
Abstract
The lack of miniaturized and cost-effective methods to control cellular excitability with dosable and temporally precise electrical perturbations represents a long-lasting and unsolved bottleneck for ion channel drug discovery pipelines. Here we developed a high-throughput–compatible fluorescent-based cellular assay that combines optogenetics and co-culture approaches to obtain spatial, temporal, and quantitative control of ion channel activity. The modularity and increased flexibility of control of this light-tandem assay, combined with contained costs and compatibility with conventional drug-screening platforms, make this system suitable for temporally precise screening of ion channel function in controlled conformations and can also be used to recapitulate other complexly regulated biological processes.
Introduction
Voltage-gated ion channels (VGIC) are transmembrane proteins that determine rapid ion flux across cell membranes upon changes of electric membrane potential near the channel. They play a crucial role in propagating electrical signals in excitable cells and in regulating ion homeostasis, and their dysfunctions are responsible for many severe human diseases. 1 Unfortunately, the identification of novel molecules acting on these important drug targets is strongly limited by the lack of a high-throughput technique able to recapitulate their physiological functioning, in particular to allow the study of ion channel properties at different fixed membrane potential values or to monitor the ion channel flux upon rapid and repetitive changes of membrane potential. In fact, even if the currently available miniaturized electrophysiology devices can deliver all these optimal physiological controls, they present severe limitations in the application to large-scale compound screening campaigns because of their considerable assay costs. In the past years, the development of the optogenetic technique has enabled the control of many biological processes, including cellular excitability, through light-controlled proteins able to monitor temporally precise perturbations in a spatially defined manner. In particular, light-activated ion channels (channelrhodopsins) have been used to deliver fast and reversible membrane depolarizations, 2 while genetically encoded fluorescent indicators have been used to monitor rapid changes of cellular parameters, such as calcium concentration or membrane potential. 3 In parallel with the fast and repetitive control of cellular excitability achievable by optogenetic manipulation, a steady-state control of membrane voltage can be obtained introducing a specific amount of a passive potassium conductance (such as an inward rectifier potassium channel Kir2.x); by this means, the level of depolarization of the cell membrane will change according to the level of expression of Kir channel protein and to the extracellular potassium concentration. Taken together, light- and potassium-controlled approaches might be used to obtain transient or steady-state perturbations of cellular excitability, thus recapitulating different voltage-clamp electrophysiology protocols in a functional fluorescent cellular assay. To validate the possibility of using such optogenetic and potassium channel-based approaches to study VGIC functions, we used a syncytium approach, based on the ability of HEK-293 cells to form cell-cell connections and to transmit electrical signals through gap junction proteins.4,5 This method combines two distinct cell lines (each expressing a component of the cellular process) to obtain a functional syncytium assay that provide many advantages: (1) recapitulate multicomponent cellular process at a cell population level, thus decreasing the experimental effort for assay development and also the metabolic burden of each single cell line; (2) enable the analysis of different combinations of VGIC cell line and light- or potassium-controlled cell lines, by simply co-culturing for few days; and (3) fine-tune the level of expression of each single component of the syncytium by varying the relative number of co-cultured cells, thus enabling a titration of their functional contribution to the biological response ( Fig. 1 ).
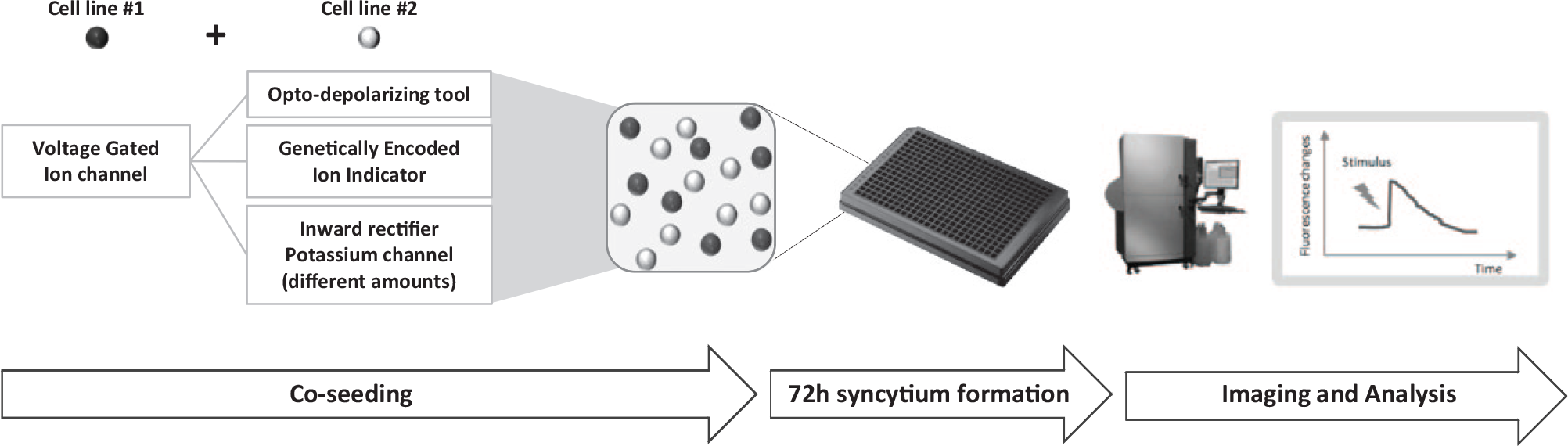
Experimental flowchart for co-culture functional cellular assays. Different combinations of two distinct HEK-293 cell lines are co-seeded in 384-well plates in 1:1 or other ratios, to obtain syncytial cell populations after 72 h of co-culturing. Thanks to ions and second messenger sharing through endogenously expressed connexin channels, the two cell lines became electrically wired and behave as a functional monoculture assay. Syncytia are analyzed using a fluorescence plate imager instrument, the FLIPR, for both the optical or chemical stimulation and measurement of the fluorescent readout response.
Materials and Methods
Gene Constructs
Full-length coding sequences, optimized for mammalian codon usage, were obtained by GeneArt, subcloned in the vector of interest under constitutive promoters, then fully sequence verified. Human Kir2.3 (NM_004981.1) was cloned in pcDNA3 vector (Invitrogen, Carlsbad, CA), human CaV3.3 (NM_021096.3) in pcDNA6-blasti vector (Invitrogen), human CaV1.2 (NM_001129843.1) in pIres2-EGFP vector (Clontech, Mountain View, CA), CaV accessory subunits (CACNB3, NM_000725.3 and CACNA2D1, NM_000722.2) in pBudCE4.1 bi-cistronic vector (Invitrogen), Chlamidomonas reinhardtii channel rhodopsin 2/chlamyopsin 4 light-gated ion channel (“ChR2 LCTC,” XM_001701673.1; amino acids 1 to 309, mut L132C, T159C) in pcDNA3.1Hygro vector (Invitrogen), and RCaMP (KC572547.1) in pBudCE4.1 vector (Invitrogen).
Cell Culture and Transfection
HEK-293 cells (ATCC CRL-1573) were maintained in EMEM (BioWhittaker) supplemented with 10% fetal bovine serum, 100 U/mL penicillin, 0.1 mg/mL streptomycin, and 2 mM L-glutamine in a 37 °C humidified incubator with 5% CO2 atmosphere. Cells were detached by trypsin-EDTA treatment (EuroClone ECB3052D, Milan, Italy) then automatically counted using Beckman Coulter Z1 particle counter. For stable transfections, 3 × 106 cells were resuspended in 800 µL Opti-MEM I (Gibco 31985070) then electroporated (300 V, 950 µF; BioRad Gene Pulser II) with 10 µg of DNA; selective antibiotic was added 48 h after transfection, and stable pools were obtained after about 10 to 14 d of growth; for multiple transfected cell lines, sequential procedures of electroporation and antibiotic selection were carried out. T-type voltage-gated calcium channel (VGCC) cell line (containing Kir2.3 and CaV3.3 constructs) was maintained in G418 0.2 mg/mL and blasticidin 5 µg/mL; L-type VGCC cell line (containing Kir2.3, CaV1.2, and CaV accessory subunits constructs) was maintained in G418 0.2 mg/mL and Zeocin 10 µg/mL; VGSC cell line (from Genionics AG, Zurich, Switzerland, expressing the human NaV1.5 protein, acc. Q14524) was maintained in gentamicin 20 µg/mL and Zeocin 100 µg/mL; ChR2 and VGSC-ChR2 are the same cell line (NaV1.5 cell line stably expressing ChR2-LCTC) and was maintained in hygromycin 20 µg/mL, gentamicin 20 µg/mL, and Zeocin 100 µg/mL; genetically encoded calcium indicator (GECI) cell line (containing RCaMP construct) was maintained in Zeocin 10 µg/mL; and Kir2.3 cell line (containing Kir2.3 construct) was maintained in G418 0.4 mg/mL.
Stable Clones Generation
After antibiotic selection, each stable pool was diluted in eight 96-well plates at three cells/well density, then the obtained clones were replicated in poly-D-lysine–coated black-clear 384-well plates (Matrix GR-4332-CPL). Functional best clones were selected at FLIPR Tetra instrument (Molecular Devices, Sunnyvale, CA), upon chemical or optical stimulation and imaging. VGCC cell lines were analyzed as potassium-induced calcium response (Fluo-8,NW assay), VGSC cell line was analyzed as veratridine-induced membrane depolarization (Membrane Potential Dye Assay), ChR2/VGSC-ChR2 cell line was analyzed as blue light–induced membrane depolarization (Membrane Potential Dye Assay), GECI cell line was analyzed as carbachol-induced calcium response (RCaMP assay), and Kir2.3 cell line was analyzed as potassium-induced membrane depolarization (Membrane Potential Dye Assay).
Syncytium Formation
Two distinct cell lines were seeded in the same well of a 384-well black-clear poly-D-Lys plate (Matrix GR-4332-CPL) in 25 µL/w of standard culture medium without selective agents (5 µM All-trans Retinal was included in ChR2 experiments), then cultivated for 72 h in a 37 °C humidified incubator with 5% CO2 atmosphere, in order to allow syncytium formation; standard cell density for 1:1 ratio experiments was 3000 + 3000 c/w, whereas 3000 c/w plus 10 to 2000 c/w density was used in the VGSC-Kir2.3 dose-response assay.
FLIPR Assay
Fluo-8,NW (AAT Bioquest, Sunnyvale, CA; cat. 36316), or Membrane Potential (Molecular Devices; cat. R8034) dye loading solutions were prepared according to the manufacturer’s instruction in Tyrode’s buffer (composition in mM: NaCl 130, KCl 5, CaCl2 2, MgCl2 1, NaHCO3 5, HEPES 20; pH 7.4; autoclaved) or K0Na150 buffer (composition in mM: KCl 0, NaCl 150, CaCl2 2, HEPES 10, glucose 10; pH 7.4; 0.22 µm filtered). Medium was manually removed from 384-well plates, and then cells were incubated at 37 °C, 5% CO2, with 20 µL/w of dye loading solution (1 h incubation) or buffer alone (GECI experiments, 15 min incubation), in the presence or absence of indicated blockers’ concentrations; 5 µM All-trans Retinal was included for ChR2 experiments. Plates were then analyzed with the FLIPR Tetra instrument (Molecular Devices) equipped with an intensified charge-coupled device camera, upon injection of 20 µL/w of the indicated stimuli (2× concentrated, in same loading buffer) and fluorescence changes monitoring (Fluo-8,NW assay: λexc/λem 470–495 nm/515–575 nm, 1 Hz; Fluo-8,NW-ChR2 assay: λexc/λem 470–495 nm/515–575 nm, 30 Hz; Membrane Potential or RCaMP assays: λexc/λem 510–545 nm/565–625 nm, 1 Hz; Membrane Potential–ChR2 assay: λexc/λem 510–545 nm/565–625 nm and 495 nm/515–575 nm, 3.5 Hz). Data were analyzed using Screenworks software (Molecular Devices; version 4.0.0.30) after applying “response over baseline,” “subtract background,” and “show as percentage” corrections; maximum peak (MAX) values were exported and analyzed with Excel software to calculate mean and standard deviations (four-well replicates) then used in PRISM software (GraphPad version 7.0) to fit sigmoidal dose-response curves (variable slope).
Chemicals
All-trans Retinal, amitriptyline hydrochloride, carbachol, and DMSO were from Sigma Aldrich (St. Louis, MO); potassium chloride was from DelChimica (Italy); veratridine and isradipine were from TOCRIS Bioscience (Bristol, UK); and selective antibiotics were from Invivogen (Thermo Scientific, Waltham, MA).
Results and Discussion
The first co-culture assay that we generated was aimed to obtain a fluorescent cellular assay where a voltage-gated calcium channel (VGCC) can be activated by fast, repetitive, and reversible membrane voltage changes, similarly to what happens in physiological environment and also to what can be done by electrophysiology technique; this temporally precise stimulation would significantly improve the study of VGCC function, as currently available methods to induce membrane depolarization in cellular assays include mainly the use of unphysiological and not reversible stimulation with high potassium buffers,6,7 whereas physiological fast stimulation protocols are exclusive of expensive electrophysiology techniques. The syncytium was composed of a cell line expressing either a T-type or an L-type voltage-gated calcium channel and another cell line expressing the fast kinetic blue light–activated cation channel Channelrhodopsin2 (ChR2, L132C T159C mutant), 2 in 1:1 ratio. Upon stimulation of the syncytium with repetitive blue light pulses, we obtained fast, robust, and specific activation of calcium flux through both the VGCCs analyzed ( Fig. 2A ). These results showed that membrane depolarization induced by light stimulation of ChR2 can be well transferred through gap junction connections of HEK-293 cells and can be used to obtain a robust activation of a voltage-gated calcium channel. Moreover, they provide a fully high-throughput screening (HTS)–compatible mean to activate VGCCs that mimics physiological electrical perturbations, thus facilitating the identification of more biologically relevant blockers’ compounds during drug-screening campaigns.
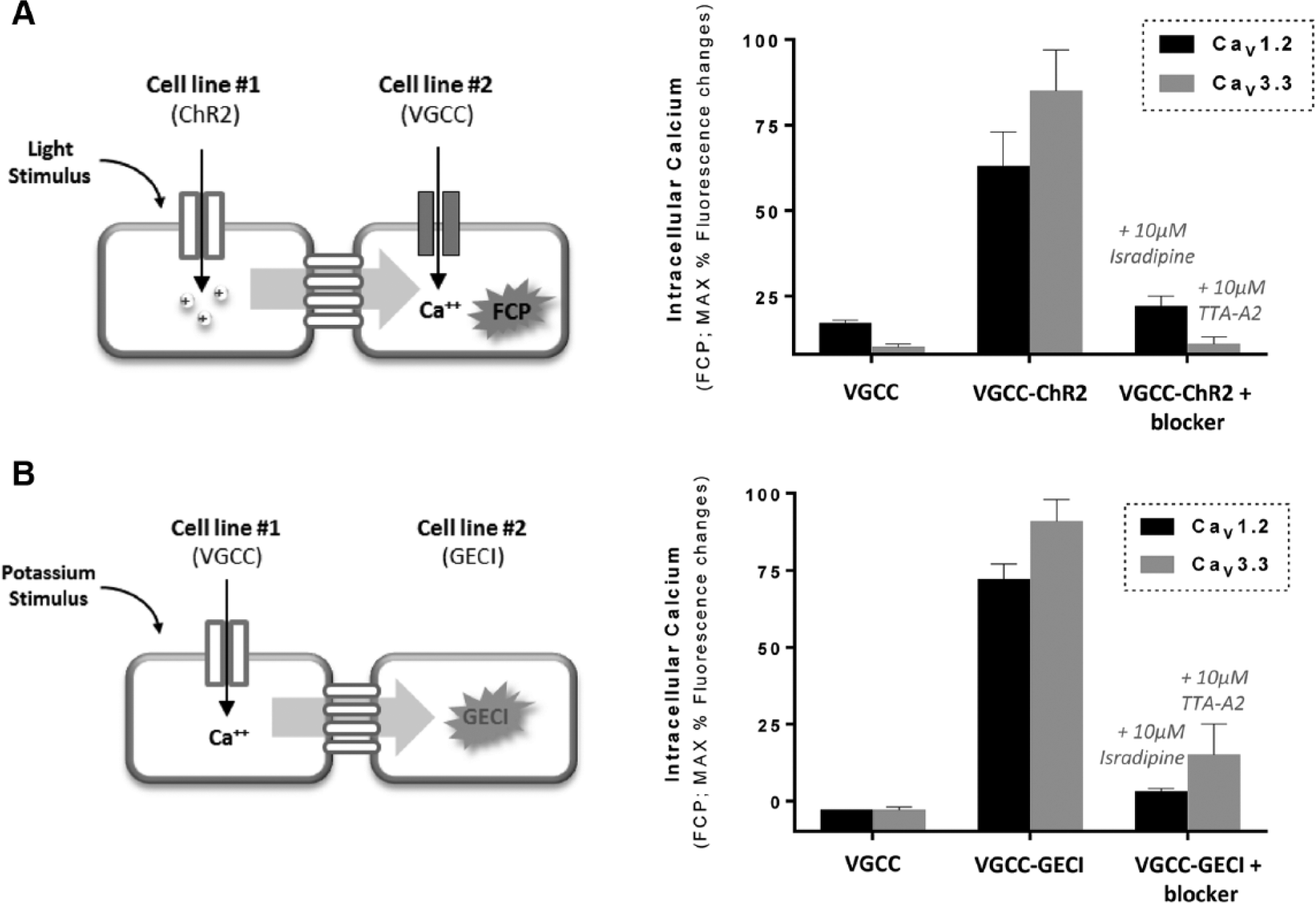
Optical control of voltage-gated calcium channel assay by Channelrhodopsin stimulation or RCaMP indicator imaging. (
Based on the validation of this optically controlled VGCC co-culture assay, different possible combinations might be explored in the future, for example, the co-culture between a different light depolarizing tool (with the desired biophysical properties of speed, photocurrent amplitude, etc.) and a VGIC of interest, thus potentially enabling the generation of various light-controlled ion channel assays suitable for temporally precise HTS operations.
To investigate if the modularity of the co-culture approach could be also applied in an opposite direction, we created a second co-culture system with the aim of measuring the activity of a VGCC through a GECI. Depending from the type of GECI used, significant improvements can be achieved over the use of organic calcium-sensitive dyes, such as faster kinetic response, lower cytotoxicity effects, and spatially defined detection of calcium oscillations. 8 We generated a syncytium assay in which either a T-type or an L-type VGCC cell line is co-cultured with a cell line expressing the well-known red fluorescent calcium indicator RCaMP 8 in a 1:1 ratio. Membrane depolarization was obtained by a high potassium buffer application to the syncytium, then calcium influx through the VGCC was transferred to the GECI cell line, generating a specific and robust fluorescent signal, well inhibited by channel reference blockers ( Fig. 2B ). These experiments demonstrated that intracellular calcium oscillations determined by VGCC activity can be well transferred through gap junction connections and can be precisely monitored using GECIs such as RCaMP. The use of different versions of genetically encoded indicators (with the desired biophysical properties of speed, ion sensitivity, spectral feature, etc.) with the ion channel or target of interest could be also explored in the future, thus enabling the generation of optically tracked cellular assays with improved spatial and temporal resolution.
After demonstrating that spatiotemporally precise optogenetic tools, such as channelrhodopsin or calcium indicators, can be used in modular multicellular assays to efficiently control VGCC activity, we sought to determine if the syncytium concept can be used in a quantitative fashion to achieve a dosable and controllable effect of single biological components expressed by the co-cultured cell lines. For example, the ability of inwardly rectifying potassium channels to maintain the membrane voltage of a cell near hyperpolarized potassium equilibrium potential, and to induce rapid membrane depolarization in response to extracellular potassium increase, is well known. 9 These properties are often used in cellular assays where a tight control of membrane voltage is needed, for example, to study VGIC 7 ; in fact, normal functioning of VGICs requires a hyperpolarized membrane potential to keep the channel in a resting (closed) state and a depolarization step to drive the channel first in a conducting (open) state and then in a nonconducting and refractory (inactivated) state. Most of the human diseases caused by ion channel dysfunctions involve an augmentation of cellular excitability that results in an increased number of VGICs in an open/inactivated state and severe ionic unbalances; thus, the possibility of identifying drug molecules selectively targeting the pathological state of the channel is strongly beneficial for efficacious disease treatment and avoidance of side effects due to action on normally functioning VGICs. 10 For this reason, it is very important to include in any drug-screening method the possibility of studying the compound effect on different VGIC biophysical states, and this aspect can be investigated by “clamping” the membrane potential to different values. To recapitulate such voltage clamp protocols used in the electrophysiology technique, we generated a co-culture assay in which the membrane voltage of a VGIC cell line is conditioned to different levels by the co-culture with different amounts of a Kir potassium channel cell line. This system exploits the concept of “quantitative” modulation of the biological effect of the Kir channel, similar to inducible expression systems but with the advantages of (1) avoiding possible pleiotropic effects associated with chemical induction; (2) enabling a tight control of the amount of Kir protein, in particular in the low level of expression range, which is often hardly achievable by inducible system modulation; and (3) providing a modular approach to explore the effect of different Kir expression levels on the activity of the VGIC of interest. The co-culture assay included a fixed amount of the cardiac NaV1.5 voltage-gated sodium channel (VGSC) cell line in combination with different amounts of a Kir2.3 cell line. To demonstrate if different amounts of Kir2.3 cells can be used to induce different levels of membrane hyperpolarization, and to control VGICs’ biophysical states, we measured the NaV1.5 response upon stimulation with veratridine, an alkaloid neurotoxin that preferentially binds the channel in its open state, profoundly delaying its inactivation. 11 Our results showed that different amounts of Kir2.3 cells are influencing dose-dependently the response of the NaV1.5 channel to veratridine, consistent with a proportional hyperpolarizing effect on the syncytium ( Fig. 3A ). In fact, when Kir2.3 cells are not present, the resting voltage of HEK-293 cells is expected to be about −30/−40 mV, 12 and this condition corresponds to a high proportion of NaV1.5 channels in the inactivated state and very few channels available for opening, resulting in a modest veratridine effect. When a moderate number of Kir2.3 cells is present, the membrane voltage can move to partially hyperpolarized values, which are ideal for NaV1.5 activity detection, as they correspond to an optimal proportion of NaV1.5 channel in the open state available for activation and resulting in an enhanced veratridine response.
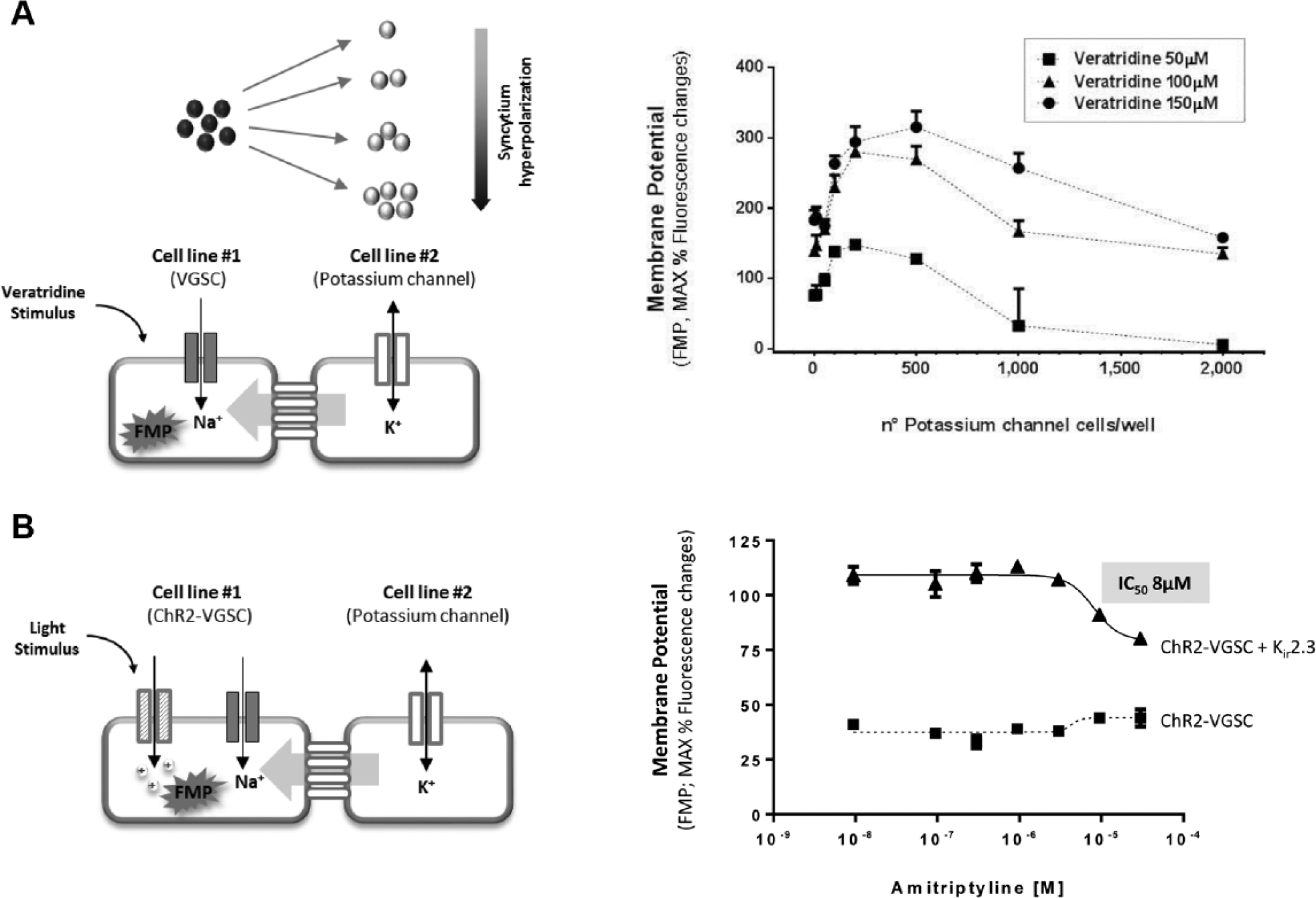
Modulation of ligand- or light-induced stimulation of voltage-gated sodium channel by Kir2.3 co-culture. (
Finally, when the Kir2.3 channel is highly expressed (similarly to recombinant expression by constitutive promoters), the membrane voltage of the cells will be strongly hyperpolarized, keeping the majority of NaV1.5 channels in a resting (closed) state, and resulting in a limited veratridine effect.
After showing with these experiments that Kir2.3 expression levels can modulate the proportion of NaV1.5 channels available for activation resulting in a quantitative control of VGSC affinity for openers, we sought to determine if this strategy may help in the detection of VGSC activity elicited by Channelrhodopsin light stimulation. In fact, when a NaV1.5-Channelrhodopsin cell line was stimulated by light, only the amitriptyline-insensitive Channelrhodopsin component was detected ( Fig. 3B ), possibly because of small amounts of NaV1.5 channels available for activation and also because of poor temporal resolution of FLIPR instrument and oxonol voltage indicators, which render tricky the detection of fast kinetics currents typical of VGSCs. We showed that when Kir2.3 cells are co-seeded in the optimal 1:15 ratio with NaV1.5-Channelrhodopsin cells, the amitriptyline-sensitive response of NaV1.5 to light stimulation became detectable, suggesting that tight membrane voltage regulation can be trivial to obtain an optimal stimulation of VGSC activity by reversible electrical perturbations. These proof-of-concept experiments are the first evidence of a fully high-throughput compatible cellular assay suitable to detect blockers’ effect on VGSC physiologically activated by fast kinetics channelrhodopsin. This strategy might be applied in the future to more temporally precise instrumentations and readouts and may enable the study of the effects of blockers on light-controlled VGIC assays in different biophysical states, thus significantly ameliorating the success rate and therapeutic relevance of drug-screening campaigns against VGSCs.
Taken together, our results demonstrated that co-culture approaches can be used in miniaturized assays to recapitulate multicomponent processes at a cell population level, delivering a modular and dosable mean to control complex cellular events, such as cellular excitability. By using a standard light-emitting diode–based fluorescence plate imager as a light source to control optogenetic tools, we obtained the first example of a cheap, precise, and fully high-throughput–compatible mean to study ion channel functions following their physiological conformation changes and electrical perturbations.
The syncytium approach is based on ion and small-molecule exchange through gap junction–connected cells and thus can also be useful for studying other cellular processes involving membrane receptors or intracellular protein signaling, avoiding difficult and time-consuming multicomponent assay development, and delivering a valuable mean to tightly dose the contribution of each single biological component.
In conclusion, by combining the spatial and temporal resolution provided by optogenetic tools with the modularity and dosable aspect of co-culture approaches, cell-based assays can become controlled in a three-dimensional manner: spatial (where), temporal (when), and quantitative (how much).
This improved content combined with contained costs can provide a remarkable solution to assessing complex cellular functions in drug discovery programs as well as in basic research investigations.
Future applications to large-scale screening campaigns or to more precise instrumentations (such as high-speed or high-content imagers that can overcome the limitations in temporal and spatial resolution of FLIPR instrument) may result in more accurate cellular interrogations and will unravel the overall utility of our findings for drug discovery purposes.
Footnotes
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.