
Abstract
Demonstration of in vitro target engagement for small-molecule ligands by measuring binding to a molecular target is an established approach in early drug discovery and a pivotal step in high-throughput screening (HTS)-based compound triaging. We describe the setup, evaluation, and application of a ligand binding assay platform combining automated affinity selection (AS)-based sample preparation and label-free matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) analysis. The platform enables mass spectrometry (MS)-based HTS for small-molecule target interactions from single-compound incubation mixtures and is embedded into a regular assay automation environment. Efficient separation of target–ligand complexes is achieved by in-plate size exclusion chromatography (SEC), and small-molecule ligands are subsequently identified by MALDI-TOF analysis. In contrast to alternative HTS-capable binding assay formats, MALDI-TOF AS-MS is capable of identifying orthosteric and allosteric ligands, as shown for the model system protein tyrosine phosphatase 1B (PTP1B), irrespective of protein function. Furthermore, determining relative binding affinities (RBAs) enabled ligand ranking in accordance with functional inhibition and reference data for PTP1B and a number of diverse protein targets. Finally, we present a validation screen of more than 23,000 compounds within 24 h, demonstrating the general applicability of the platform for the HTS-compatible assessment of protein–ligand interactions.
Introduction
High-throughput screening (HTS) of large compound libraries against molecular targets has proven itself an efficient approach for early drug discovery campaigns focusing on the identification of novel chemical matter for lead optimization. While sophisticated assay technologies have been developed to enable assessment of test substances for their biological or biochemical effects, the HTS-compatible measurement of protein–ligand interaction has proven to be challenging. Because demonstration of small-molecule target engagement in a primary or secondary assay format is an integral part of HTS-driven drug discovery campaigns, innovative and HTS-capable binding assay formats would be beneficial. This is further emphasized by an increasing demand for binding assays aiming at the de-orphanization of unprecedented targets or the identification of multivalent ligands for targeted protein degradation [e.g., proteolysis-targeting chimeras (PROTACs)]. One major challenge has been the selection of unbiased screening formats and detection principles, because effects attributable to the readout technology, functional labels, and detection reagents can result in assay-specific hit sets with poor overlap among individual assay technologies. 1
Isothermal titration calorimetry (ITC), surface plasmon resonance (SPR), nuclear magnetic resonance (NMR) spectroscopy, and X-ray crystallography (XRC) represent established and label-free biophysical reference methods, which provide valuable information on structural, thermodynamic, and kinetic aspects of the macromolecule–ligand interaction but are not yet compatible with cycle times required for HTS of compound libraries commonly exceeding 100,000 molecules. Historically, displacement assays, in which the competition between a test compound and a labeled tracer molecule for binding to the target of interest is monitored, have been the method of choice for larger screening campaigns.2–4 Labeling strategies for a tracer ligand of known affinity comprise chemical introduction of functional labels or replacement of specific atoms by their respective radioisotopes for direct or indirect downstream detection. While competitive binding formats usually provide the required throughput, are easy to miniaturize, and can be fully automated, their dependence on a labeled tracer molecule necessitates the availability of a known ligand and constrains the exploration of binding epitopes on the target protein to the respective tracer binding site. An alternative HTS-capable format that has been widely used for detecting in vitro target engagement by small molecules is differential scanning fluorimetry (DSF). 5 Here, the ligand-dependent stabilization of a protein against thermally induced denaturation is measured with the aid of a hydrophobic dye that emits a fluorescent signal on binding to hydrophobic surfaces on denatured protein. Although this assay does not require a target-specific tracer and is potentially capable of detecting ligands of different binding epitopes, its information value is limited, because the correlation between protein stabilization and ligand binding is not generally valid. 6 Furthermore, unspecific compound-mediated protein perturbation, due to constraints in buffer composition, and signal interference can exacerbate data interpretation and analysis.
To circumvent such limitations, the use of mass spectrometry (MS) for label-free analyte detection combined with an affinity-based selection procedure, termed affinity selection mass spectrometry (AS-MS), has been suggested for identifying target ligands.7,8 A significant advantage of this technique is its label-free readout, which is independent of tracers and detection reagents and enables screening of orphan targets, in which no function or endogenous ligand is known. Furthermore, the isolation of protein–ligand complexes from nonbinding buffer components prior to analysis removes constraints in buffer composition and minimizes ion suppression caused by buffer components during MS analysis. Sophisticated applications of AS-MS are the Automated Ligand Identification System (ALIS)9,10 and the SpeedScreen 11 platform, which couple size exclusion chromatography (SEC)-based isolation of protein–ligand complexes to unambiguous detection of bound ligand via liquid chromatography/electrospray ionization MS (LC/ESI-MS). Although technical improvements of ESI instrumentation have shifted analysis cycle times to a few seconds per sample, this throughput is still inadequate for traditional large compound library screens. 12 The above-mentioned technologies therefore use compilations of test substances in compound pools comprising up to 2500 members per experiment. 9 While this is a powerful approach for screening specially designed mass-encoded libraries, technical implementation of large-scale compound pooling, complex library deconvolution, and ensuring compound integrity and solubility can be cumbersome. In addition, required elevated DMSO concentration (≥2%) might not be compatible with certain targets, and mutual binding competition of compound pool members could decrease assay sensitivity, thereby complicating the identification of low-affinity binders. The integration of AS-MS into regular HTS infrastructure therefore requires the use of MS technology with faster cycle times.
Compared to ESI-based MS, matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) MS provides the sampling speed required for HTS and has been established as a valuable readout strategy for large-scale screening campaigns against the catalytic activity of enzymatic targets.13–17 The adoption of the technology to interrogate protein–ligand interaction has been the subject of a number of proof-of-principle studies using ultrafiltration or affinity capture-based workflows to isolate protein–ligand complexes from incubation mixtures.18–20 Although these studies have demonstrated the general feasibility of using MALDI-TOF MS for label-free small-molecule detection, a report of its combination with an HTS-capable automation platform is unprecedented.
In this work, we present how the speed of MALDI-TOF MS combined with fully automated sample preparation and software-aided compound tracking can be leveraged to perform label-free protein–ligand binding assays in standard single-compound incubation HTS format on a regular automation platform. We describe the setup and validation of an automated SEC-based workflow in 384-well format for efficient and reproducible sample preparation, as well as the optimization of sample/MALDI matrix co-crystallization for MALDI-TOF small-molecule detection. Using the protein phosphatase PTP1B as a model system, we cross-compare the obtained AS-MS results to two orthogonal binding assay formats and a functional activity-based readout, and further demonstrate the capability of the platform in a validation study screening >23,000 small molecules on a single day.
Materials and Methods
Materials
Disposable HTS MALDI target plates (no. 1847006) and HTS MALDI Adapter (no. 8283496) were purchased from Bruker Daltonics (Billerica, MA). 384-well polypropylene (PP) microwell plates (no. 784201) were obtained from Greiner Bio-One (Frickenhausen, Germany). Recombinant human PTP1B_CA (PTP1B_C215A_h2-321_NHis8TEV) was purchased from Trenzyme GmbH (Konstanz, Germany). The catalytic domain of PTP1B (PTP-1B_h2-321_NHis8TEV) was produced in-house at Boehringer Ingelheim. α-Cyano-4-hydroxycinnamic acid (4-HCCA; no. 70990), acetonitrile (no. 34851), DMSO (no. D5879), ammonium acetate (no. 73594), bovine gamma globulin (BGG; no. G5009), HEPES (no. H3375), isopropanol (no. 34863), and SYPRO Orange (no. S5692) were obtained from Sigma Aldrich (St. Louis, MO). Trifluoroacetic acid (TFA; no. 6957) was purchased from Carl Roth (Karlsruhe, Germany). Bovine serum albumin (BSA; no. 11945.03) and dithiothreitol (DTT; no. 20710.04) were obtained from Serva (Heidelberg, Germany). Bio-Gel P10 Media (Fine: 45–90 µm; no. 150-4144) was purchased from BioRad (Feldkirchen, Germany). Multiscreen HTS 384-well filter plates [0.45 µm polyvinylidene fluoride (PVDF) membrane; no. MZHVN0W10], Tris (no. 108382), and MgCl2*6 H2O (no. 105833) were obtained from Merck Millipore (Burlington, MA). Peptides (custom synthesis), KCl 2M solution (no. AM9640G), and TCEP [tris(2-carboxyethyl)phosphine; no. 20491] were purchased from Thermo Fisher Scientific (Waltham, MA). AlphaScreen reagents (Streptavidin Donor beads, no. 676002; and anti-6xHis AlphaLISA Acceptor beads, no. AL128M) were obtained from PerkinElmer (Waltham, MA).
MALDI-TOF AS-MS Screening Workflow
The setup of incubation mixtures, the in-plate SEC-based separation process, and the transfer of 384-well plates into 1536-well format were performed on a fully automated system featuring a modular design and robotic plate transportation. At the beginning of each iterative cycle, a 384-well compound plate was transported to a CyBio Well vario liquid-handling system (Analytik Jena, Jena, Germany) equipped with a 384-well format capillary head to transfer 50 nL of 5 µg/µL or 10 mM compound solutions or DMSO to the wells of a 384-well PP assay plate. The compound plate was then put back into storage, and the assay plate was transported to a Thermo Multidrop Combi (Waltham, MA) dispenser, where 5 µL of 10 µM (0.40 mg/mL) PTP1B_CA in 50 mM HEPES (pH 7.4), 100 mM NaCl, 2 mM DTT, 0.01% BGG, 0.001% Tween20, and 25 µM quality control (QC) peptide (assay buffer) were added to every well. The assay plate was then vortexed for 30 s at 1000 rpm on a Bioshake 3000 (Quantifoil Instruments, Jena, Germany) device, covered with a plastic lid, and incubated at 24 °C for 40 min in a humidified chamber. Just before the completion of the incubation step, a loaded 384-well filter plate was stacked onto an empty 384-well PP plate, and both were loaded into a centrifuge. After centrifugation for 2 min at 800 rcf, the PP plate was discarded, and the filter plate was placed in a CyBio Well vario liquid-handling system (Analytik Jena) equipped with a 384-well format and 40 µL head. After transportation of the assay plate to the multichannel pipettor, incubation solutions were quantitatively transferred to the filter plate. On completion of this step, the filter plate was immediately stacked onto an empty 384-well PP plate (elution plate), and the combined plates were loaded into the centrifuge and spun at 800 rcf for 2 min. Subsequent to the separation step, the filter plate was restored, and the elution plate containing the eluted samples was transported to a CyBio FeliX liquid-handling system (Analytik Jena). There, 3 µL of every well was transferred from the 384-well elution plate to one quadrant (Q1/Q2/Q3/Q4) of a 1536-well plate. After four consecutive transfers from four individual elution plates (elution plate 1 → Q1, elution plate 2 → Q2, etc.), 3 µL of a mixture containing internal standard (500 nM final concentration) and the standard for the SEC quality control (QCref; 250 nM final concentration) in acetonitrile were added to every well of the 1536-well plate using a Certus Flex Micro Dispenser (Gyger, Gwatt, Switzerland). Plates were then sealed and stored until preparation of the MALDI target plates. A flowchart of the automated binding assay and a Gantt chart of the process sequence are provided in the online supporting information (
Hit Confirmation Workflow
Process steps and plate handling were executed as described above. For hit confirmation experiments, 50 nL of 5 µg/µL or 10 mM compound solutions were transferred to the wells of two individual 384-well PP assay plates (sample plate and low control plate). A third plate received 50 nL DMSO per cavity (high control plate). Sample plates and high control plates were further processed by dispensing 5 µL of 0.4 mg/mL PTP1B_CA in assay buffer into every well of the plates, while low control plates received 5 µL BGG (0.4 mg/mL) instead. After incubation, mixtures were transferred to assay-ready filter plates using automated liquid handling. Filter plates containing samples or low controls were then stacked onto 384-well PP plates containing 50 nL DMSO per cavity, while high control–containing filter plates were stacked onto plates containing 50 nL of 10× diluted (0.5 µg/µL or 1 mM) compound solutions in DMSO. Plates were then centrifuged, and eluates were subsequently transferred to quadrants of a 1536-well microtiter plate (samples → Q1, low controls → Q2, and high controls → Q3). Q4 received 3 µL of 10 mM NH4Ac buffer (pH 7.3) as negative control. Addition of standards and further plate processing were conducted as described above.
MALDI Target Preparation
4-HCCA was dissolved at 3 mg/mL in a 1:1 mixture of acetonitrile and 5 mM NH4H2PO4/0.1% TFA (v/v) with the aid of vigorous vortexing. The CyBio Well vario liquid-handling system (Analytik Jena) equipped with ceramic tips and operated in 1536-well format was used to conduct dried droplet spotting, providing highly homogeneous spot shapes. Here, assay plates were centrifuged at 1000 rpm for 60 s, and the seals were removed before 100 nL matrix solution and 100 nL sample were aspirated successively from the matrix reservoir and the assay plate, respectively, and dispensed together onto the plain steel MALDI target plates. The target plates were dried in a vacuum chamber before the spotting process was repeated. Between the first and second spotting steps and after the second spotting step, the ceramic tips were washed three times with isopropanol/0.1% aqueous TFA (70/30, v/v). Following successful transfer and drying of matrix–analyte mixtures, an on-target washing procedure was applied using 10 mM NH4H2PO4/0.3% TFA. Processed MALDI target plates were stored until analyzed. After every preparation cycle, three repetitive washing cycles using 0.1 M NH4OH followed by three cycles of 70% isopropanol and 30% water/0.1% TFA (v/v) were carried out on the liquid-handling system to prevent carryover and clogging of the tips.
MALDI-TOF-Based HTS Readout
Mass spectra were acquired with a rapifleX MALDI-TOF/TOF instrument from Bruker Daltonics, including a Smartbeam 3D laser. FlexControl (v. 4.0), flexAnalysis (v. 4.0), and rapifleX MALDI PharmaPulse (MPP; v. 2.2), all from Bruker Daltonics, were used for MS acquisition and data analysis. Target plates were loaded onto an Orbitor RS (Thermo Fisher Scientific) robotic system controlled by the laboratory automation software Momentum (v. 4.2.3, Thermo Fisher Scientific) and automatically inserted into the MALDI-TOF device. Mass spectra were acquired in the mass range of m/z 200–1100 to cover target analyte, internal standard, and controls of the binding assay. Therefore, 2500 laser shots per sample spot were accumulated in 20-shot increments across a small spiral spot raster in positive or negative ionization mode using a 160 ns pulsed ion extraction with a 10 kHz laser frequency, a digitizer setting of 5.00 GS/s, and an M5 defocus Smartbeam parameter at a 50×50 µm scan range, which resulted in a 124×124 µm field size. The laser power was adjusted manually before every start of a batch process to reach a sufficient signal intensity for the internal standard (~2×105 cts in positive ionization mode; ~5×104 cts in negative ionization mode). The acquired spectra were processed in MPP2.2 Synthesis Screening mode or using flexAnalysis with a centroid peak detection set to a signal-to-noise ratio (S/N) ≥ 15 and a peak width of m/z 0.05.
Data Analysis
For data evaluation, signal area values [area under the curve (AUC)] extracted from flexAnalysis result files were referenced against corresponding standard signals for small molecules [internal standard (IS)] and quality controls (QCref), and reported as ratio values. When spectra were processed by MPP2.2 Synthesis Screening mode, ratio values for detected compound signals and QC signals were extracted straight from result files. In all cases, signals measured with an m/z value deviating more than 0.032 from the calculated theoretical value were rejected and not used for further calculations. The ratio of QC peptide signal and QC standard signal had to stay lower than the threshold of 1.0 (representative for 1% SEC leakage) for measured samples to be accountable for further analysis. For signals within the mass deviation threshold, the proportional increase of the signal ratio in sample wells compared to the mean of negative control–containing wells was calculated and expressed as Fold Change (FC) values, according to the following equation:
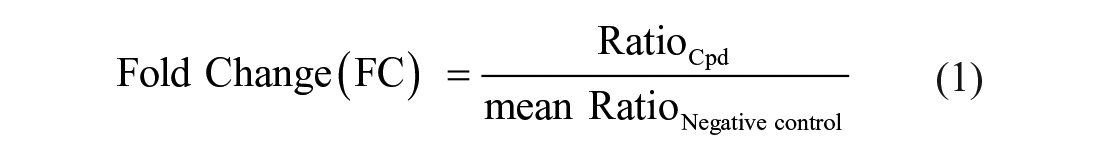
Small-molecule analyte signals reaching FC values of ≥2.5 were considered as hits in the screening workflow or in compound detectability analysis. In cases in which signals at compound m/z were observable in sample measurements but not in negative controls, the FC value calculation was omitted, and the compound was counted as an assay hit. Measurements failing the QC threshold or not containing any measurable IS were reported together with compound information and aggregated in a dropout list to enable retesting in a separate assay run.
Data from hit confirmation workflow experiments were analyzed in a similar fashion as described for screening workflow data. Ratio values were calculated for compound and QC signals from Exp. A (sample), Exp. B (low control), and Exp. C (high control) measurements. In addition, a FC value was calculated from high control compound signals and the mean of observable negative control ratio values. Relative binding affinities (RBAs) for specific compound–protein interactions were calculated via the following equation:
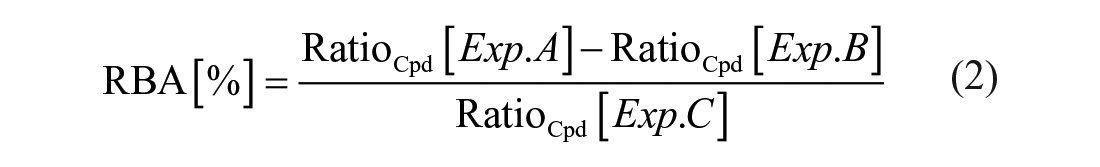
To qualify as a hit in the hit confirmation workflow, test compound data had to fulfill the following criteria: m/z deviation ≤ 0.032, internal standard detected in all three experiments (A–C), QC passed (QC ratio ≤ 1.0) in all three experiments, FC ≥ 2.5, and RBA ≥ 5%. Data were further processed using GraphPad Prism (v. 8.30; GraphPad Software, La Jolla, CA). A more detailed description on data handling and processing is provided online in the supporting information. The assignment of compounds to the corresponding measurements was achieved by software-aided deconvolution of every 1536-well assay plate to the corresponding 384-well substance plates in the course of data analysis, as we have described in Ref. 21 .
Further Methods
A summary table compiling information on components and controls of the presented MALDI-TOF AS-MS concept targeting PTP1B (
Results and Discussion
MALDI-TOF AS-MS Workflows
SEC-based separation performed directly in microtiter plates is a straightforward and effective approach that is amenable to automation on current screening platforms. To enable screening of small-molecule libraries for noncovalent binding to a protein target, two workflows were designed based on an in-plate SEC-enabled separation procedure in 384-well format. A schematic depiction of both workflows is provided in Figure 1 . In the first workflow (Screening workflow), individual incubations of protein and test compounds are transferred to SEC resin-filled 384-well filter plates, and the components are separated according to the size exclusion cutoff on centrifugation. Compounds interacting with the target pass the SEC resin and can be detected in subsequent analysis. To control for efficient cutoff separation, a quality control (cutoff QC) is added to the assay buffer at a fixed concentration, which allows for quantification of SEC leakage. To account for intrinsic MALDI-TOF signal variabilities, two reference standards are added to every sample after elution from the filter plate. While the IS is referenced against compound signals (Ratiosample), the quality control standard (QCref) defines the threshold for acceptable SEC leakage and is used to precisely quantify the amount of QC in eluted sample fractions. Compounds detected at levels higher than certain thresholds in sample wells compared to DMSO control wells are considered a screening workflow hit. Because this first workflow provides only a qualitative measure of protein binding and does not account for unspecific interactions, a second assay (Hit confirmation workflow) including controls for every test compound was implemented. Here, in addition to the regular sample incubation of test compound and protein target, a low control that detects unspecific binding and a high control that resembles the maximum achievable compound concentration after elution are included. While for low controls the protein of interest is substituted for an unrelated protein or an unfavored protein isotype, the high control is performed with the target but in the absence of test compound. After elution from the filter plate, dilutions of test compounds are spiked into high controls at concentrations that mimic full 1:1 target saturation. After analysis, controls are used to calculate the specific RBA, which is a quantitative measure of specific protein binding. Combining the two presented workflows conceptually enables screening of small-molecule compound libraries for qualitative target binding followed by stringent hit verification and relative quantification of ligand affinity of primary screening hits using a label-free MALDI-TOF MS readout. Following workflow design, we evaluated critical method parameters for the SEC-based in-plate separation process and for MALDI-TOF MS-enabled small-molecule detection.
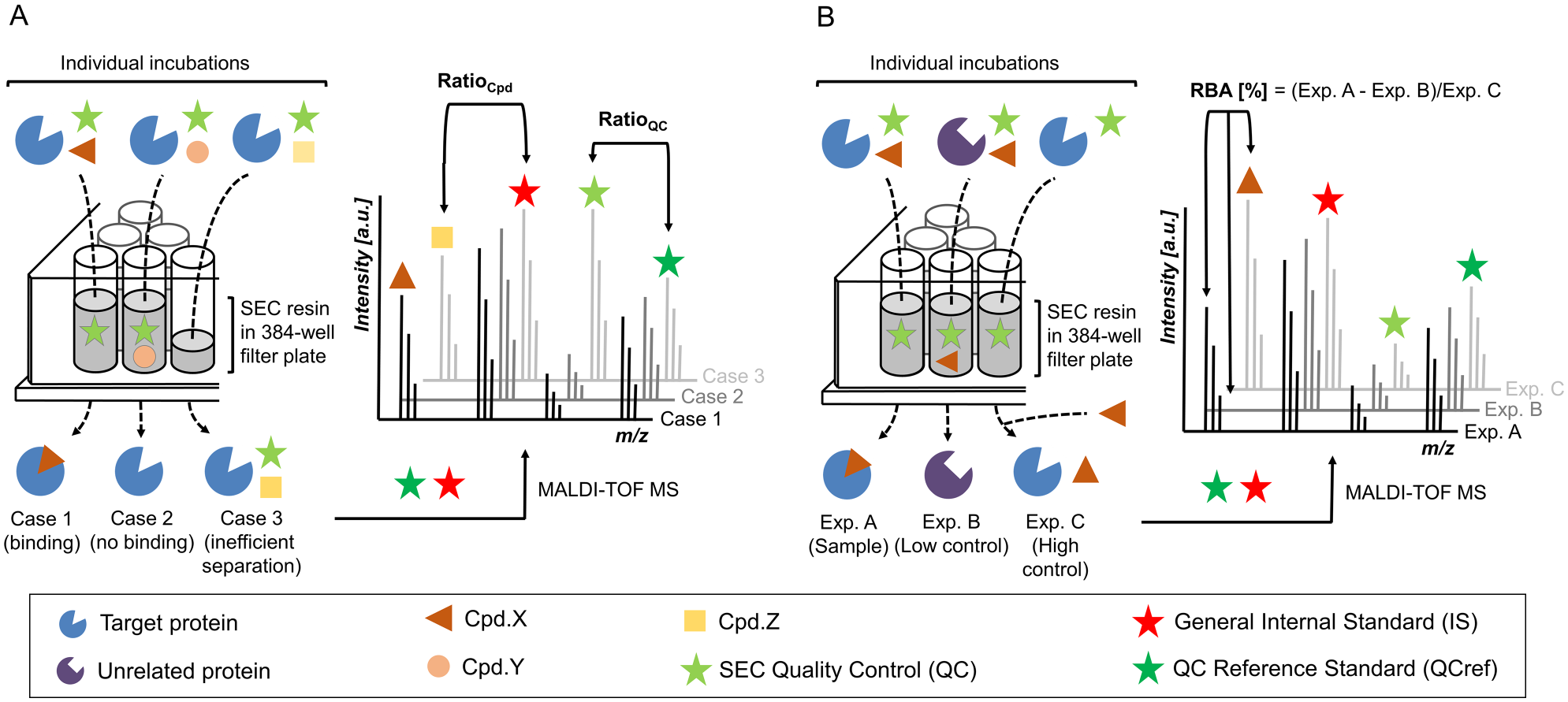
Scheme of matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) affinity selection mass spectrometry (AS-MS) assay workflows. (
SEC-Based In-Plate Separation Process
Efficient retention of unbound ligand and buffer components is a prerequisite for robust data generation by the two presented workflows. Optimization of SEC separation efficiency was carried out with buffer containing fluorescein as a tracer. A bed volume of 65 µL SEC resin per well caused >99.5% retention of the fluorescent tracer and was used for further experiments (
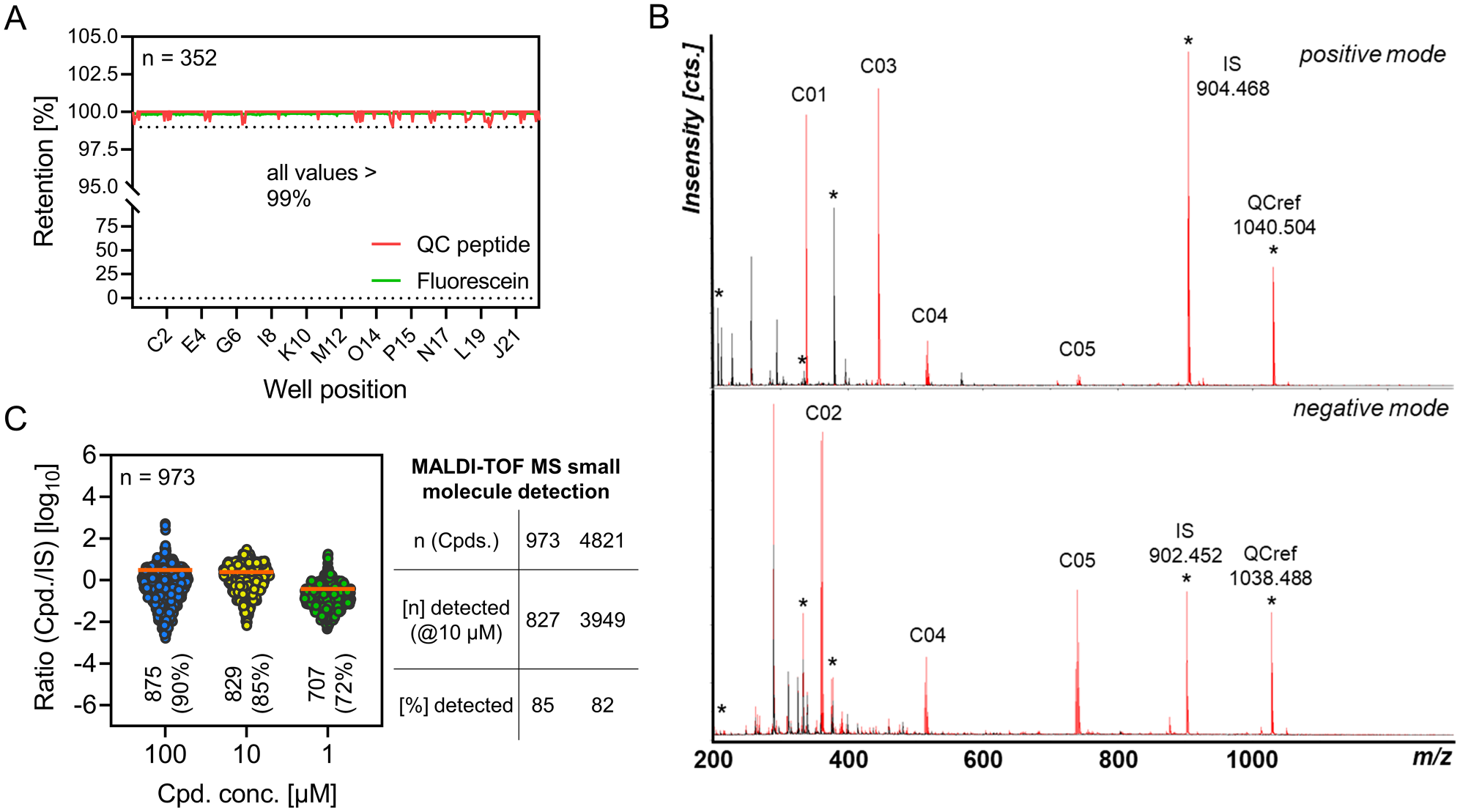
Evaluation of in-plate size exclusion chromatography (SEC) and matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) mass spectrometry (MS) small-molecule detection. (
Automation of the in-plate SEC procedure was a prerequisite for handling larger compound numbers and enabling HTS-capable sample processing. We therefore transferred the screening workflow to our in-house HTS system comprising components of regular automation platforms for process steps that include liquid handling, sample mixing, incubation, centrifugation, robotic plate handling, plate storage, and plate sealing. A flow chart of the automated process is provided in the supporting information (
Optimization of MALDI Matrix Composition and Deposition for Small-Molecule Detection
Interferences in the low mass region arising from background signals caused by the matrix and chemotype-dependent low ionization efficiency have been major issues in MALDI-TOF MS analysis of small molecules. A multitude of approaches to minimize background interference while maximizing compound ionization have been reported, suggesting a variety of matrix constituents, additives, solid supports, and preparation procedures.22–26 Many of these improvements, however, are valid only for specific analyte chemotypes, require special treatment of MALDI target plates, or interfere with automation processes. Therefore, a set of 10 different MALDI matrices, which have proven to be compatible with the applied automated spotting procedure, were evaluated for suitability. Co-crystallization with α-cyano-4-hydroxycinnamic acid (HCCA) produced acceptable S/N values for each of eight chemically diverse small molecules and the QC peptide (1 µM in demineralized H2O) in positive and/or negative ionization mode (
ESI-based liquid chromatography (LC)/MS methods operating in positive ionization mode have been reported to be capable of detecting >85% of library compounds under screening conditions.
11
To achieve a better estimate of the analytical coverage accessible by MALDI-TOF MS, a chemically diverse selection of 973 compounds was tested for detectability at three different concentrations ranging from 1 to 100 µM (
Comparison of MALDI-TOF AS-MS to Alternative Assay Formats
We have previously demonstrated that MALDI-TOF MS is a versatile drug discovery readout for targeting the enzymatic activity of human protein tyrosine phosphatase PTP1B.
27
To investigate whether the technology can be used as a readout strategy for in vitro target engagement analysis, we selected PTP1B as a model system to set up a MALDI-TOF MS-enabled binding assay, according to the described workflows. Here, an active site mutant of PTP1B (PTP1B_CA) was used as the target of interest, and BGG was chosen as an unrelated protein for low control experiments. Two previously published PTP1B inhibitors were used as reference compounds (Ref_Cpd.), and their binding toward PTP1B_CA was confirmed by ITC and NMR experiments (
First, the elution profile of a 384-well filter plate was recorded for 5 µL fractions after applying 100 µM Ref_Cpd. 1 alone or in mixture with 10 µM PTP1B_CA (
To assess data quality and to compare assay hits identified by MALDI-TOF AS-MS to orthogonal HTS-capable binding assay formats, the diversity set of 973 small molecules (971 compounds + 2 reference compounds) was subjected to the testing scheme outlined in Figure 3A . All assays were performed in biological duplicates on two individual days to ensure concise and representative data.
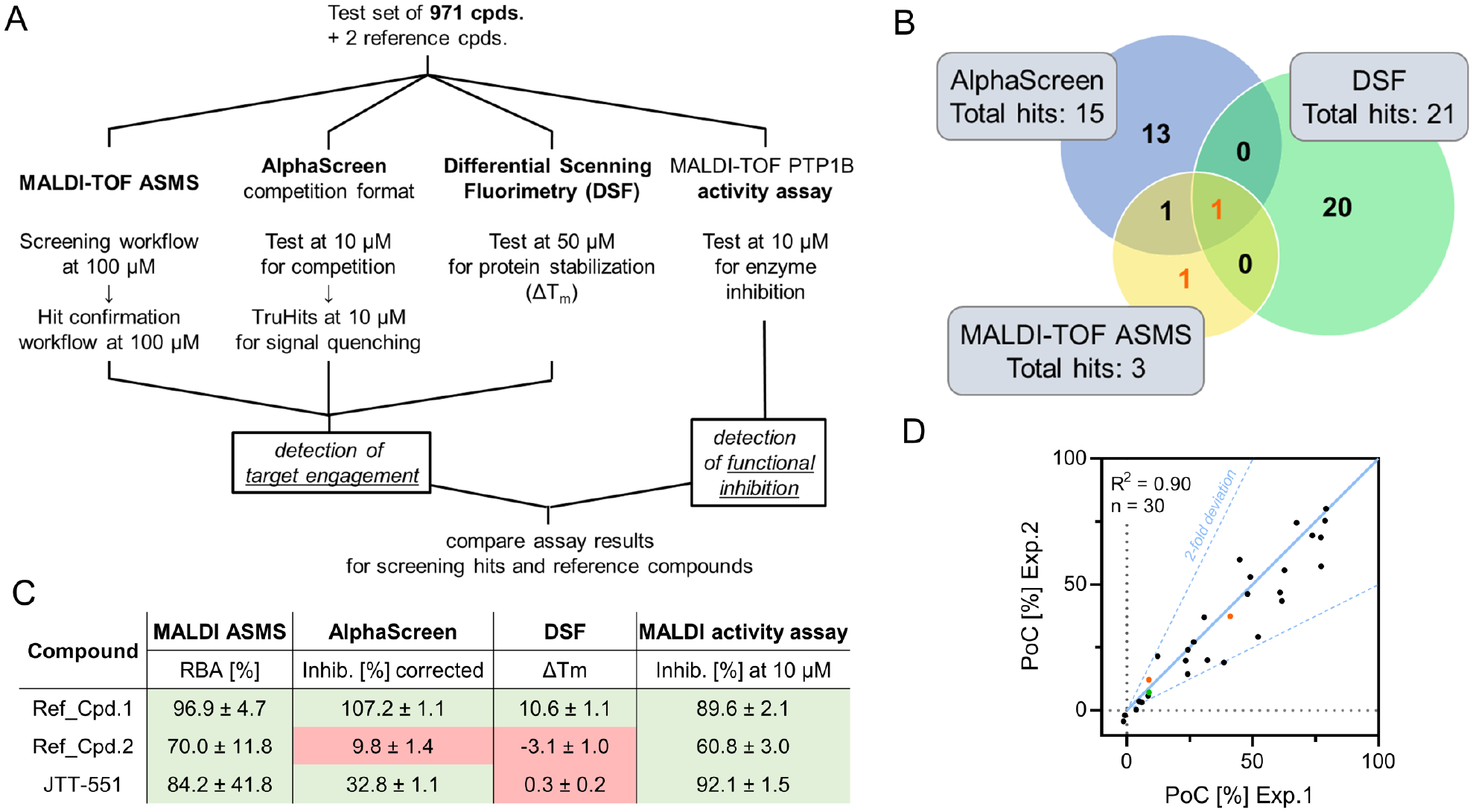
Matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) affinity selection mass spectrometry (AS-MS) comparison to alternative assay formats using protein tyrosine phosphatase 1B (PTP1B) as a model system. (
For MALDI-TOF AS-MS, a total of 19 primary hits (1.95% hit rate) were identified, and ratio values showed acceptable correlation for individual experiments (
To compare results to a displacement assay format, an AlphaScreen technology-based assay was used, using a biotinylated tracer based on the structure of active-site ligand Ref_Cpd. 1. Compounds were also tested in a TruHits format that allowed for quantification of compound-induced signal quenching by using a biotinylated His-tag as a tracer–protein complex surrogate. AlphaScreen competition data were corrected for average signal quenching, and a hit threshold of 80% of controls [percentage of controls (PoC)] was defined. With this approach, 15 compounds were identified as hits (1.54% hit rate), among which JTT-551 and the orthosteric binder Ref_Cpd. 1 but not the allosteric binder Ref_Cpd. 2 were found (
As a second orthogonal binding assay, DSF was performed in detergent-free buffer using the hydrophobic SYPRO Orange dye. Test substances were tested for their effect on thermally induced protein denaturation, indicated by a shift of the protein melting temperature (ΔTm). Screening the 973-compound library with a hit threshold of ΔTm > 0.5 °C produced 21 hits (2.15% hit rate;
To test whether protein binding also translates into functional inhibition, we performed a MALDI-TOF-based PTP1B activity assay for the compound test set. Substances were tested for inhibition of PTP1B-mediated tyrosine dephosphorylation. From the set of 973 compounds, 30 hits impeded catalytic activity by more than 20% (3.08% hit rate), including the two reference inhibitors (
Figure 3B
shows a comparison of the hit sets generated by MALDI-TOF AS-MS, AlphaScreen, and DSF. Little agreement was found for hits identified by the three different technologies, with Ref_Cpd. 1 as the only mutual hit. Notably, allosteric protein binding by the known ligand Ref_Cpd. 2 was detectable solely by MALDI-TOF AS-MS, but not by alternative binding assay formats. The published inhibitor JTT-551 showed sufficient signal inhibition in the AlphaScreen assay and the functional readout, but did not promote any ΔTm shift in DSF analysis. A possible reason could be insufficient compound solubility, because DSF analysis prohibits the use of detergents in buffer solutions. Together with the results from the functional assay, summarized in
Figure 3C
, the compiled data illustrate the capability of MALDI-TOF AS-MS to identify functional PTP1B ligands originating from different modes of action, which further underscores the competitive advantages of the presented platform. Comparing the distribution of MALDI-TOF AS-MS hits to the hit set of the functional enzyme activity assay showed reproducible inhibition in line with reported values (Ref_Cpd. 1 IC50 = 0.12 µM;
28
Ref_Cpd. 2 IC50 = 8 µM;
29
JTT-551 Ki = 0.22 µM;
31
Because the MALDI-TOF AS-MS platform can conceptually be adjusted to any target with a size greater than the SEC cutoff, we assessed the versatility of the assay by simply exchanging the protein of interest for either cyclic guanosine monophosphate–adenosine monophosphate synthase (cGAS), carbonic anhydrase II, human serum albumin, or T-cell PTP1B isotype (TcPTP1B). Determining RBA values for corresponding reference compounds demonstrated apparent protein binding and allowed comparison with literature values (
HTS Validation Study
To evaluate the performance of the assay under HTS conditions and in a fully automated environment, we performed a 24-h screening run on our in-house automation platform with subsequent MALDI target spotting and MALDI-TOF MS analysis.
A total of 23,444 compounds were tested in the automated screening workflow at 100 µM for binding to 10 µM PTP1B_CA, including reference compounds.
Figure 4A
shows the distribution of ratio values for positive controls (Ref_Cpd. 1) on every 1536-well plate. Signals scattered around an average ratio value of 0.96 with a CV of 23.0%. This signal variation was slightly increased compared to our findings from plate uniformity analysis, which could be attributable to the limited number of controls per plate and the combination of four individual assay plates into 1536-well format. Two positive controls (<0.19%) were not detected during MALDI-TOF MS analysis due to flawed sample spotting. The predefined QC threshold of <1% SEC leakage failed in one instance (<0.01%), and the corresponding spectrum was excluded from analysis. Overall, robust and stable detection of qualitative protein binding was demonstrated throughout the campaign. 240 primary hits (1.02% hit rate) were identified from the compound library and subjected to the hit confirmation workflow. Here, primary hits were tested in duplicates, and the amount of PTP_CA binding was quantified against high controls (eluates spiked with 7 µM compound) and low controls (BGG instead of PTP_CA). From the pool of 240 hits, 229 were rejected because of interfering low control signals. Two compounds did not reproduce their assay response among duplicates and were therefore also excluded from the hit pool. From the remaining nine substances, four had relative binding affinities lower than 5% and were not considered significant. This left us with five confirmed hits (0.021% final hit rate) exhibiting conclusive and reproducible target engagement (
Fig. 4B
). Comparing determined RBA values for two independent experiments further demonstrated good correlation (R2 = 0.97) of assay results (
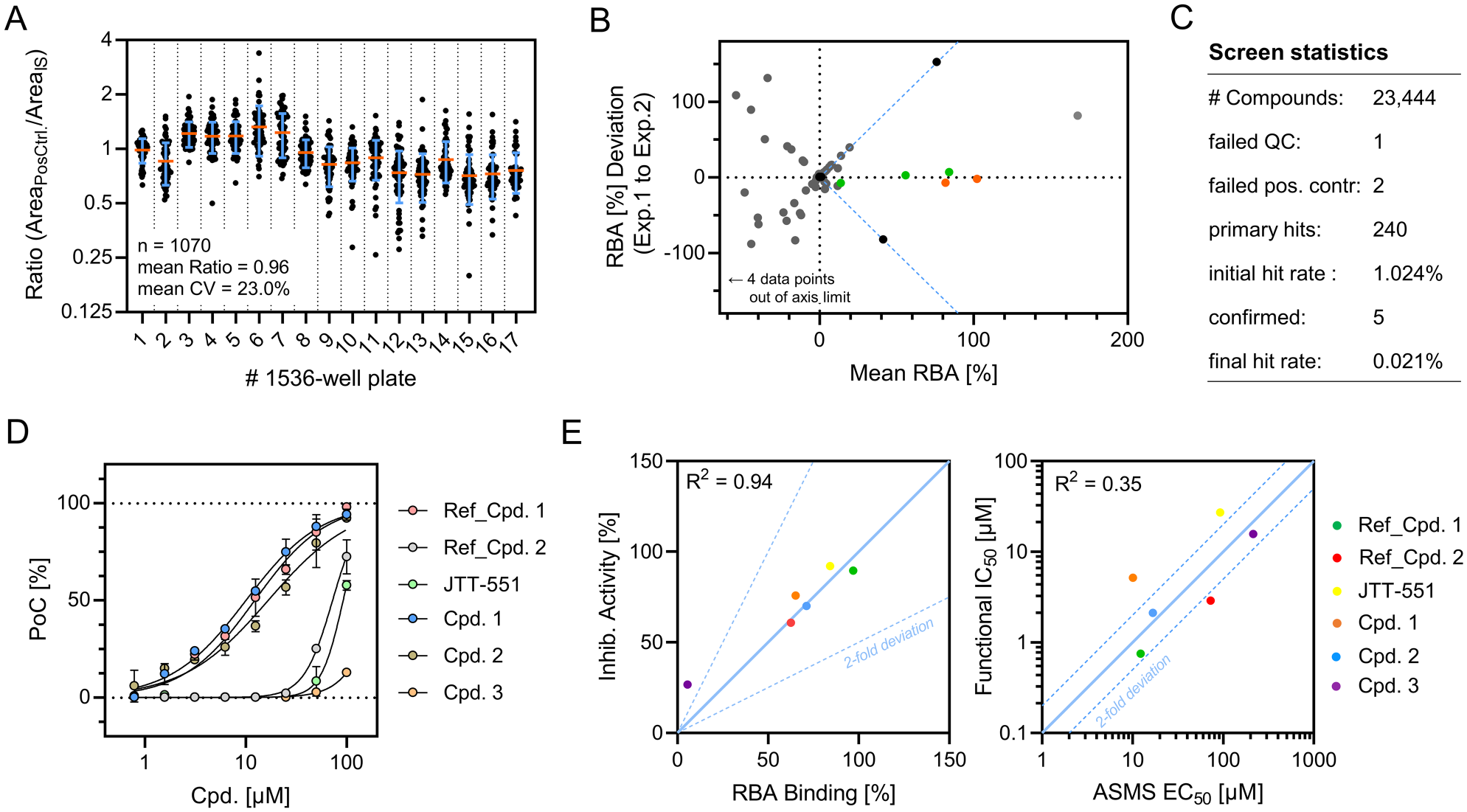
Results from the matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) affinity selection mass spectrometry (AS-MS) validation project. (
In summary, the validation study demonstrated automated screening of more than 23,000 compounds for PTP1B target engagement per day by MALDI-TOF AS-MS. Stable and robust separation of assay components was achieved by automated in-plate SEC on a regular unmodified automation platform, as indicated by monitoring QC controls and reference compounds. The so-identified PTP1B ligands exhibited reproducible target binding, determined by RBA values, which correlated with functional inhibition of PTP1B enzymatic activity. Screening single-compound incubation mixtures allowed for the convenient integration of the platform into our regular HTS environment. Together, we provide evidence that the presented assay platform is an HTS-compatible tool, built exclusively from commercially available components, for providing high-quality data on small-molecule target engagement. MALDI-TOF AS-MS is easy to adapt, as demonstrated for a number of diverse proteins, and does not require any knowledge about target function or endogenous ligands, which makes it especially suited for addressing targets for which no detection reagents are available or multiple binding epitopes could be captured. We aim to use the entire concept mainly for filtering large compound sets for their interaction with a selected target protein. The presented data suggest that ligand ranking by RBA might be possible, but detailed characterization by biophysical reference methods (e.g., ITC, SPR, and NMR spectroscopy) should follow the AS-MS concept for accurate ranking of binding affinities. As demonstrated, the hit rate of the AS-MS concept is quite low, so the biophysical methods can cope with the resulting ligand numbers.
Challenges of the MALDI-TOF AS-MS platform are the potential occurrence of false-negative assay results due to the incompatibility of ~15% of analytes with MALDI and micromolar analyte concentrations required for stable hit detection, consequently resulting in a high protein demand of the assay. While the use of focused compound libraries with proven MALDI amenability would simply exclude potential false negatives, possible enhancements of detection rates and method sensitivity could be achieved by the further development of matrices and additives. In addition, the commercialization of matrix-independent MALDI target plates, which omit the occurrence of interfering background signals, holds great potential for improving small-molecule detection by MALDI-TOF MS.
37
Furthermore, the detectability of binding ligands is dependent on the koff constant of the respective protein–ligand complex, which requires fast separation of assay components to minimize this bias. By demonstrating the transferability of the presented concept to other protein targets, we were able to show specific binding of known ligands with binding affinities in the low-double-digit micromolar range (
At the moment, MALDI-TOF AS-MS is a fast methodology to screen in high-throughput fashion for protein binders, whereas the suitability of ESI-based high-resolution MS coupled with emerging HTS-capable sampling technology as a readout strategy will be the subject of future investigations.38–40 Higher method sensitivity would likely result in a concurrent reduction of protein demand, which could be further decreased by assay miniaturization beyond the 384-well format. For projects in which protein supply is limited, screening with less protein-intensive technologies followed by MALDI-TOF AS-MS-enabled hit confirmation is an attractive option for ensuring the identification of high-quality hits with proven target engagement, which is particularly applicable when addressing enzymatic targets.
Conclusively, MALDI-TOF AS-MS is a versatile expansion of our existing MALDI-TOF MS screening platform and will be helpful in upcoming drug discovery campaigns by providing the capability to perform automated single-incubation HTS for in vitro target engagement to native targets in a label-free fashion and embedded in regular HTS infrastructure.
Supplemental Material
Supplemental_Material_MALDI_TOF_ASMS_Simon_et_al – Supplemental material for MALDI-TOF-Based Affinity Selection Mass Spectrometry for Automated Screening of Protein–Ligand Interactions at High Throughput
Supplemental material, Supplemental_Material_MALDI_TOF_ASMS_Simon_et_al for MALDI-TOF-Based Affinity Selection Mass Spectrometry for Automated Screening of Protein–Ligand Interactions at High Throughput by Roman P. Simon, Martin Winter, Carola Kleiner, Lucie Wehrle, Michael Karnath, Robert Ries, Markus Zeeb, Gisela Schnapp, Dennis Fiegen, Tim T. Häbe, Frank Runge, Tom Bretschneider, Andreas H. Luippold, Daniel Bischoff, Wolfgang Reindl and Frank H. Büttner in SLAS Discovery
Footnotes
Acknowledgements
We thank Meike Hamester and Arndt Asperger from Bruker Daltonics (Bremen, Germany) for their valuable input and helpful discussion. Furthermore, we thank Margit Bauer for the preparation of the PTP1B catalytic domain and Christofer Tautermann for his valuable support.
Declaration of Conflicting Interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: All authors are employees of Boehringer Ingelheim Pharma GmbH & Co. KG and completed this work within the scope of their employment.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: All work was completed as work-for-hire for the employer Boehringer Ingelheim Pharma GmbH & Co. KG.
Supplemental material is available online with this article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.