
Abstract
Voltage-gated ion channels produce rapid transmembrane currents responsible for action potential generation and propagation at the neuronal, muscular, and cardiac levels. They represent attractive clinical targets because their altered firing frequency is often the hallmark of pathological signaling leading to several neuromuscular disorders. Therefore, a method to study their functioning upon repeated triggers at different frequencies is desired to develop new drug molecules selectively targeting pathological phenotype. Optogenetics provides powerful tools for millisecond switch of cellular excitability in contactless, physiological, and low-cost settings. Nevertheless, its application to large-scale drug-screening operations is still limited by long processing time (due to sequential well read), rigid flashing pattern, lack of online compound addition, or high consumable costs of existing methods. Here, we developed a method that enables simultaneous analysis of 384-well plates with optical pacing, fluorescence recording, and liquid injection. We used our method to deliver programmable millisecond-switched depolarization through light-activated opsin in concomitance with continuous optical recording by a fluorescent indicator. We obtained 384-well pacing of recombinant voltage-activated sodium or calcium channels, as well as induced pluripotent stem cell (iPSC)-derived cardiomyocytes, in all-optical parallel settings. Furthermore, we demonstrated the use-dependent behavior of known ion channel blockers by optogenetic pacing at normal or pathological firing frequencies, obtaining very good signal reproducibility and accordance with electrophysiology data. Our method provides a novel physiological approach to study frequency-dependent drug behavior using reversible programmable triggers. The all-optical parallel settings combined with contained operational costs make our method particularly suited for large-scale drug-screening campaigns as well as cardiac liability studies.
Introduction
Ion channels are pore-forming membrane proteins with a gate that is controlled by diverse stimuli, such as binding of extracellular ligands, changes in membrane potential, mechanical stress, temperature, or intracellular second messengers. Their transitions from closed (not conducting) to open (conducting) and then to inactivated refractory states happen within milliseconds, and in some cases are catalyzed by well-defined conformational changes. The frequency of this cycling orchestrates many critical biological processes, from gene expression to hormone secretion, heart beating, and neurotransmission. Importantly, an increased frequency of ion channel activation because of altered membrane potential contributes to the accumulation of channels in a refractory inactivated state; this altered channel population influences the downstream controlled processes and is associated with several nerve, muscle, and endocrine disorders. 1
Chemically diverse molecules acting on ion channels were shown to be effective in the treatment of “channelopathies,” human disorders including neuronal, cardiac, or respiratory diseases that are caused by excess or reduced ion channel function. 2 In particular, for voltage-gated channels, use-dependent compounds typically bind to and inhibit a particular kinetic state that is induced by a specific number and frequency of voltage changes; the ability of these molecules to preferentially act on channels with altered firing frequency provided therapeutic selectivity for the pathological state and made them highly desirable for new drug development. Nevertheless, ion channels still are targets with great potential, and identification of new drugs is difficult because methods need to be developed to recapitulate, in in vitro settings, the dynamic changes of ion channel states with a technique suitable to screen large compound collections typical of early phases of drug discovery. Among others, two parameters are particularly challenging for such large-scale screening campaigns: first, a precise and reversible control of membrane voltage (necessary to study voltage-activated ion channels under normal or pathological conditions of firing), and then, amenability for high-throughput configuration (e.g., miniaturized assay format, reduced assay component costs, and reduced experimental time). The coexistence of these features in available systems is still rare: In fact, they are limited by assay component cost and scalability (e.g., for automated electrophysiology), experimental recording time (e.g., for well-by-well detection in some optical instruments), or physiological stimulation (e.g., for irreversible chemical means used as voltage triggers).
Automated electrophysiology provided a valuable combination of millisecond-switchable voltage control with a miniaturized 384-well assay format; nevertheless, this technique still requires expensive consumables that significantly increase the cost per test-point. This is the main reason why automated electrophysiology is often not pursued in the screening of large compound collections (typically, more than 1 million) used in the early drug discovery phase: because the overall cost of the screening campaign is hardly affordable.
In recent years, the use of optogenetic methods (i.e., millisecond-switchable photo-controlled depolarizing proteins, e.g., channelrhodopsin), combined with relatively low-cost fluorescent indicators, has provided means to achieve control of the cellular membrane potential in a physiologically relevant manner while offering a drastic reduction in assay component cost compared to electrophysiology. These all-optical systems have already been validated in miniaturized assay format (e.g., 96- or 384-well plates3–6), but their applications are still limited by the long instrument reading time required for sequential processing of single wells, or the lack of simultaneous liquid injection for online monitoring of potential drug effect (see Table 1 ). As a consequence, these systems are not fully suited to study paced cellular processes, such as frequency-dependent drug effect, in large-scale screening activities.
Common Instruments for Ion Channel Screening in Miniaturized Format with Indicative Features, Performance, and Selected Examples.
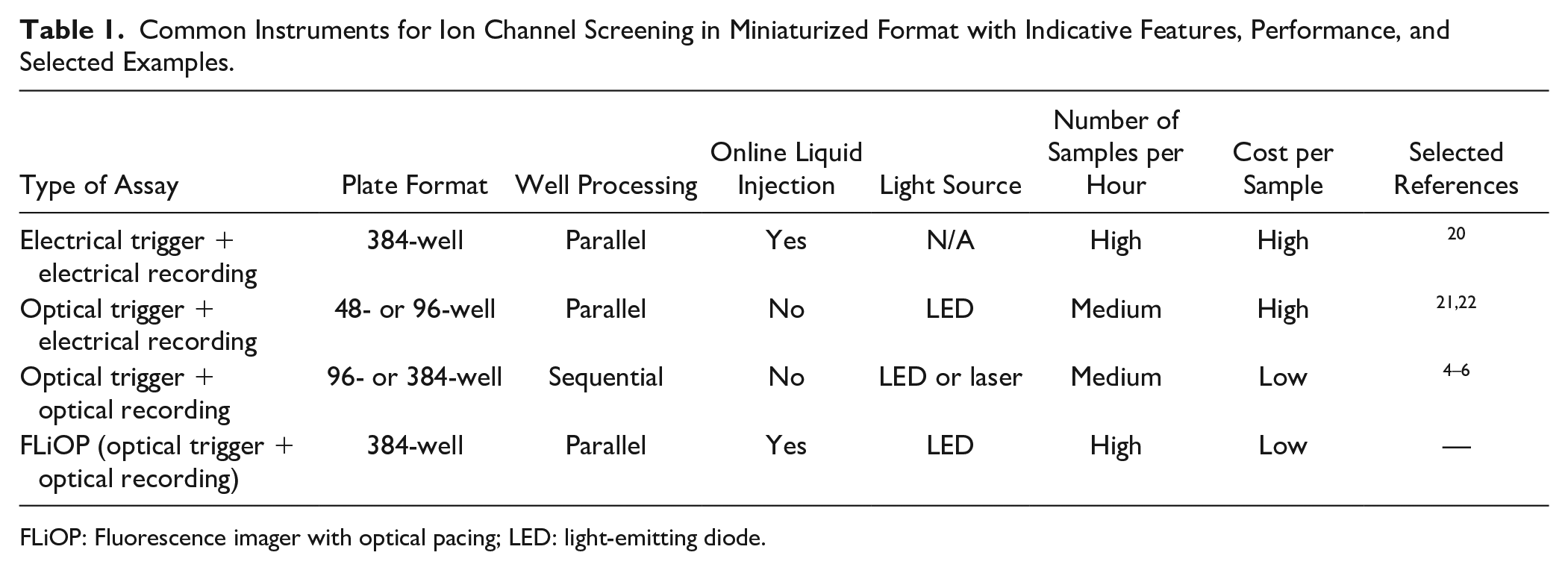
FLiOP: Fluorescence imager with optical pacing; LED: light-emitting diode.
Here, we developed a method that enables millisecond-programmable stimulation of optogenetic tools concomitant with subsecond fluorescence imaging and online liquid injection; in particular, our system performs all these operations in parallel in 384 wells simultaneously. We demonstrated proof of concept for the screening of use-dependent voltage-gated calcium channel blockers in both recombinant and induced pluripotent stem cell (iPSC)-derived cellular systems. The contained operational costs of our system together with enriched temporal control can significantly advantage high-throughput identification of drug molecules selectively targeting pathological target function.
Materials and Methods
Recombinant Cell Line Generation
HEK-293 cells (ATCC CRL-1573; ATCC, Baltimore, MD) were maintained in a 37 °C, 5% CO2 incubator with Eagle’s minimum essential medium (EMEM; BioWhittaker, Walkersville, MD) supplemented with 10% fetal bovine serum (FBS), 100 U/ml penicillin, 0.1 mg/ml streptomycin, and 2 mM L-glutamine; the subcultivation ratio was 1:6 twice per week. Full-length, mammalian codon usage optimized sequences of ChR2 (Chlamydomonas reinhardtii channel rhodopsin 2 / chlamyopsin 4 light-gated ion channel, XM_001701673.1, amino acids 1–309, T159C mutant), KIR2.3 (KCNJ4, NM_004981.1), CaV1.3α (CACNA1D, NM_000720.2), α2δ1 (CACNA2D1, NM_000722.2), β3 (CACNB3, NM_000725.3), TASK1 (KCNK3, NM_002246.2), and NaV1.5 (SCN5A, NM_198056.2 with IFM-QQQ mutations) were synthesized (GeneArt, Regensburg, Germany), then subcloned in mammalian expression vectors downstream to the EF1alpha (α2δ1 subunit) or cytomegalovirus (CMV; all the other genes) constitutive promoter, and sequence verified. Stable pure clones were obtained upon electroporation (300 V, 950 µF) in HEK-293 cells, selected with antibiotic, and growth at low density followed by the functional selection of best-performing clones, through fluorescence imaging and electrophysiology analysis. The CaV1.3 cell line stably expresses KIR2.3, together with CaV1.3α, β3, and α2δ1 subunits.
Mono- and Co-Culture Assays
Recombinant cell lines were seeded as monocultures (20,000 cells/well) or co-cultures (10,000 cells/well for each cell line) in black-clear poly-D-lysine-coated 384-well cell culture plates (Twin Helix, Milan, Italy) 24 h before analysis in culture medium without selective antibiotics and supplemented with 5 µM all-trans retinal (ATR). Human iPSC-derived cardiomyocytes Cor.4U (Ncardia, Gosselies, Belgium) were thawed in Cor.4U medium (Ncardia) and immediately seeded (14,000 cells/well) in black-clear 384-well cell culture plates (Greiner Bio-One, Monroe, NC) coated with 0.01 mg/ml fibronectin, following the manufacturer’s instructions. Five days after thawing and seeding, Cor.4U medium was substituted with BMCC medium (Ncardia) supplemented with 2% FBS (cat. no. 30-2020; ATCC) and 5 µM ATR, and containing HEK-293/ChR2 cells (1,400 cells/well, corresponding to a 10:1 ratio between Cor.4U and ChR2 cells); functional analysis of the Cor.4U–ChR2 co-culture was performed 72 h after ChR2 cell seeding.
Cell Preparation for Imaging
Calcium-sensitive (Rhod-4,NW, AAT Bioquest, Sunnyvale, CA) or membrane potential–sensitive (MPdye, Molecular Devices, San Jose, CA) fluorescent dye loading solutions were prepared according to manufacturers’ instructions at 0.5× concentration in K0 buffer [in millimolars (mM): NaCl 150, KCl 0, CaCl2 2, glucose 10, HEPES 20; pH 7.4], K10 buffer [in micromolars (mM): NaCl 140, KCl 10, CaCl2 2, glucose 10, HEPES 20; pH 7.4], or BMCC medium, supplemented with 5 µM ATR. In recombinant cell experiments, the culture medium was removed, then plates were immediately incubated with dye loading solution prepared in K0 or K10 buffer for 45–60 min at 37 °C and 5% CO2 protected from the light. In Cor.4U cell experiments, the culture medium was removed and substituted with BMCC medium supplemented with 5 µM ATR, then, after 2 h incubation at 37 °C and 5% CO2 protected from the light, dye loading solution was added to the medium, and it was further incubated for 60–90 min at 37 °C and 5% CO2 protected from the light. When indicated, 1× concentrated compounds were included in the dye loading solution.
Fluorescence Imaging and Optical Pacing with FLiOP
Dye fluorescence changes over time were monitored by a FLIPR (fluorescence imaging plate reader) equipped with an intensified charge-coupled device (ICCD) camera (FLIPR TETRA; Molecular Devices) through embedded ScreenWorks software (version 4.0.0.30; Molecular Devices) and the embedded fluorescence imaging optics [in particular, light-emitting diode (LED) banks with an excitation wavelength of 510–545 nm, combined with a filter set with an emission wavelength of 565–625 nm]. Instrument parameters of Excitation % and Gain were set following the manufacturer’s instruction. ChR2 light stimulation was delivered simultaneously to dye imaging through a custom-built apparatus [Tetra LED controller (TLC)] controlling the flashing of the second embedded optics of the FLIPR instrument, in particular the LED banks of excitation wavelength 470–495 nm. The parameters of duration, intensity, repetitions, and interval between light pulses are operated by a controller unit (comprising an Arduino microcontroller and a 12-bit digital-to-analog converter), which powers a driver unit (LEDdynamics, Randolph, VT) connected to the controlled LED bank through a non-inverting amplifier (Texas Instruments, Dallas, TX), to flash with programmed duration (from 1 ms to 400 s, resolution 0.1 ms) and intensity (0% to 100% driver power, resolution 0.1%), with timing synchronization based on each FLIPR camera-opening event. Flashing program parameters are entered through a Windows-based graphical user interface (UI) that operates the controller unit functioning, while the fluorescence imaging experiment is run by FLIPR embedded ScreenWorks software.
Data Analysis
Relative fluorescence units (RFUs) recorded by the FLIPR instrument were analyzed with ScreenWorks software as percentage fluorescence changes (ΔF%) by setting “Response over baseline,” “Subtract background” (using initial fluorescence value as baseline), and “Show as percentage” corrections. Peak amplitudes were calculated as Maximum ΔF% value in between each subsequent test pulse (TP) delivered by the TLC apparatus throughout fluorescence measurement. The mean and standard deviation of at least a four-well replicate were calculated. The percentage of activity was computed for each test pulse as peak amplitude in the presence of a given compound concentration divided by peak amplitude in the absence of treatment. Sigmoidal dose–response curve fitting was obtained using GraphPad Prism software (variable slope). Normalized activity for a given compound concentration is calculated as the percentage of activity at a given test pulse divided by the percentage of activity at the first test pulse. Z’ factor was calculated according to the following formula: 1 − [(3 SD max + 3 SD min) / (MEAN max − MEAN min)], where SD is the standard deviation, MEAN is the arithmetic average, max refers to wells treated with DMSO (0.5%), and min refers to wells treated with 30 µM amitriptyline. The percentage coefficient of variation (CV%) was calculated according to the following formula: SD / MEAN × 100.
Chemicals
All-trans retinal, fibronectin, amitriptyline hydrochloride, nicardipine, amlodipine besylate, diltiazem hydrochloride, and verapamil were from Sigma Aldrich (St. Louis, MO). Mibefradil was from Alomone Labs (Jerusalem, Israel). Pimozide was from Tocris Bioscience (Bristol, UK).
Results and Discussion
Our work aimed to combine the advantageous assay times and cost of fluorescence plate imagers (which use low-cost reagents and simultaneously process all wells of miniaturized plates) with the temporal control of photoactivated tools to create a solution that allows physiological, rapid, and low-cost analysis of drug effect on ion channels and use dependency. Our goal was to provide a physiological mean to study processes activated rapidly and at a specific frequency, such as action potential propagation or cardiac muscle contraction, for which existing methods lack in physiology, throughput, or temporal control.
We developed a method that applies programmable millisecond-switched light perturbations in concomitance with kinetic fluorescence plate imaging via simultaneous 384-well processing. We called our system FLiOP (fluorescence imager with optical pacing). We used FLiOP to study cellular processes paced by photoactivated proteins, such as channelrhodopsin, in both recombinant and iPSC-derived systems. The newly developed method allowed us to study voltage-activated targets at different frequencies of stimulation, and to investigate the use dependency of blocker molecules in a miniaturized all-optical setting that is well suited for large-scale drug-screening activities.
Setup of FLiOP
We investigated the feasibility of applying repeatable optogenetic challenges to large-scale drug-screening instrumentations by combining flexible programming of the light-pacing sequence with the instrument operation of fluorescence imaging. In particular, we aimed to develop a new method to identify drug molecules selectively targeting the pathological frequency of target functioning without acting on the normal cell signaling; to this aspect, we developed a method able to provide transient triggers at frequencies relevant for normal or pathological target functioning. FLiOP was created by design and assembly of a TLC apparatus to be connected to a FLIPR instrument to control its light-flashing operations. The TLC apparatus controls the temporal pattern of flashing of one of the two embedded LED optics of FLIPR, while the second embedded optics records the fluorescent probe response through the native plate imager functionalities. The TLC includes: LED driver modules (to control the flashing of one LED bank), a photodiode (to register the light flashing of LED banks), an electrical connection to the FLIPR camera (to allow the synchronization of the controlled LED flashing with the functioning of the imager), an Arduino controller (to monitor the photodiode output and manage LED driver parameters), and a graphical user interface (a software control module configured to run on a computer cooperating with the imager) (
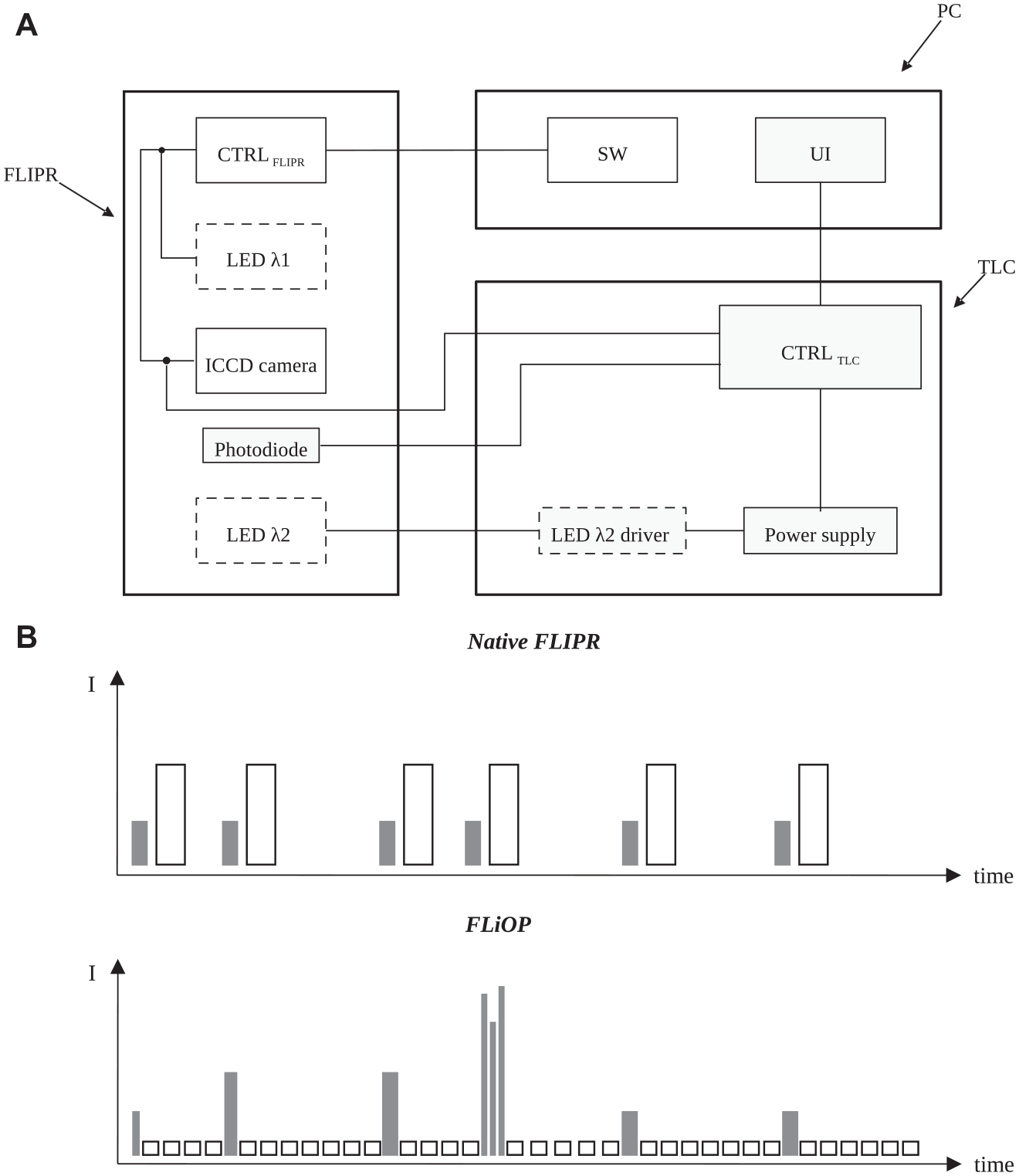
The fluorescence imager with optical pacing (FLiOP) method. (
We designed FLiOP to be well suited for the investigation of use dependency in voltage-activated targets using light-activated depolarizing tools. In particular, (1) we reduced the native minimum excitation time down to 1 ms (this is important to trigger targets that suffer from fast inactivation, such as voltage-activated sodium channels); (2) we enabled independent programming of the two FLIPR optics: the controlled LED and the LED used for fluorescence-recording flash with different intensity, number, duration, and frequency (this is crucial to design a flashing pattern to stimulate the voltage-activated target without affecting the continuous recording of the fluorescence indicator response) (
The main novelty of our method is that optogenetic trigger and fluorescence recording are programmed with different temporal settings and are applied simultaneously in 384 wells of miniaturized plates, using instrumentation that can also inject compounds by simultaneous 384-tip-head operation. This combination of features is ideal to perform large-scale pharmacological investigations, because a fast and low-cost assay is mandatory to process millions of compounds, as well as a physiological means to mimic fast-paced biological processes and the possibility to monitor online the effect of test compounds. In particular, the use of optical readouts can be very advantageous for its contained operational costs, and it has already been applied to develop optogenetic ion channel assays in miniaturized fluorescence-screening settings. Compared to existing methods, the main implementation of our method is that the light used for optogenetic stimulation can be switched on for a very short time (in particular, we reduced the minimum excitation time of about 10 ms of the FLIPR instrument down to 1 ms): This aspect can be fundamental to study fast-inactivating targets (such as, e.g., voltage-activated sodium channels) because they may accumulate in the inactive refractory state when triggered for more than a few milliseconds, rendering difficult the study of target behavior upon repeated activation cycles.
Moreover, compared to existing plate imagers, our method enables researchers to deliver the optogenetic trigger only at the programmed time, and not during the functioning of fluorescence imaging; in fact, to avoid the continuous activation of the target of interest, the optical actuator (ChR2 in our experiments) and optical indicator (calcium- or voltage-sensitive dye in our experiments) should be excited with well-separated wavelengths; and, more importantly, the light used for optogenetic stimulation should be switched off during the recording of target response by the optical indicator to avoid continuous activation of the optical actuator. To our knowledge, our method is the first example of a temporally flexible all-optical assay performed with a parallel 384-well recording instrument suitable for online compound addition. Future improvements of our method might include the increase of acquisition speed and light source power of the optical reader: These two features would enable the study of submillisecond processes (such as neuronal action potentials or fast-inactivating sodium channel currents) by the use of faster but dimmer voltage indicators (such as QuasAr archaerhodopsin-based voltage indicators). 7 Also, the possibility to diversify light intensity, color, and temporal pattern applied to every single well might enable the study of an array of different conditions simultaneously in a single assay plate, further reducing the assay operational costs while increasing its multiplexing capacity.
Optogenetic Pacing of Recombinant Voltage-Activated Ion Channels
We first validated the use of FLiOP by pacing the activity of recombinantly expressed voltage-gated ion channels (VGICs) with a millisecond-switchable photoactivated cation channel, channelrhodopsin 2 (ChR2). 8 We chose both sodium- and calcium-permeable VGICs for these first proof-of-concept experiments, because their targeting by selective use-dependent drugs represents a largely unmet challenge with great therapeutic potential. 1 Although the electrophysiological technique has adequate resolution power to elucidate the use dependency of VGIC blockers, study of these channels in a homogeneous high-throughput format is currently limited to nonphysiological and nonreversible chemical triggers.
We studied NaV1.5 and CaV1.3 voltage-activated channels in HEK-293 cells stably expressing TASK1 or KIR2.3 potassium channels. Indeed, wild-type HEK-293 cells exhibit a plasma membrane potential around −20 mV, a potential at which most of the NaV1.5 and CaV1.3 are in the inactivated state and therefore do not conduct. The presence of a potassium channel produces an electrochemical gradient for K+ adjusting the plasma membrane close to its reversal potential (i.e., around −80 mV) in the absence of added external potassium and keeping the VGICs in their closed resting state. As shown in
Figure 2
, both NaV1.5 and CaV1.3 targets were efficiently and specifically activated by repetitive light stimulation of ChR2, either recombinantly coexpressed or provided by a “cell delivery” strategy.
9
Ion conductance, measured by spectrally separated fluorescent indicators, showed very good signal reproducibility among different well replicates, maintained response amplitude throughout subsequent test pulses, and sensitivity to specific target inhibitors, in either hyperpolarized (K0 buffer;
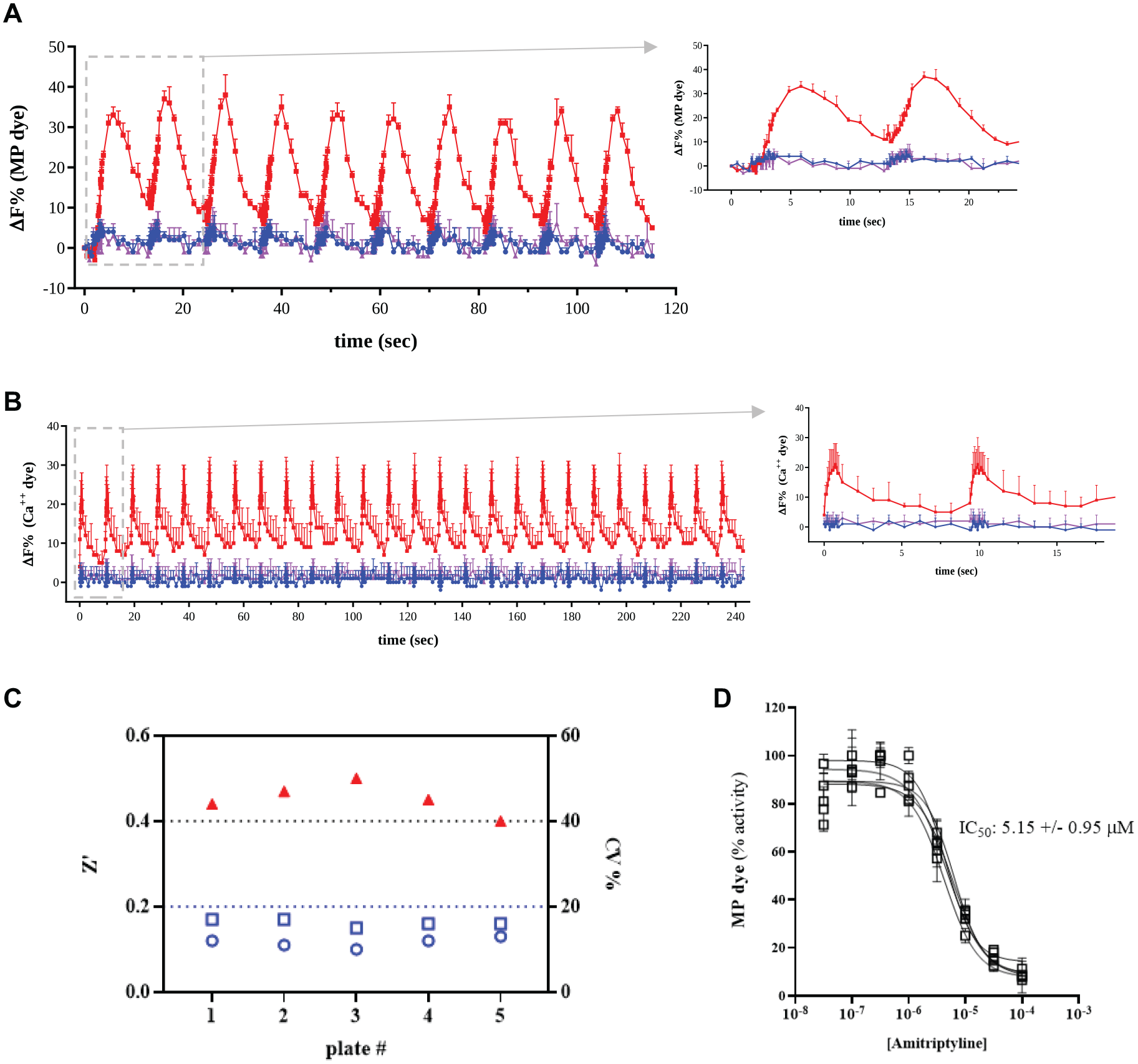
Repetitive stimulation of voltage-activated ion channels by a fluorescence imager with optical pacing (FLiOP). (
Taken together, these first results demonstrate that FLiOP is a robust and reliable method to study voltage-activated targets by repeated transient triggers, thus suggesting the possibility to test use dependency in drug-screening operations.
To further address the suitability of FLiOP for large-scale compound testing, we have determined the assay robustness and reproducibility in a multiplate test on a NaV1.5 assay. Our data showed good assay performance (Z’ value higher than 0.4 and CV% lower than 17% in all five tested plates;
In our experiments, we used the T159C mutant of ChR2 as the optical actuator, which shows an intense and fast kinetic photocurrent combined with high light sensitivity. 8 The former is important to achieve a robust photoactivation of the voltage-sensitive target and to avoid its inactivation by prolonged stimulation; the latter is very important for the intensity of the light source used: In our case, the LED of the FLIPR instrument has a native power of about 0.1 mW/mm2, 10 which we increased by about 100% via our apparatus for LED control (data not shown). As supported by previous data, other light actuators (such as bPAC enzyme) can be triggered by a parallel 384-well FLIPR imager, 11 thus suggesting that FLiOP can be used to explore other optically triggered cellular processes.
As optical readout, we used in our experiments calcium- or voltage-sensitive organic indicators, because they provide good light sensitivity and a big assay window, even if their response kinetic is moderately slow. Nevertheless, previous data also showed that genetically encoded indicators (such as GCaMP or ArcLight) can be well suited for all-optical studies in a parallel 384-well FLIPR imager:9,11 These indicators have faster kinetic responses, which can provide a better temporal resolution in the studied process. Moreover, their genetic targeting can be a big advantage to avoid the cytotoxic drawbacks of organic dyes and to facilitate stable long-term cellular studies.
With these first proof-of-concept experiments, we have shown the utility of FLiOP to transiently trigger voltage-gated calcium or sodium channels, one of the most medically relevant target classes, by means of ChR2 photocurrent. Nevertheless, the exploitation of our method to voltage-gated potassium channels would be strongly desired for drug discovery applications; to this aspect, the opposite direction of ChR2 and K+ channel current should be taken into account when setting up the proper cellular assay.
Optogenetic Pacing of iPSC-Derived Cardiomyocytes
We further investigated the use of FLiOP for pacing the activity of iPSC-derived cardiomyocytes, with the purpose of establishing a biological and methodological procedure to reliably evaluate the potential cardiac liability of drug candidates in a high-throughput configuration. To obtain optogenetic pacing of cardiac cells, we chose a “cell delivery” approach, co-seeding iPSC-derived cardiomyocytes and HEK-293 cells recombinantly expressing ChR2; this approach does not alter the genetic background of cardiomyocytes (as occurs with viral or other “gene delivery” strategies), and it has been shown to be feasible for the pacing of cardiac systems12,13 and adaptable to a miniaturized 96-well plate format analyzed by all-optical sequential well readers. 14 Thanks to endogenous gap junction expression, HEK-293 cells and iPSC-derived cardiomyocytes can connect and exchange ions and small-molecule second messengers, 13 thus generating a multicellular system in which both membrane potential setting and cellular excitability are transferred from cell to cell, as in a functional cardiac syncytium.
Similar to the results obtained with recombinant cells, iPSC-derived cardiomyocytes closely followed the light-pacing pattern with very high reproducibility among different well replicates, and with a response amplitude and shape very similar to those of native unsynchronized wells and very well maintained throughout subsequent test pulses (
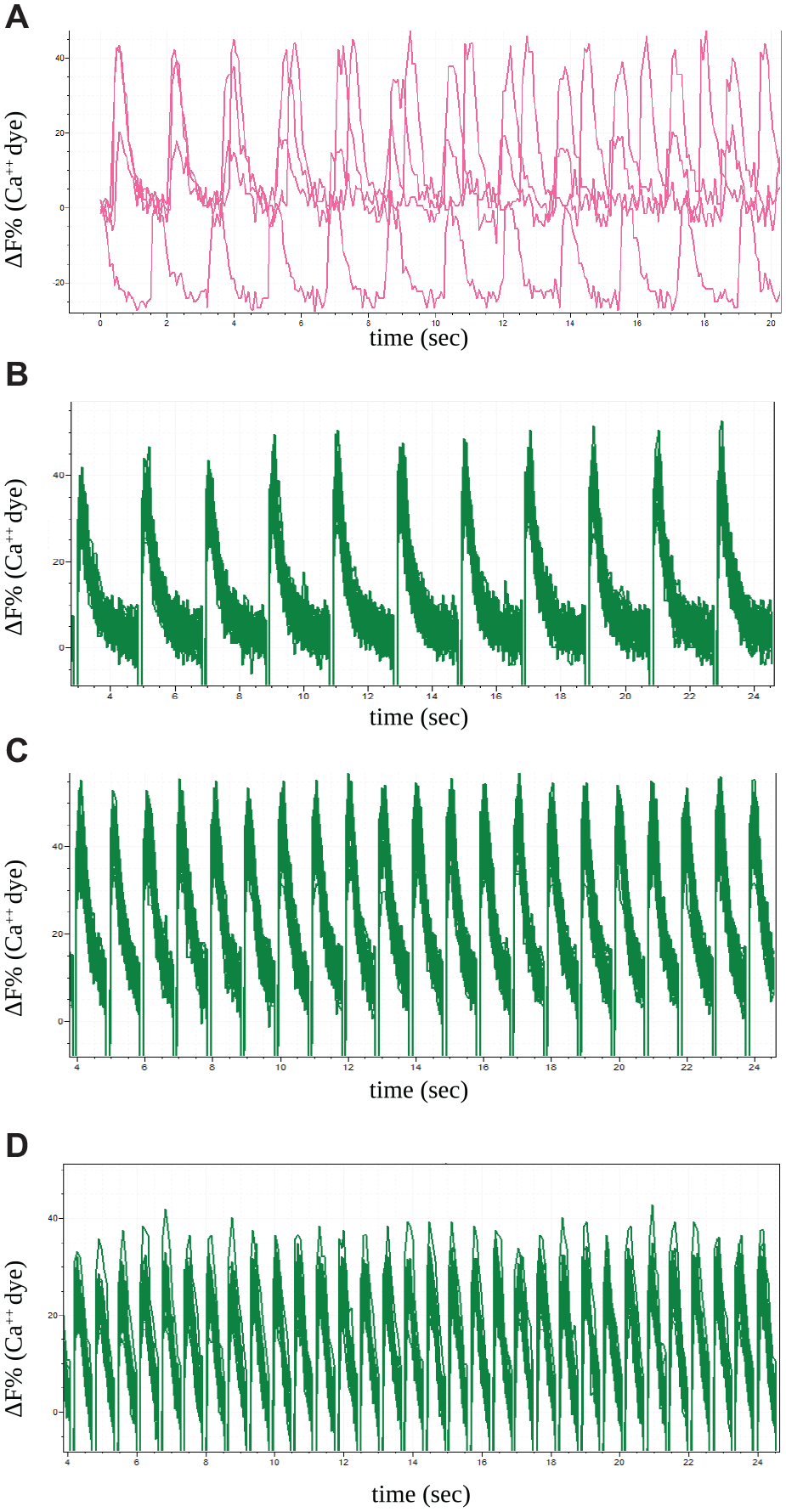
Optical pacing of induced pluripotent stem cell (iPSC)-derived cardiomyocytes by fluorescence imager with optical pacing (FLiOP). iPSC cardiomyocytes: (
Screening of Use-Dependent Calcium Channel Blockers
After setting up the conditions for optical pacing of cellular excitability by FLiOP, we investigated a small collection of compounds for their ability to inhibit in a use-dependent manner CaV1.3, an L-type voltage-gated calcium channel mainly represented in the brain and involved in several human disorders. 15 We selected six compounds that have been previously described to inhibit the activity of the L-type calcium channels subtype with various use dependency properties,16–19 with the aim of demonstrating that our method can well overlap results obtained by the gold standard electrophysiology technique.
In our experimental conditions, we observed different degrees of use dependency, from moderate (mibefradil) to massive (nicardipine) with five of the tested compounds, while no use dependency was observed for amlodipine (
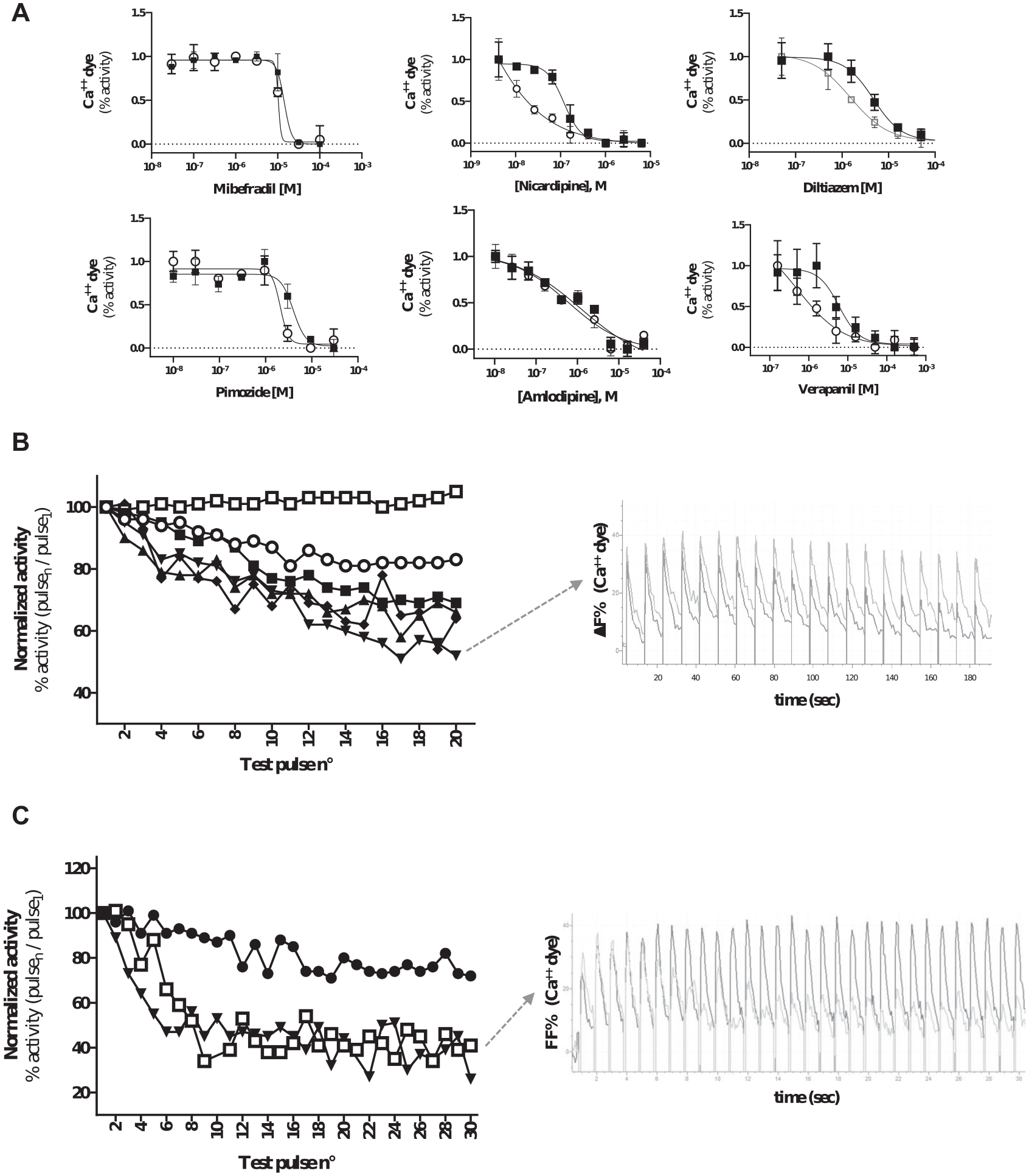
Screening of use-dependent drug effect by fluorescence imager with optical pacing (FLiOP). (
Among the six profiled compounds, we then selected pimozide to be used for validation of our cardiotoxicity FLiOP assay, because this potent neuroleptic drug is known to cause cardiac arrhythmic side effects.
19
Optogenetic pacing of iPSC-derived cardiomyocytes showed that pimozide acts as a use-dependent inhibitor of cardiac calcium transients, in particular at physiological beating rates [60 and 90 beats per minute (bpm);
Taken together, our results represent the first all-optical assay in parallel 384-well format that synchronizes iPSC-derived cardiac cell beating. The possibility to evaluate drug effect at different beating frequencies by a cost- and time-effective method could be of significant support for cardiac liability studies and for basic research investigations.
With our method, we explored for the first time drug behavior at different pacing frequencies in parallel 384-well all-optical settings, obtaining use-dependent profiles in good agreement with published pharmacological data produced with the electrophysiology technique. For this aspect, our method provides superior utility compared to classical chemical triggers, since it enables comparative studies on targets analyzed at their physiological or pathological functioning.
Thanks to its contained operational costs and assay time, our method could be already applied to the first steps of large-scale screening campaigns: By immediate identification of drug molecules acting on the pathological frequency of ion channel firing, the safety and specificity of the developed drug could be substantially improved.
Footnotes
Declaration of Conflicting Interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: All the authors currently work for AXXAM S.p.A biotechnology company. Methods and applications of FLiOP were deposited as Industrial Invention Patent (application nos. 18174217.2 and PCT/EP2019/063174) and are currently under evaluation.
Funding
This research was partially funded by the Ministero dello Sviluppo Economico (EuroTransbio grant no. F/0001/01/X29), which provided financial support for the conduct of the research. The funding source was not involved in the study design; the collection, analysis, and interpretation of data; the writing of the report; or the decision to submit this article for publication.