
Abstract
Cell viability assays fulfill a central role in drug discovery studies. It is therefore important to understand the advantages and disadvantages of the wide variety of available assay methodologies. In this study, we compared the performance of three endpoint assays (resazurin reduction, CellTiter-Glo, and nuclei enumeration) and two real-time systems (IncuCyte and xCELLigence). Of the endpoint approaches, both the resazurin reduction and CellTiter-Glo assays showed higher cell viabilities when compared directly to stained nuclei counts. The IncuCyte and xCELLigence real-time systems were comparable, and both were particularly effective at tracking the effects of drug treatment on cell proliferation at sub-confluent growth. However, the real-time systems failed to evaluate contrasting cell densities between drug-treated and control-treated cells at full growth confluency. Here, we showed that using real-time systems in combination with endpoint assays alleviates the disadvantages posed by each approach alone, providing a more effective means to evaluate drug toxicity in monolayer cell cultures. Such approaches were shown to be effective in elucidating the toxicity of synthetic lethal drugs in an isogenic pair of MCF10A breast cell lines.
Introduction
In vitro viability assays are essential tools for drug development, allowing for the assessment of drug efficacy prior to subsequent in vivo analyses. Whether performed as a single-plate experiment or as part of a high-throughput screen, the concept remains the same—cells are incubated with a particular compound(s), then assessed for viability to quantify drug-induced cell toxicity.
Numerous commercial cell viability assays that exploit different cellular processes to quantify cytotoxicity are now available, each highlighting the variability that can be obtained from different methodologies.1 –3 Consequently, the selection and application of an effective assay(s) should be a major consideration in any drug-based experimental design.
A widely used approach to determine drug-induced cytotoxicity involves measuring cellular metabolic activity at the conclusion of an experiment. Such approaches include the 3-[4,5-dimethylthiazolyl-2]-2,5-diphenyltetrazolium bromide (MTT) assay, resazurin reduction, and the CellTiter-Glo assay, each using a different aspect of cellular metabolism as a means of quantifying live cells. The MTT assay relies on the mitochondrial activity of live cells to convert a yellow MTT substrate into purple formazan crystals, detectable via spectrophotometry. 4 The resazurin reduction assay, used in alamarBlue and CellTiter-Blue assays, is a more sensitive alternative to MTT and uses the intracellular reduction potential of living cells to convert resazurin to the fluorescent product resorufin. 5 CellTiter-Glo adopts the use of firefly luciferase, which reacts with available cellular adenosine triphosphate (ATP) to produce a bioluminescent signal proportional to the number of live cells present in the assay. 6 The nuclei counting method, which is a direct measure of viability, is considered to be the most accurate; 1 however, the ease of mix-and-measure metabolic-based approaches makes them a common feature in high-throughput drug screens.
Unlike endpoint approaches, real-time assay systems allow for the tracking of cellular growth over the entire time course of an experiment. This is particularly effective for assessing the impact of cytostatic compounds, where subtle growth inhibitory effects are easily noticeable but may be missed using endpoint-based methods. Real-time assays are typically performed using equipment capable of capturing images at regular intervals and quantifying cellular surface area coverage as a measure of proliferation (e.g., IncuCyte FLR; Essen BioScience, Ann Arbor, MI). Such methods also facilitate visualization of drug-induced cell morphology changes. Alternatives to this approach include the xCELLigence (ACEA Biosciences, San Diego, CA), which uses electrical impedance to measure both cellular adhesion strength and surface area coverage as a combined proxy of cellular proliferation.
In this study, we compared the performance of five different cell-based viability assays. Three endpoint assays (resazurin reduction, CellTiter-Glo, and nuclei enumeration) and two real-time assays (IncuCyte and xCELLigence) were used to investigate the effectiveness of each approach for the validation of candidate synthetic lethal drugs in an isogenic pair of MCF10A breast cell lines.
Materials and Methods
Cell Culture
MCF10A and the derived CDH1-negative isogenic line (MCF10A CDH1−/−) were purchased from Sigma-Aldrich (St. Louis, MO). Cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM)/F12 (1:1; Life Technologies, Carlsbad, CA) with 5% horse serum (Life Technologies), 10 µg/ml insulin (Novo Nordisk Pharmaceuticals Ltd, Bagsvaerd, Denmark), 20 ng/ml human epidermal growth factor (Peprotech, Rocky Hill, NJ), 100 ng/ml cholera toxin (Sigma-Aldrich), and 500 ng/ml hydrocortisone (Sigma-Aldrich). 7 The cells were cultured in exponential growth phase at 37 °C and 5% CO2.7,8 An isogenic MCF10A cell line pair was selected to demonstrate drug-induced synthetic lethal phenotypes against CDH1 in a nonmalignant cell background.
Vorinostat was purchased from SelleckChem (Houston, TX), and paclitaxel (Taxol) was purchased from Sigma-Aldrich. Both drugs were reconstituted in 100% DMSO, stored at −80 °C, and individual aliquots were diluted to working stocks in complete growth medium prior to use in an experiment. Vorinostat, a histone deacetylase inhibitor, was selected because it has previously demonstrated selective lethality toward CDH1-deficient MCF10A cells. 9 Taxol was chosen as a chemotherapeutic agent that demonstrated nondiscriminate lethality in the MCF10A isogenic cell line pair.
Endpoint Assays
MCF10A and MCF10A CDH1−/− cells were seeded at 4×103 cells per well in 96-well, black-walled, clear-bottomed tissue culture plates (Corning, Corning, NY) in 100 µL complete growth medium and left to equilibrate at room temperature for 30 min before incubation at 37 °C. After overnight incubation, cells were treated in triplicate with drug or DMSO for controls. Endpoint assays were performed at 48 h post drug treatment. For resazurin reduction assays, resazurin (Sigma-Aldrich) was made to 440 µM stock solutions in phosphate buffered saline (PBS) and aliquoted for storage at −20 °C. Resazurin solution was added to cells at 20% final concentration, and plates were incubated for 3 h at 37 °C prior to reading fluorescence at 550 nm excitation and 590 nm emission using a POLARstar Optima (BMG Labtech, Ortenberg, Germany). For luminescent assays, CellTiter-Glo (Promega, Madison, WI) was added at 20% final concentration. Earlier optimization had shown this concentration to consistently reproduce the manufacturer’s recommended 1:1 vol/vol ratio (results not shown). Luminescent readings were obtained using a POLARstar Optima after 10 min incubation at room temperature with shaking. For nuclei counting assays, Hoechst 33342 (Life Technologies) was added at 1 µg/mL final concentration and incubated for 30 min in the dark at 37 °C with shaking. Plates were then imaged at four fields per well under 4× magnification using the Cytell Cell Imaging System (GE Healthcare, Buckinghamshire, United Kingdom), and imaged nuclei were enumerated using CellProfiler 10 to obtain a total cell count. For direct comparison between total cell count and measured confluency performed in the IncuCyte FLR, SYTOX (Life Technologies) was used in place of Hoechst for nuclei enumeration because this IncuCyte model only has a single, green fluorescence filter. A one-step, no-wash, mild permeabilization and fixation protocol was adopted from Chan et al. (2013), 1 using a final concentration of 0.25% paraformaldehyde, 0.075% saponin, and 10 nM SYTOX. Because the SYTOX dye stains only membrane-compromised cells, permeabilization was required to obtain a total cell count.
xCELLigence Assays
Experiments conducted on the RTCA-MP xCELLigence system (ACEA Biosciences, San Diego, CA) were performed in accordance with the instructions of the supplier. Complete growth medium (100 µL) was added into each well of the E-plate 96 (ACEA Biosciences), followed by a brief background impedance measurement on the RTCA-MP station. MCF10A and MCF10A CDH1−/− cells were seeded at 4×103 cells per well in 100 µL complete growth medium, and, after 30 min equilibration at room temperature, the E-plate was placed in the RTCA-MP station. The RTCA-MP station was housed in a humidified cell culture incubator at 37 °C and 5% CO2. Cell proliferation, as determined by electrical impedance, was recorded at 15-min intervals. After overnight incubation, the assay was paused, 10 µL medium was removed from each well, and cells were treated with 10 µL drug or 0.1% DMSO for controls. The assay was then resumed, taking impedance measurements every 15 min for a further 48 h. All xCELLigence experiments were performed in duplicate.
IncuCyte Assays
MCF10A and MCF10A CDH1−/− cells were seeded at 4×103 cells per well in 96-well, black-walled, clear-bottomed, tissue culture plates in 100 µL complete growth medium and left to equilibrate at room temperature for 30 min before 37 °C, 5% CO2 incubation. After overnight incubation, cells were treated with 10 µL drug or 0.1% DMSO for controls, and the plate was inserted into the IncuCyte FLR for real-time imaging, with three fields imaged per well under 4× magnification every 2 h for a total of 48 h. Data were analyzed using the IncuCyte Confluence version 1.5 software, which quantified cell surface area coverage as confluence values. All IncuCyte experiments were performed in triplicate.
Results and Discussion
Metabolic and Nuclei-Counting Endpoint Assays
First, we compared the performance of two metabolic-based assays alongside a nuclei-counting one. The resazurin reduction and CellTiter-Glo assays were chosen ahead of the MTT assay because previous studies have reported reduced sensitivity with MTT compared to other endpoint methods.11,12 To compare the efficacy of each assay, MCF10A and MCF10A CDH1−/− cells were treated with vorinostat for 48 h and assessed for cell viability using each method. A dose-dependent effect was observed in all three methods with increasing vorinostat concentration in both cell lines. In both MCF10A and MCF10A CDH1−/− cells, 0.63 µM vorinostat treatment showed negligible viability inhibition with no marked differences observed between the two metabolic assays. At dosages of 1.25 and 2.5 µM vorinostat, however, the CellTiter-Glo assay gave significantly higher viabilities than the resazurin reduction assay (P < 0.05; Fig. 1A ). Both metabolic-based approaches gave significantly higher viabilities than the nuclei-counting approach for all three vorinostat concentrations in both cell lines (P < 0.05; Fig. 1A ), suggesting that resazurin reduction and CellTiter-Glo were overrepresenting cell viability. In addition, viability as measured by nuclei counting was more comparable to that measured using resazurin reduction, suggesting CellTiter-Glo gave less sensitivity to the other endpoint methodologies for cells treated with vorinostat. This is contrary to previous literature reports, which indicate that CellTiter-Glo can provide higher sensitivity than resazurin-based assays.13,14
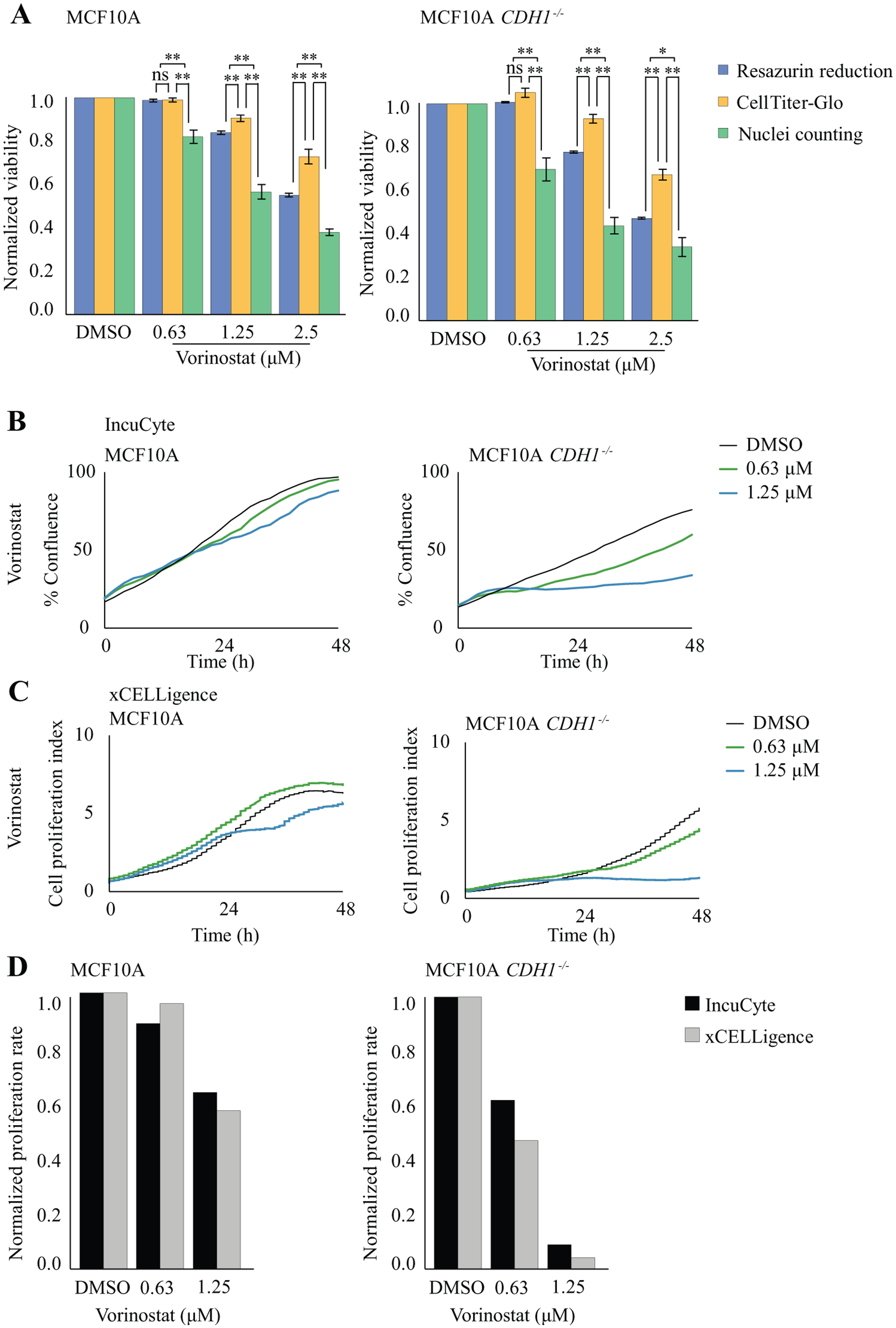
Comparison of endpoint and real-time cell viability assays. (
In addition to reduced sensitivity, the two metabolic assays also have other potential limitations. For example, the resazurin reduction assay requires a 37 °C incubation step of several hours, which has been reported to cause morphological changes in cells. 13 However, we did not observe resazurin-induced morphology changes in both cell lines (data not shown). In comparison, CellTiter-Glo, which has a shorter incubation phase to permeabilize cells and release their ATP for measurement, has a considerably greater cost, which can be a drawback in high-throughput screening. Furthermore, it is possible that drugs affecting cellular metabolic processes could interfere with the performance of both the resazurin reduction and CellTiter-Glo assays, giving rise to inaccurate viability measurements. 2
Overall, the nuclei-counting method is still considered to be the most accurate measure of cell viability. 1 However, its application to high-throughput screening requires efficient automated imaging systems with built-in enumeration software, which can be cost prohibitive. Fortunately, more affordable entry-level systems, such as the Cytell (GE Healthcare, Buckinghamshire, UK), Cytation 5 (BioTek, Winooski, VT), and EVOS (Thermo, Waltham, MA), are available to provide such technologies at a reduced cost. Alternatively, more standard imaging systems without accompanying enumeration software can be used with free open-sourced applications such as ImageJ 15 or CellProfiler. 10
Real-Time Assays
To complement the endpoint assays, real-time IncuCyte and xCELLigence assays were performed on MCF10A and MCF10A CDH1−/− cell lines treated with vorinostat over 48 h ( Fig. 1B and 1C ). The IncuCyte uses automated imaging to determine cellular confluence at designated intervals over the time course of an experiment as a measure of viability. The xCELLigence uses gold-plated plates to measure cell surface area coverage and adhesion strength via electrical impedance, combining these factors as a measurement of cell viability.
From the IncuCyte and xCELLigence platforms, both cell lines showed a dose-dependent inhibitory response to vorinostat, although this effect was more pronounced in MCF10A CDH1−/− cells ( Fig. 1B and 1C ). To compare the two real-time systems, we determined the proliferation rate at logarithmic growth phase (taken from 12 to 36 hours post drugging in Fig. 1B and 1C ) between control and drug treatment within the respective MCF10A and MCF10A CDH1−/− cells. In both systems, the proliferation rates of vorinostat-treated MCF10A cells were quite comparable and differed by no more than 10% across each tested concentration ( Fig. 1D ). MCF10A CDH1−/− cells showed slower proliferation rates in the xCELLigence than in the IncuCyte. From 0.63 and 1.25 µM vorinostat doses, 24% and 53% smaller measurements were observed, respectively ( Fig. 1D ). This difference could possibly be attributed to the compromised adhesion previously characterized in MCF10A CDH1−/− cells, 8 which may have been further exacerbated by vorinostat treatment. As a result, the xCELLigence, which measures adhesion impedance, would have registered a greater reduction in MCF10A CDH1−/− cell viability compared to the IncuCyte, which is incapable of detecting adhesion strength. MCF10A cells showed no marked difference between the two systems, presumably because this cell line does not exhibit compromised cellular adhesion. 8 Unfortunately, the gold-plated xCELLigence plates used in this study lacked clear bottoms, which prevented imaging analysis. Newer E-plates with partially clear-bottomed sections for cell visualization are now available; however, these were not released prior to our investigation. Conversely, the IncuCyte system was able to provide images that showed no substantial morphological changes over the time course of vorinostat treatment in both cell types ( Fig. 2A ). Overall, both real-time platforms showed comparable performance, except for measuring MCF10A CDH1−/− cells treated with 0.63 µM vorinostat.
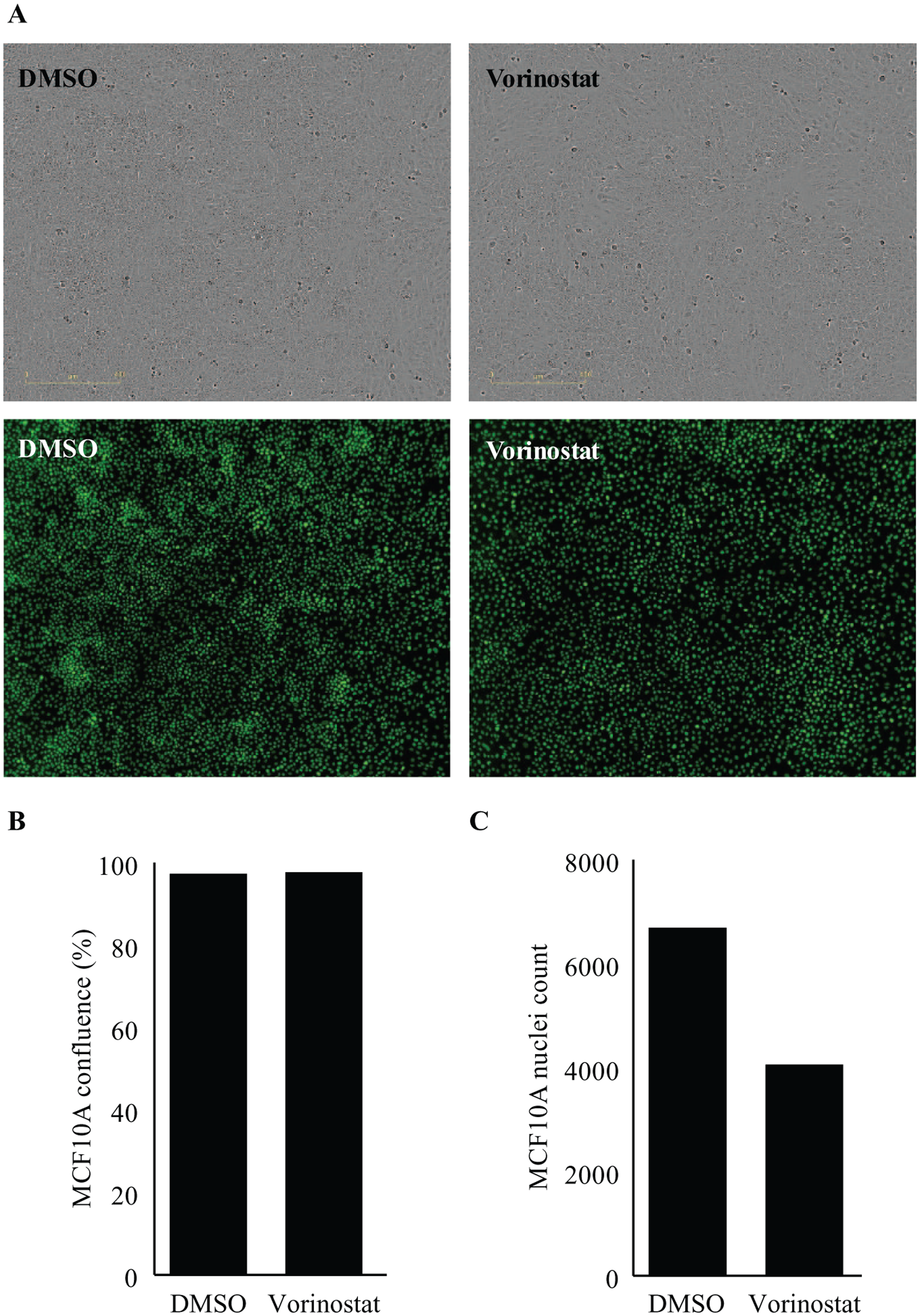
Cellular confluence measurements do not reflect cell densities at full surface area coverage. (
At the conclusion of each assay, both real-time platforms showed that vorinostat-treated MCF10A cells had achieved viability values very similar to those of DMSO-treated controls ( Fig. 1B and 1C ). This was contrary to data from our endpoint methods ( Fig. 1A ), which had shown that each vorinostat concentration had produced lower viabilities than control-treated cells, particularly in the nuclei-counting assay. A closer inspection of representative phase-contrast and fluorescent images from the IncuCyte revealed an observable difference in cell density between control and vorinostat-treated MCF10A cells ( Fig. 2A and 2C ). Even though both control and vorinostat (1.25 µM) treated MCF10A cells showed full growth confluence covering the entire surface area of each respective well ( Fig. 2B ), subsequent nuclei counting confirmed significant differences in cell numbers, whereby 39% fewer cells were present following drug treatment compared to control treatment ( Fig. 2C ). This key observation demonstrated the IncuCyte’s inability to discriminate between differing cellular densities when cells had covered the entire surface area of their respective wells. As such, caution should be taken when analyzing data at full cellular confluence because further validation is required from direct cell counting. Nevertheless, the IncuCyte still produced valuable data during sub-confluent growth phases, which was comparable to nuclei-counting data (data not shown). These results demonstrate that a combination of distinct methodologies provides a more comprehensive and accurate assessment of drug efficacy than singular assays.
Real-Time Assay and Endpoint Assay Multiplexing
To mitigate the shortfalls observed in the endpoint and real-time assays, we combined the IncuCyte real-time assay with both the resazurin reduction and nuclei-counting assays. The resazurin reduction and nuclei-counting methods were selected as endpoint assays because they have been reported to multiplex together effectively. 16 This multiplexed approach also allowed for more data to be gathered from one drug-treated experiment. We also wanted to investigate if the combined approach was capable of evaluating synthetic lethal properties of two different drugs, in which assay sensitivity is essential to distinguish preferential targeting of one cell type over another. In this case, a synthetic lethal effect would involve the selective growth inhibition of MCF10A CDH1−/− cells but not MCF10A cells. To test this, we subjected MCF10A and MCF10A CDH1−/− cells to either vorinostat or Taxol treatment over 48 hours, with cellular growth being tracked in the IncuCyte, followed by resazurin reduction and nuclei counting at the conclusion of the real-time analysis.
At 48 h following vorinostat treatment (0.63, 1.25, 2.5 µM), the confluence measurements from the IncuCyte showed that MCF10A cells were marginally inhibited and proliferated similarly to control treated cells ( Fig. 3A ). However, in MCF10A CDH1−/− cells, a significant dose-dependent inhibitory response was observed in which drug-treated cells did not reach the confluency of control-treated cells ( Fig. 3B ). Following the IncuCyte assay, the same plate was then subjected to the resazurin reduction assay. Increasing vorinostat treatment caused a more marked reduction in MCF10A CDH1−/− cell viabilities (93%, 71%, and 43%; Fig. 3E ) compared to the corresponding MCF10A treated cells (98%, 83%, 55%; Fig. 3E ). Similarly, the nuclei-counting analysis, performed immediately after the resazurin reduction assay, also showed increasing vorinostat treatment causing a more marked effect on MCF10A CDH1−/− cell viabilities (77%, 47%, and 26%; Fig. 3F ) compared to MCF10A cells (79%, 57%, and 37%; Fig. 3F ). These results infer synthetic lethality, which in the context of cancer therapeutics allows for greater target specificity toward tumor cells with reduced side effects.
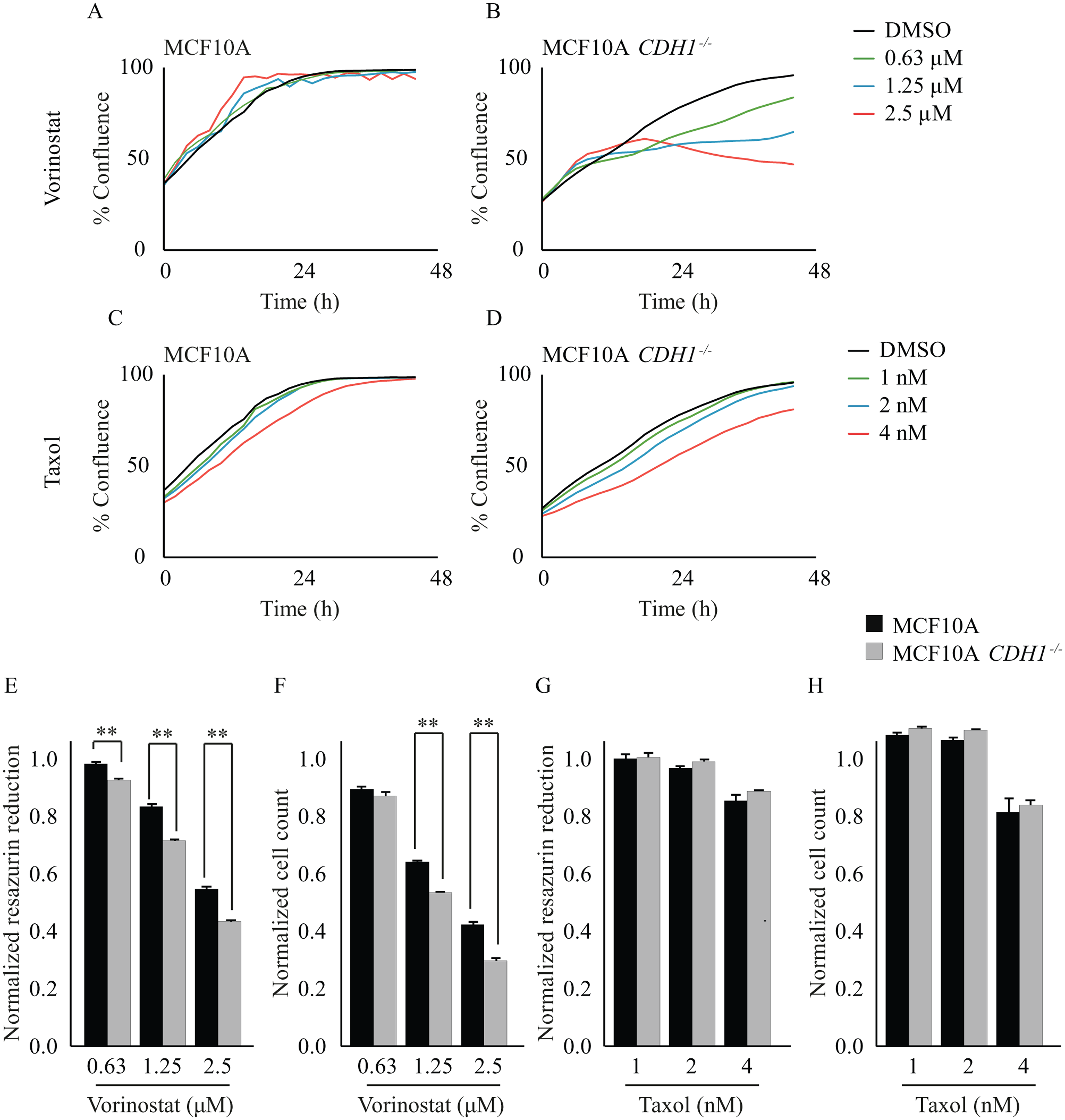
Combined real-time and endpoint assays facilitate the evaluation of drugs for synthetic lethal properties in MCF10A isogenic cells. (
As another measure of synthetic lethality, we calculated the viability ratio of MCF10A CDH1−/− cells to MCF10A cells, whereby a ratio of less than 1 indicated an increased susceptibility of MCF10A CDH1−/− cells to drug treatment, concordant with a drug-induced synthetic lethal phenotype. Both the resazurin reduction and nuclei-counting assays produced comparable viability ratios for 0.63, 1.25, and 2.5 µM vorinostat treatment between the isogenic cell lines (resazurin reduction: 0.95, 0.85, 0.78; nuclei counting: 0.97, 0.82, 0.70). This result is consistent with the IncuCyte confluence analysis, although the extent of this differential was more marked in real time. Overall, the combined assays demonstrated an increased susceptibility of MCF10A CDH1−/− cells compared to MCF10A cells with increasing vorinostat dose.
IncuCyte analysis showed that MCF10A cells treated with 1 and 2 nM Taxol exhibited negligible inhibition. When treated with 4 nM Taxol, MCF10A cell viability was affected within the first 36 hours but eventually attained confluence measurements similar to those of controls at the conclusion of the real-time assay ( Fig. 3C ). A similar effect was seen in Taxol-treated MCF10A CDH1−/− cells, although the highest concentration (4 nM) gave rise to growth inhibition that prevented full confluency observed in control treatment ( Fig. 3D ). The resazurin reduction and nuclei-counting assays showed that Taxol treatment also produced a dose-dependent effect in both MCF10A and MCF10A CDH1−/− cells, without showing preferential inhibition in either cell type at the tested concentration range ( Fig. 3G and 3H ). Furthermore, the viability ratios determined from both the resazurin reduction and nuclei-counting assays were not less than 1 (resazurin reduction: 1.00, 1.02, and 1.05; nuclei counting: 1.02, 1.03, and 1.03), indicating no synthetic lethality. Taxol treatment at higher concentrations (up to 16 nM) yielded a dose-dependent effect in both isogenic cells lines, although no synthetic lethal phenotype was observed at these concentrations (data not shown). Here, we have shown that our combined real-time and endpoint assay approach reliably identified drug-induced synthetic lethal effects in the tested MCF10A isogenic cell lines. We have also previously used the combined IncuCyte and endpoint method to uncover other drugs that induce synthetic lethality in MCF10A CDH1−/− cells. 9 The IncuCyte and nuclei counting were used to show that drugs such as crizotinib, LY2784544, and saracatinib each caused significantly reduced viabilities in MCF10A CDH1−/− cells compared to MCF10A cells. 9
Our current study has shown that the IncuCyte system is well suited for tracking sub-confluent cell growth phases, which can be followed up by nuclei counting to assess drug efficacy when cells are at full confluence. In the absence of a real-time system like the IncuCyte, nuclei counting should be performed because it provided the most accurate measurement of viable cells in our analysis. Overall, we have demonstrated the utility and strengths of five viability assays and have adapted a robust real-time and endpoint multiplexed method for the investigation of synthetic lethal drugs.
Footnotes
Acknowledgements
We thank Dr. Michelle McConnell and Ms. Clare Fitzpatrick (Department of Microbiology and Immunology, University of Otago) for assistance with the xCELLigence real-time system; Dr. Adele Woolley and Mr. Michael Algie (Department of Pathology, University of Otago) for the use of the IncuCyte FLR; and Dr. Kenny Chitcholtan (Department of Obstetrics and Gynaecology, University of Otago) for providing the resazurin dye recipe and associated assay recommendations.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Health Research Council of New Zealand and the University of Otago (PhD scholarships to A. Single, H. Beetham, and B. Telford).