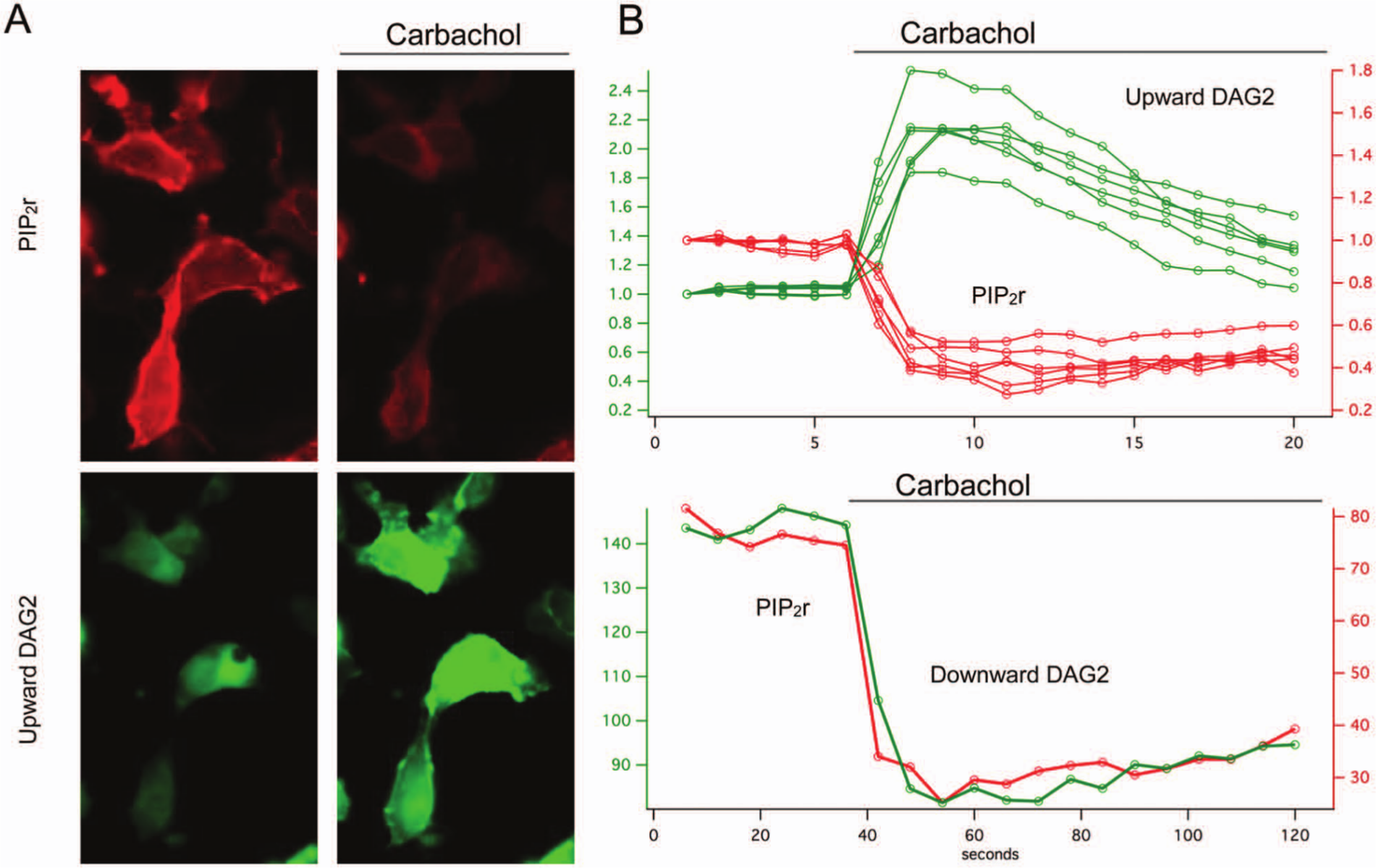
The red fluorescent phosphatidylinositol 4,5-bisphosphate (PIP2r) and diacylglycerol (DAG) sensors can be coexpressed and measured simultaneously. Stimulation of phospholipase C cleaves PIP2 and produces DAG, which is clearly seen in living cells as the red fluorescence of the PIP2r vanishes and the Upward DAG2 sensor increases in fluorescence (
White and colleagues 28 have reported that adenosine triphosphate (ATP), acting at the P2Y11 receptor, produces inositol phosphate turnover and transient Ca2+ signaling consistent with Gq signaling, whereas uridine triphosphate (UTP) acting at the same receptor only triggers a Ca2+ response. To explore whether multiplex sensors could be used to detect this distinct signaling pattern, we expressed the human P2Y11 receptor with combinations of the Downward DAG2 or Upward DAG2 and R-GECO1 sensors. In HEK293 cells, both ATP and UTP triggered a Ca2+ response that was identical in terms of kinetics ( Fig. 5 ). The Upward and Downward DAG2 sensors, however, revealed that the ATP triggers signaling via the phospholipase C pathway, whereas the UTP is causing a Ca2+ transient in a very different way. This UTP effect could be seen in cells that expressed only the sensor, without the P2Y11 receptor, so these results are likely to be due to the action of UTP on a receptor intrinsic to this cell line, unlike what White and colleagues saw with a different cell line.
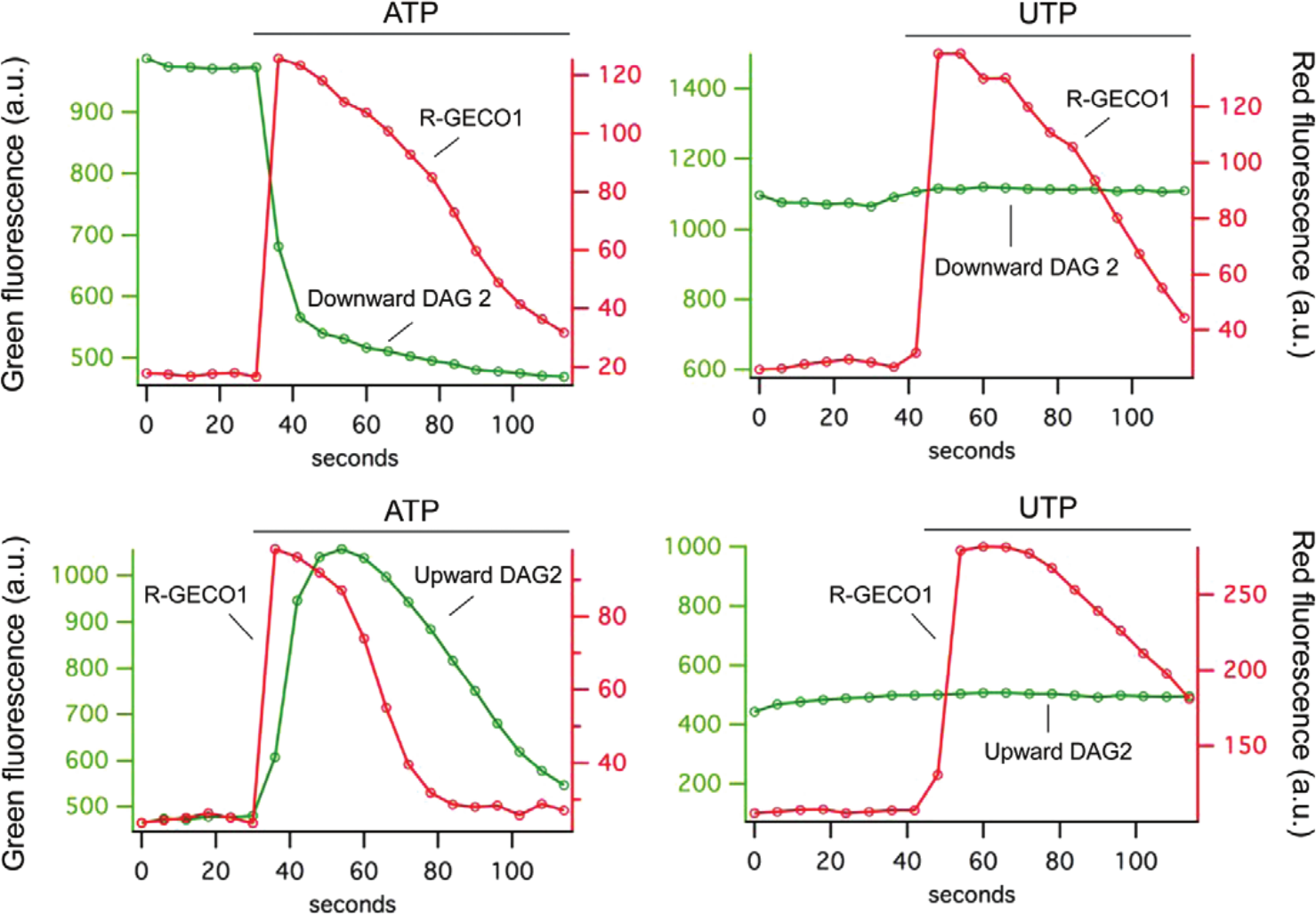
In HEK cells expressing the human P2Y11 receptor, the addition of adenosine triphosphate (ATP) or uridine triphosphate (UTP) produces a transient increase in Ca2+ that is consistent with receptor activation. However, the simultaneous recording of the Upward or Downward DAG2 sensors reveals that the ATP is activating the phospholipase C pathway, whereas UTP is producing a Ca2+ transient through a different pathway.
Protein-based, fluorescent biosensors have often worked at the microscope, under exacting experimental control, and failed to make an impact on the field of laboratory automation and screening. To test whether the fluorescent DAG sensors described here would be suitable for routine applications and automated screening, we coexpressed the M1 or P2Y11 receptors with the Downward DAG2 sensor in HEK293T cells plated on a 96-well Corning Co-Star polystyrene plate. Media were replaced with phosphate-buffered saline, and the fluorescence of each well before and after the addition of drug was measured using a standard plate reader. The change in fluorescence was measured for addition of vehicle alone as well as vehicle carrying carbachol or ATP. Using only the signal provided by Downward DAG2, we were able to obtain Z′ values 29 of 0.6 or greater ( Fig. 6A ).
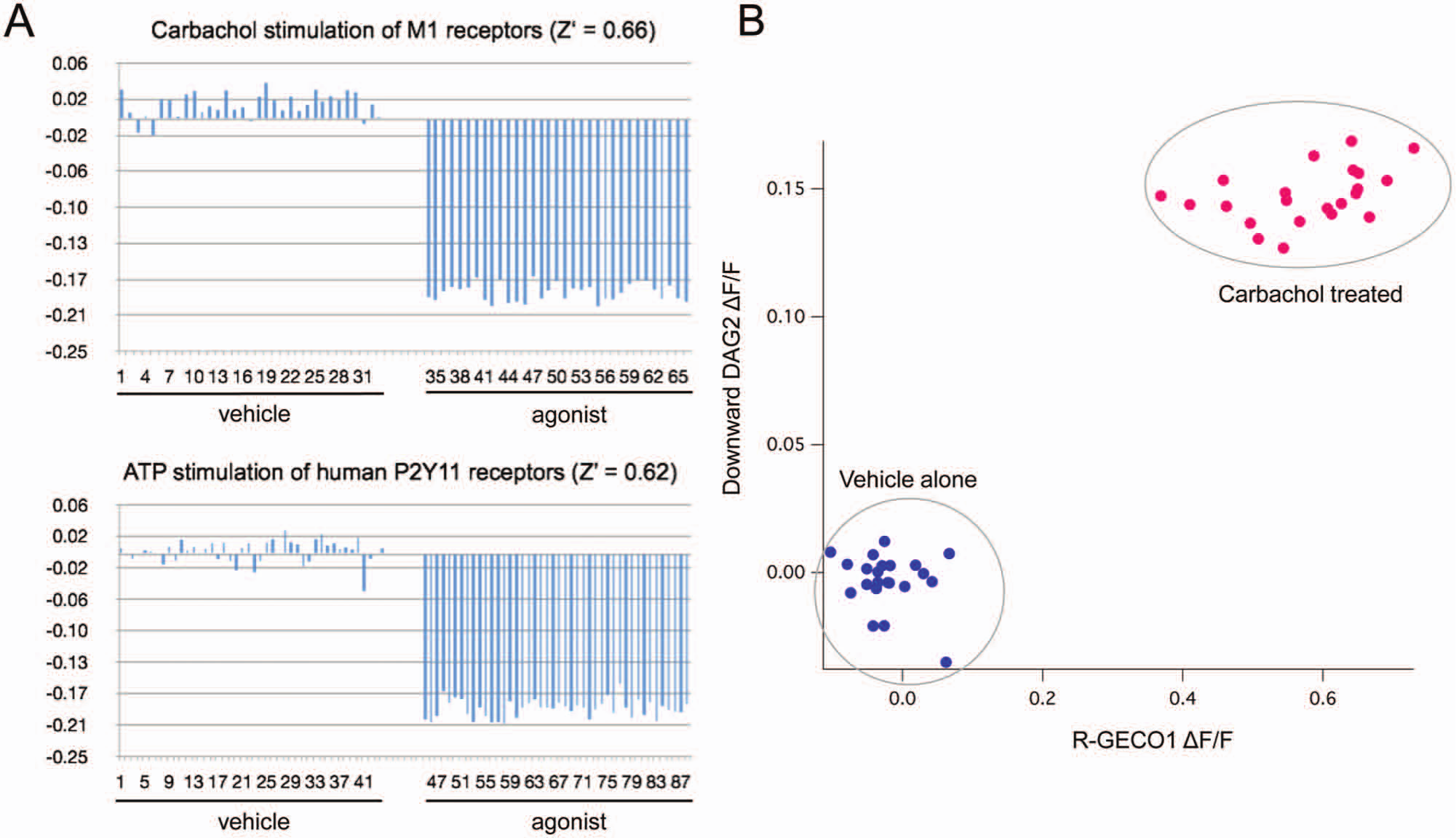
The diacylglycerol (DAG) sensors described here are compatible with automated drug discovery. The Downward DAG2 sensor coexpressed with the M1 or P2Y11 receptor produces a consistent, reproducible signal (Z′ > 0.6) on a standard fluorescence plate reader (
Multiplex sensors offer the opportunity to improve an assay by making multiple, simultaneous, independent measurements. When both the green and red fluorescence measurements were captured from wells of cells expressing both the R-GECO1 Ca2+ sensor and the Downward DAG2 sensor, it was possible to plot the response to M1 receptor activation in terms of both sensors. This reveals that there is a strong correlation between the amplitude of the two signals and, even more important, that the two independent signals can be used to increase the stringency of the assay, as well as separation between stimulated and unstimulated cells ( Fig. 6B ).
Discussion
The DAG sensors described here produce large changes in fluorescence that can be readily detected even on simple fluorescent plate readers. Because the sensors are based on single fluorescent proteins, they can be readily multiplexed with other fluorescent protein-based sensors such as the R-GECO series 30 or the PIP2r sensor described here, as well as with fluorescent dyes. Such multiplexing can improve the quality of the information produced in a screen in several ways. First, the simultaneous detection of multiple components of a signaling pathway provides an unambiguous readout for a particular pathway. Second, detection of two different signals can be used to improve assay performance/reliability. Finally, the use of multiplex sensors such as these has the potential to provide new views of agonist-biased signaling by providing relative ratios of the activity of different signaling components. 7
The multiplex sensors described here offer new opportunities for live-cell assays by producing large, reproducible changes in fluorescence that can be detected on standard fluorescence plate readers used in laboratory automation. These live-cell assays require no additional reagents, cell lysis, or complex liquid handling steps. The results obtained here ( Fig. 6 ) indicate that reasonable Z′ values can be reliably obtained from transiently transfected HEK293 cells. It is quite likely that even better values can be obtained by using stable cell lines expressing uniform levels of the sensor. Since this sensor system is genetically encoded and carried by relatively short coding regions of DNA, it should be possible to deliver the sensor using a variety of viral transduction strategies or the BacMam system. 31 Indeed, transduction or transgenic expression with tissue or cell type–specific promoters can deliver these sensors to appropriate cell types so that screening can be done in the most relevant cellular contexts. Finally, although Z′ is the appropriate metric for single sensors measuring single analytes, the multiplex sensors here introduce multiple, independent measurements where the separation of background and signal occurs in multidimensional space.
Although our experiments demonstrate that these fluorescent protein-based sensors are ready for routine use on standard equipment, it is quite likely that even better signals will be obtained with plate readers that can measure the response of the sensors in every well over time. The advent of multiplex sensors for both Ca2+ and cAMP, for example,2,4 shows that cells can produce antiphase, cyclic patterns of signaling that can only be detected by collecting the responses of the two sensors over time. Similarly, in the experiments described here, the Ca2+ and DAG/PIP2 responses are quite different, with different rates of onset and return to baseline. The only way to capture these interesting and biologically relevant patterns of signaling in microplate format is to measure multiple signals over several time points at 0.1 to 5 Hz, making the Molecular Devices (Sunnyvale, CA) Fluorescent Imaging Plate Reader (FLIPR) and Hamamatsu (Hamamatsu City, Japan) FDSS32,33 ideal instruments for these assays.
The multiplex sensors described here provide just a glimpse of the near future. They focus on the Gqα limb of GPCR signaling, and they can be coexpressed with a wide variety of Gq-coupled GPCRs in a screening environment. Although they could potentially be used to detect the activation of other GPCRs by coexpressing hybrid Gα subunits, 34 this would be quite artificial. A better solution would be new, analogous multiplex sensor systems for other G-proteins such as Gi and Gs. Beyond detecting Gα activity, new multiplex sensors capable of resolving Gβγ and/or arrestin signaling would provide new insights into agonist-biased signaling. 7 Finally, although the signals generated by the sensors described here are robust enough for use on standard fluorescence plate readers, the rapid evolution of ever better signals in the analogous Ca2+ sensors14,30 indicates that even better multiplex sensors are certain to become available in the very near future.
Footnotes
Acknowledgements
We thank Drs. Cathy Berlot and Tom Hynes for experimental advice and Dr. Bryan Roth, Director of the National Institute of Mental Health Psychoactive Drug Screening Program, for his advice and encouragement. Many thanks to Drs. Flori Sassano and Xi-Ping Huang in the Roth Lab at UNC for advice on cell culture and high-throughput screening. We are grateful to Dr. Robert Campbell for generously sharing his GECO Ca2+ sensors and constructs. The engineers at Autopilot in Bozeman, Montana, contributed expertise in the design and production of specialized plate reader components.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the National Institute of Mental Health (1R43MH096670- 01A1) and The Montana SBIR Matching Funds Program (12-50-RC SBIR-006) to A.M.Q.