
Abstract
Label-free, mass spectrometric (MS) detection is an emerging technology in the field of drug discovery. Unbiased deciphering of enzymatic reactions is a proficient advantage over conventional label-based readouts suffering from compound interference and intricate generation of tailored signal mediators. Significant evolvements of matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) MS, as well as associated liquid handling instrumentation, triggered extensive efforts in the drug discovery community to integrate the comprehensive MS readout into the high-throughput screening (HTS) portfolio. Providing speed, sensitivity, and accuracy comparable to those of conventional, label-based readouts, combined with merits of MS-based technologies, such as label-free parallelized measurement of multiple physiological components, emphasizes the advantages of MALDI-TOF for HTS approaches. Here we describe the assay development for the identification of protein tyrosine phosphatase 1B (PTP1B) inhibitors. In the context of this precious drug target, MALDI-TOF was integrated into the HTS environment and cross-compared with the well-established AlphaScreen technology. We demonstrate robust and accurate IC50 determination with high accordance to data generated by AlphaScreen. Additionally, a tailored MALDI-TOF assay was developed to monitor compound-dependent, irreversible modification of the active cysteine of PTP1B. Overall, the presented data proves the promising perspective for the integration of MALDI-TOF into drug discovery campaigns.
Introduction
Lead discovery for drug targets is a key step within the process of drug development, which is predominantly achieved by high-throughput screening (HTS). A prerequisite of these technologies is the ability to target up to hundreds of thousands of compounds per day. Conventional HTS screening approaches are mainly driven by fluorescence- and chemiluminescence-based readouts. A comprehensive review of different label-based assay readouts has been published previously by Inglese et al. 1 These provide common features, including sensitive, reproducible, and fast analysis, combined with largely universal applicability. In turn, they are susceptible to compound-dependent screening artifacts leading to false positives or negatives 2 and require demanding assay development, including the generation of artificial, nonphysiological probes and/or specific antibodies. 1 Especially HTS development toward hitherto intractable drug targets requires alternative concepts to the conventional label-based technologies. 3 This is expressed by the growing landscape of label-free, mass spectrometry (MS)–based screening approaches in the field of drug discovery research.4–6 These offer the possibility to simultaneously track several molecules, without preliminary labeling strategies, enabling quantitative analysis of physiologically relevant substrates and/or products of an enzymatic reaction. Comprehensive deciphering of a biological system offers the generation of additional information that was formerly inaccessible for HTS readouts and even provides the opportunity to track multiple enzymatic reactions within one assay. 7
So far, MS-based readouts in drug discovery were largely dominated by instruments comprising online solid-phase extraction (SPE) and electrospray ionization (ESI) MS, such as the RapidFire (RF-MS) system of Agilent (Santa Clara, CA). This hyphenated MS technology enables cycle times of less than 10 s, which is considerably faster than conventional high-performance liquid chromatography (HPLC)–MS instrumentation. 8 However, for HTS and ultra-HTS applications the RF-MS cycle time is still too slow in comparison with fluorescence-based readouts (<1 s/sample). For this reason, RF-MS is primarily applied in compound profiling, 9 lead optimization campaigns, 6 and ADME assays. 10
An emerging alternative is provided by recent advancements in matrix-assisted laser desorption/ionization (MALDI) offering speed and robustness of conventional label-based technologies, accompanied by the advantages of an MS-based readout (e.g., label-free, interference-resistant, comprehensive assay assessment). The rapifleX MALDI PharmaPulse, recently launched by Bruker Daltonics (Bremen, Germany), offers HTS suitability by its ultrafast laser repetition rate (10 kHz) and direct compatibility with common automatization equipment. This evolvement pushes the MS-based readout closer to full-diversity HTS campaigns and has raised efforts to integrate MALDI time-of-flight (TOF) as an in-line reader into the HTS environment.11–15 Major challenges are the adjustment of assay buffer composition to match MS compatibility while enzymatic activity remains unaffected, 11 as well as tailored integration into the automated liquid handling process with a focus on the challenging assay transfer onto the MALDI target plate. Various concepts exist for the MALDI-TOF integration into an HTS environment, such as the acoustic liquid handler Echo 550 (Labcyte, Sunnyvale, CA) 12 and the Mosquito (TTP Labtech, Hertfordshire, UK) nanoliter dispenser.13,14 We selected the multichannel pipettor CyBio Well vario (Analytik Jena, Jena, Germany) and demonstrate its applicability for HTS screening. Especially the capability to simultaneously transfer matrix and samples in 384- and 1536-well format, as well as the precious possibility to perform on-target washing procedures, emphasizes its suitability for automated MALDI target plate preparation.
As a proof of concept, we selected the protein tyrosine phosphatase 1B (PTP1B) as a model enzyme to demonstrate possibilities and challenges to dissect enzymatic reactions by MALDI-TOF and its sophisticated utilization for HTS campaigns. Specific inhibition of PTP1B activity is of major interest in cardiometabolic research due to its regulatory role in insulin and leptin signaling.16,17 Increased resistance to weight gain, as well as elevated insulin sensitivity, was observed in PTP1B–/– mice. 18 Additionally, PTP1B inhibition was shown to reduce plasma glucose level as well as body weight gain, emphasizing its therapeutic potential to treat patients with diet-induced obesity and type 2 diabetes mellitus. 19 Although PTP1B is a validated therapeutic target, 20 identification of a potent small-molecule inhibitor is hampered by adverse chemical properties of the active site. 21 Particularly, the highly conserved sequence surrounding the active site (e.g., 72% sequence identity of the catalytic domain to T-cell protein tyrosine phosphatase [TCPTP] 22 ) hinders the development of selective and efficient PTP1B inhibitors. A new concept is the identification of allosteric inhibitors that target unique sites on PTP1B distinct from its active site. 21 An AlphaScreen assay was previously established in this context and was applied for the primary screen. MALDI-TOF offers an orthogonal readout to confirm structural classes determined in the primary screen, combined with the demonstration of feasibility and performance characteristics of the MS-based readout. We adapted the assay conditions to provide MALDI-TOF compatibility while preserving pharmacologically relevant assay characteristics.
An additional aspect in the hit-finding campaign is compound-dependent irreversible modification of PTP1B at the catalytic cysteine (Cys215) constituting an unwanted mode of action (e.g., compound-induced sulfonic acid generation 23 ). The exclusion of compounds inducing these modifications is elusive for conventional label-based screening technologies. On the contrary, we established a MALDI-TOF-based assay that is capable of providing this information in an HTS-compatible format, emphasizing its capabilities to support drug discovery with more than the basic HTS readout. This introduces a new dimension for the design of drug discovery campaigns.
Overall, we demonstrate the feasibility of MALDI-TOF integration into the HTS environment. Compound profiling for the phosphatase PTP1B was used as a model system to match the luminescence-based AlphaScreen with the MS-based MALDI-TOF assay. We demonstrate high correlation between the two assay technologies and provide convincing data to use the CyBio Well vario liquid handling system for MALDI target plate preparation. Further, we prove the feasibility of parallelized on-target washing, which improves the detection capabilities of target molecules in complex assay buffers and thus facilitates assay development. Finally, we demonstrate the versatile applicability of MALDI-TOF in drug discovery per se by the screening of compound-dependent modifications of the catalytic center of PTP1B.
Materials and Methods
Materials
α-Cyano-4-hydroxycinnamic acid (4-HCCA; no. 8201344), HTS MALDI disposable target plates (no. 1847006), and an HTS MALDI Adapter (no. 8283496) were purchased from Bruker Daltonics. The catalytic domain of PTP1B (PTP-1B_hA3_2-321_N_His8TEV) was produced in-house at Boehringer Ingelheim. Insulin receptor (INSR) substrate (ETDpYYRKG-amidated) and product peptide (ETDYYRKG-amidated), as well as the labeled AlphaScreen peptide (Biotin-Ahx-ETDpYYRKG-amidated), were purchased from Biosyntan (Berlin, Germany). Internal standard peptide ETDYY
PTP1B AlphaScreen Assay
For label-based detection of PTP1B inhibition, we applied the AlphaScreen technology24,25 composed of streptavidin donor beads and protein A acceptor beads. The assay was set up as a competition between a biotinylated substrate peptide (biotin-Ahx-ETDpYYRKG-amidated) and its unbiotinylated counterpart (ETDpYYRKG-amidated) for the antibody binding (anti-phosphotyrosine antibody 4G10). Within the assay, PTP1B dephosphorylates the unbiotinylated substrate, preventing its recognition by the antibody and thus increasing the AlphaScreen signal.
Enzyme PTP1B and substrate solutions were prepared in 50 mM HEPES buffer (pH 7.4) supplemented with 100 mM NaCl, 2 mM DTT, 0.001% Tween 20, and 0.001% BSA. Orthovandate, as well as donor and acceptor bead mixtures, was prepared in 50 mM HEPES buffer (pH 7.4) supplemented with 100 mM NaCl, 0.05% Tween 20, and 0.1% BSA.
To preserve the AlphaScreen beads, all experiments were conducted under green light conditions at room temperature. Ascending compound concentrations (final screening concentration between 0.01 and 100 µM) in 100 nL DMSO were added to 384-well microplates (no. 781075, Greiner, Friggenhausen, Germany) using a CyBio Well vario (Analytik Jena) liquid handling unit equipped with a capillary head. Next, compounds were incubated with 400 pM PTP1B (5 µL/well) for 10 min at 24 °C prior to the addition of 1.4 µM substrate peptide (5 µL/well; ETDpYYRKG-amidated). The enzyme reaction was executed for 2.5 h in a humidified incubator at 24 °C and finally quenched by the addition of 200 µM orthovanadate (5 µL/well). Each assay plate contained high (no compound) and low (no enzyme) controls to follow the enzyme reaction, as well as a substrate standard curve (range 2 nM–60 µM) to calculate the actual turnover of the phosphorylated peptide.
Five microliters of streptavidin donor beads (final: 5 mg/mL) mixed with biotinylated substrate peptide (biotin-Ahx-ETDpYYRKG-amidated; final: 2.5 nM) were added to the sample, followed by the addition of 5 µL of protein A acceptor beads (final: 5 mg/mL) mixed with anti-phosphotyrosine antibody 4G10 (final: 500 pM). This mixture was incubated for 2 h at 24 °C in a humidified incubator. Finally, the generated luminescence signal was read on an EnVision multilabel plate reader (PerkinElmer).
PTP1B MALDI-TOF Screening Assay
Dephosphorylation of the substrate peptide (ETDpYYRKG-amidated, m/z 1110.46) was monitored using MALDI-TOF. For this purpose, we determined the ratio of the product peptide (ETDYYRKG-amidated, m/z 1030.50) to the heavy labeled (
13
C6,
15
N4-arginine at peptide position 6) internal standard peptide (ETDYY
CatCysTide Oxidation Assay
Compound-dependent covalent modification of the catalytic center of PTP1B was tracked by MALDI-TOF analysis after tryptic digestion of the enzyme. The peptide surrounding the active site Cys215 (CatCysTide; 200ESGSLSPEHGPVVVHCSAGIGR221, m/z 2175.06) was analyzed together with the irreversibly oxidized form of this peptide (200ESGSLSPEHGPVVVH(3oxC)SAGIGR221, m/z 2223.05). For accurate judgment of peptide intensity, we evaluated the ratio to the isotope-labeled internal standard peptide (200ESGSLSPEHGPVVVHCSAGIG
PTP1B and trypsin solutions were prepared in buffer containing 50 mM HEPES buffer (pH 7.4) supplemented with 100 mM NaCl, 2 mM DTT, 0.001% Tween 20, and 0.001% BSA. Compounds were dissolved in DMSO and transferred in 100 nL fractions (final reaction concentration: 100 µM/compound) to 384-well microplates. PTP1B was added to a final concentration of 200 nM (10 µL/well) and incubated for 2.5 h at 24 °C in a humidified incubator.
In order to control this assay, three independent control experiments (n = 8) were added to each assay plate: (1) a positive control for the oxidation process being incubated with 200 µM H2O2 (without compounds), as well as (2) a high control (without compounds) and (3) a low control (without PTP1B and compounds). The combination of these controls enables the identification of enzyme-modifying compounds, as well as enzyme-oxidizing compounds.
For tryptic digest, the samples were mixed with 10 µL of trypsin solution (final concentration: 0.01 µg/µL) supplemented with the internal standard peptide (final concentration: 100 nM) and incubated for 4 h at 37 °C. Enzymatic digestion was quenched by the addition of 5 µL of TFA (final concentration of 0.1%), followed by brief vortexing.
MALDI Target Spotting
4-HCCA was diluted to a final concentration of 10 mg/mL in 30% acetonitrile and 70% water/0.05% TFA (v/v) prior to vigorous vortexing to provide a saturated matrix solution. Just before the spotting procedure, assay plates were briefly mixed for 30 s at 1.000 rpm. Spotting was conducted using the liquid handling system CyBio Well vario in either 384- or 1536-well format using ceramic tips. For highly reproducible and homogenous spot shapes, we applied double-layer spotting. 26 Saturated matrix solution (100 nL) was spotted onto plain steel MALDI target plates and dried in a desiccator containing phosphorus(V) oxide to aid drying. Subsequently, a 100 nL matrix and a 100 nL sample were aspirated successively and dispensed together onto the dried matrix spot. This sample–matrix mixture was again dried in a desiccator and stored covered until analysis. Between spotting steps, ceramic tips were washed three times with 70% isopropanol and 30% water/0.1% TFA (v/v) in order to prevent carryover. Selected samples were washed on target using the CyBio Well vario liquid handler, transferring 0.3 µL of 0.1% TFA (v/v)/10 mM ammonium dihydrogenphosphate onto each spot, with subsequent removal after 1 s of incubation. The preparation time for the semiautomatic spotting procedure was approximately 12 min per MALDI target plate. The CatCysTide oxidation assay was prediluted with matrix solution (4-HCCA in TA30) 1:10 to reduce ion suppression.
MALDI MS Data Acquisition
Mass spectra were acquired with a rapifleX MALDI TOF/TOF PharmaPulse instrument from Bruker Daltonics composed of a Smartbeam 3D laser. FlexControl (v4.0), flexAnalysis (v4.0), and MALDI PharmaPulse (v2.0) were used for MS acquisition and data analysis. Measurement of multiple target plates was facilitated by automatic handling of target plates using the robot system Orbitor RS (Thermo Scientific) controlled by the laboratory automation software Momentum (v4.2.3, Thermo Scientific). Mass spectra were acquired in the mass range of m/z 1000–1160 to cover substrate, product, and internal standard peptide of the compound profiling assay. A pulsed ion extraction of 120 ns was used, and the digitizer was set to 1.25 GS/s. The laser was operated at a 10 kHz frequency applying an M5 defocus Smartbeam parameter at a 25 × 25 µm scan range. The initial laser power was set to 65% and was automatically raised to 90% by the fuzzy control algorithm to achieve sufficient sensitivity for less abundant samples. The thresholds for this algorithm were a resolution of 5000 and a very low minimal signal intensity (requiring 2 counts per laser shot). To ensure fast analysis times, spots should be left after 10 subsequently failed judgments. At most, 10,000 shots were summed up in 1000 shot steps (movement: spiral small) controlled by the early termination algorithm to stop when an intensity threshold of 10,000 counts was exceeded (resulting in an average analysis time of 4 min per 384-well MALDI plate and 15 min per 1536-well plate, respectively).
The CatCysTide oxidation assay was analyzed with minor modifications. Mass spectra were acquired in the mass range of m/z 1000–4000. Laser power was set to 85%, and the fuzzy control algorithm and early termination were switched off. On the contrary, 20,000 shots were summed in 5000-shot steps to gain higher sensitivity (resulting in an average analysis time of 31 min per 384-well MALDI plate).
Prior to each campaign, external calibration was performed once with Peptide Calibration Standard II (Bruker Daltonics) containing seven peptides with corresponding [M+H]+ masses: angiotensin II = 1046.5418, angiotensin I = 1296.6848, substance P = 1347.7454, bombesin = 1619.8223, adrenocorticotropic hormone (ACTH) clips 1–17 = 2093.0962, ACTH clips 18–39 = 2465.1983, and somatostatin 28 = 3147.10. Additionally, internal calibration was performed using the monoisotopic peaks of substrate [M+H]+ = 1110.4616, product [M+H]+ = 1030.4952, and internal standard [M+H]+ = 1040.5035 for the PTP1B compound profiling, and using the peaks of CatCysTide [M+H]+ = 2175.0611 and internal standard [M+H]+ = 2185.0694 for the compound-dependent modification assay.
Data Analysis
MALDI-TOF data, processed with flexAnalysis (v4.0) or MALDI PharmaPulse (v2.0), as well as AlphaScreen data, generated on an EnVision reader, were exported as a comma delimited (.csv) file and further processed with either Microsoft Excel (Microsoft, Redmond, WA), GraphPad Prism (v7.03; GraphPad Software, La Jolla, CA), or in-house software MegaLab.
PTP1B activity was tracked by analyzing dephosphorylated product peptide and the corresponding heavy labeled internal standard peptide. Their ratio was calculated in order to diminish variations ascribed to the MS measurement according to the following equation:

The AlphaScreen signal was transformed into the substrate concentration with the substrate standard curve by fitting the data to a four-parameter logistical calibration curve implemented in MegaLab.
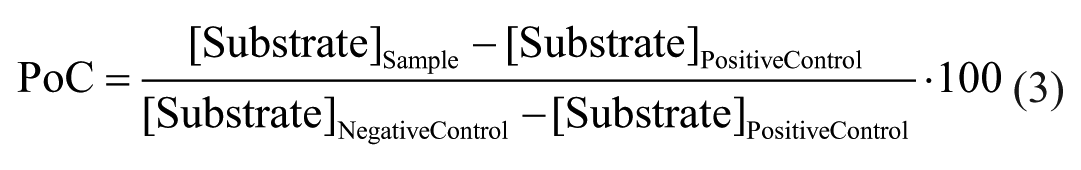
Both transformed AlphaScreen and MALDI-TOF data were normalized likewise and expressed as percentage of control (PoC) according to the following equations:
MALDI-TOF data:
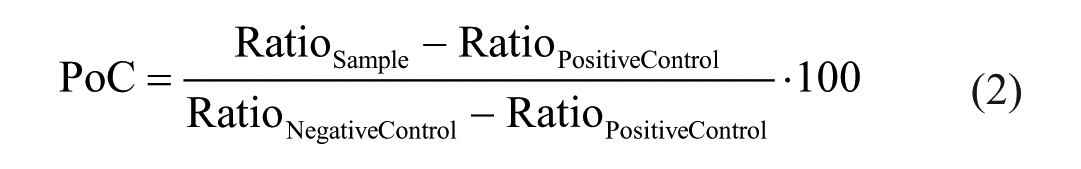
AlphaScreen:
IC50 calculation was performed in MegaLab by fitting the data to the following four-parameter logistical equation:
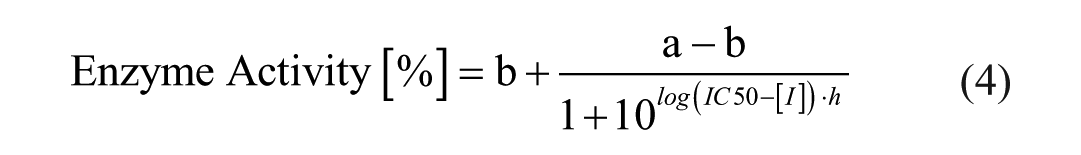
where a is the uninhibited value, b is the completely inhibited one, [I] is the inhibitor concentration, h is the Hill slope, and IC50 is the half maximal inhibitory concentration. GraphPad Prism was utilized for graphical representation of the data.
Basic appraisal of the assays was reviewed by the statistical quality parameter Z′ with the following equation:
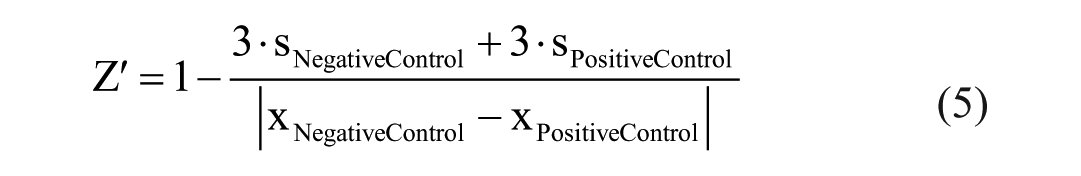
where s is standard deviation and x are the mean readouts of the AlphaScreen and MALDI-TOF assays, respectively.
Oxidation of the catalytic cysteine (Cys215) was evaluated by tracking the loss of CatCysTide after incubation with compounds by calculating the ratio of CatCysTide and oxidized CatCysTide to the corresponding heavy labeled internal standard peptide, respectively. Additionally, the efficacy of the tryptic digest was controlled by the ratio of PTP1B peptide14–24 and the internal standard peptide.

Results and Discussion
HTS requires robust and fast assay workflows with low variability and precise readouts. Recent developments introduced MALDI-TOF as a highly valuable tool in this research field. So far, MALDI-TOF’s applicability was hampered by speed and a lack of reproducible target analysis being overcome by the fast and precise rapifleX mass spectrometer from Bruker Daltonics. This improvement, accompanied by the recently developed liquid handling equipment, offers a new valuable alternative to traditional HTS readouts, omitting the necessity of laborious and sophisticated assay development, including the generation of specific antibodies or tagged substrates. Additionally, MALDI-TOF offers the unique opportunity for unbiased and simultaneous substrate/product detection, as well as the availability of an alternative HTS approach for hitherto inaccessible targets.
As a proof of concept, we evaluated the applicability and performance characteristics of MALDI-TOF as an HTS readout. For this purpose, we tracked the PTP1B-dependent dephosphorylation of the INSR (pY1189) as a model system ( Fig. 1A ) representing an important enzymatic mediator of INSR signaling. The application of MALDI-TOF to monitor PTP1B activity is an approaching concept to uncover drug candidates that might have been missed in label-based HTS efforts due to screening artifacts. To assess the suitability of MALDI-TOF for this assay, we cross-compared generated results with the chemiluminescence-based AlphaScreen technology being schematically described in Figure 1B . This recently established AlphaScreen assay comprises a competition between enzymatically generated and artificially biotinylated substrate peptides. Remaining phosphorylated substrate peptide interferes with antibody binding of the artificial biotin-labeled peptide, resulting in reduced chemiluminescence signal (generated by a singlet oxygen diffusing from acceptor to donor beads in direct proximity) that can be attributed to diminished enzymatic activity. 25 MALDI-TOF offers an orthogonal technology to confirm identified drug candidates, and the assay simultaneously serves as an attractive model to disclose the capabilities for MALDI-TOF HTS integration. In the MALDI-TOF assay ( Fig. 1C ), substrate dephosphorylation was directly tracked by analyzing the enzymatic product peptide with the occurring mass shift of 80 Da. This was analyzed in parallel with the isotope-labeled peptide composed of a 10 Da mass shift ( 13 C6, 15 N4-arginine).
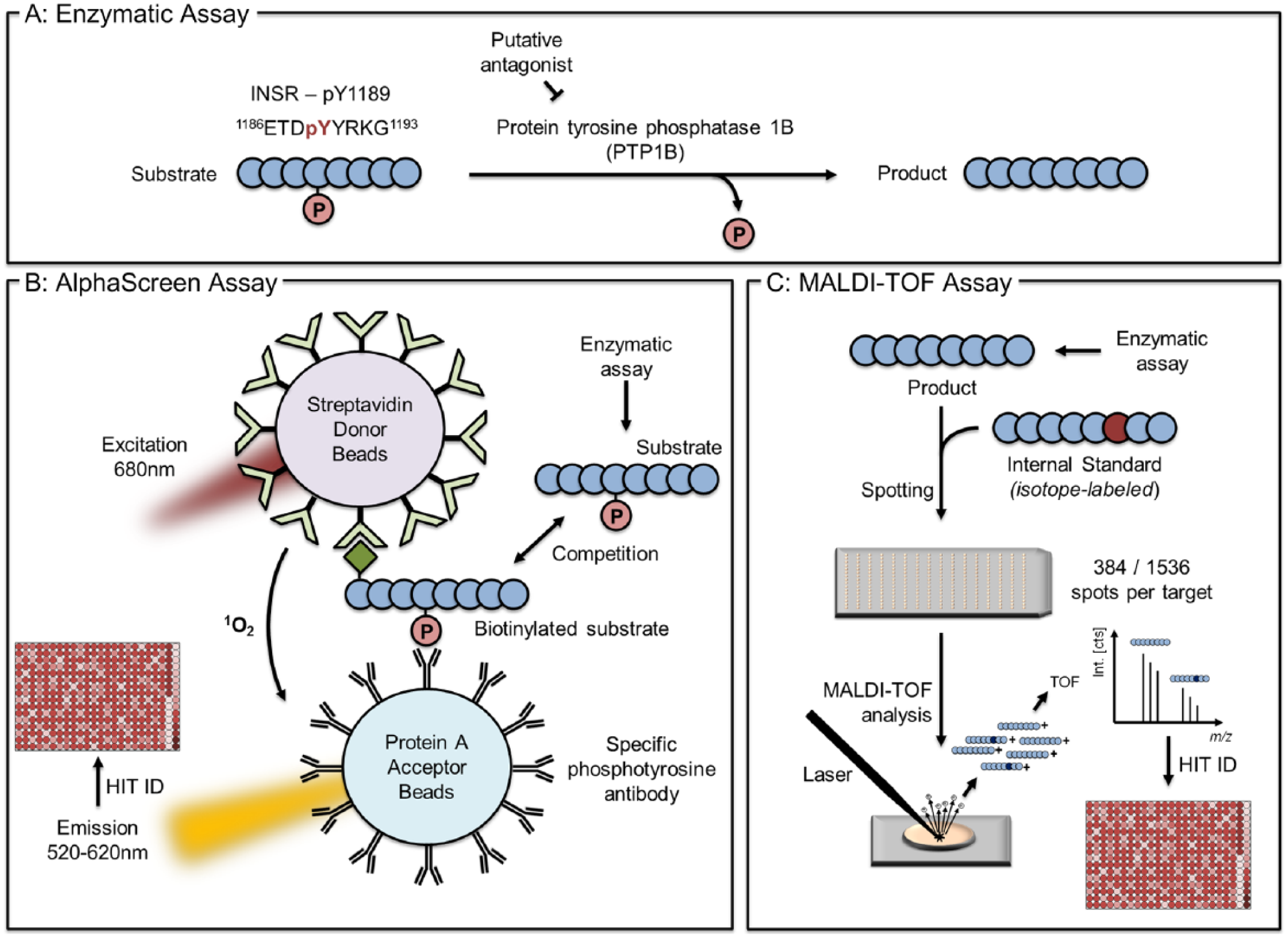
Descriptive representation of workflows established to uncover PTP1B inhibitors. (
Establishment of the MALDI-TOF PTP1B Assay
We selected the 4-HCCA matrix due to its good compatibility with small peptide detection, as well as the generation of extremely homogenous sample spots being an important prerequisite for fully automated and reproducible MALDI-TOF measurements. In order to guarantee homogenous spot shapes, double-layer spotting was applied, ensuring highly reproducible analyses (
One big advantage of MALDI-MS-based analysis is simultaneous detection of multiple assay components. As presented in
Figure 2A
, signals for the phosphorylated substrate, dephosphorylated product, and internal standard peptide were detected simultaneously and offer a direct measure of enzymatic activity. MALDI-TOF suffers from intrinsic fluctuating signals hampering quantitative analysis. These fluctuations were vastly reduced by the application of the internal standard peptide (0.4 nM in final assay solution), compensating variations within subsequent sample preparation steps and ionization/desorption processes. We demonstrate that the combined analysis of the product and internal standard peptide reduced the coefficient of variation from more than 40% (evaluation without internal standard) to 5% (
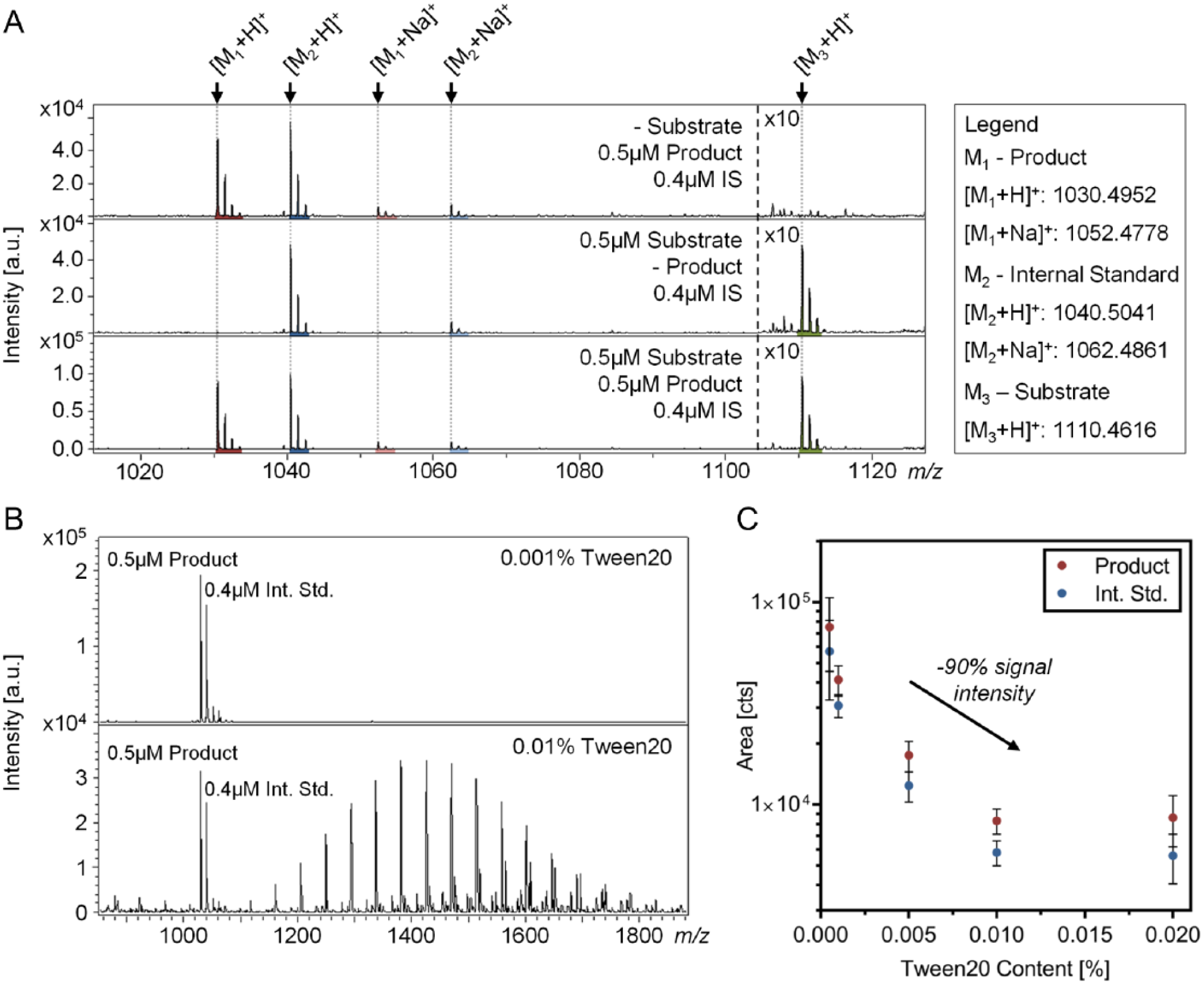
MALDI-TOF analysis and buffer adjustment. (
The signal intensity of MALDI-TOF is generally hampered by interfering components within the assay buffer. Particularly salts and detergents diminish ionization efficiency within the MALDI process, requiring the reassessment of used buffer components and adjustment of respective concentrations. Thus, we adapted the AlphaScreen assay buffer composition to be compatible with the MALDI-based readout. In this context, assay components (50 mM HEPES [pH 7.4], 100 mM NaCl, 0.01% Tween 20, 2 mM DTT, and 0.001% BSA) were concisely verified with respect to MALDI-TOF compatibility. Here, Tween 20 was identified as a pivotal ingredient for ionization efficiency ( Fig. 2B ). Due to this finding, the Tween 20 content was reduced down to 0.001%, which in turn increased the MS signal by approximately 90% ( Fig. 2C ). Detergents are known to disturb ionization efficiency; 27 however, they are essential for enzymatic assays by reducing unspecific binding, increasing the solubility of enzymes, and preventing the precipitation of compounds. The suitability of the modified assay buffer was briefly verified using the AlphaScreen assay, which produced consistent results (data not shown).
Other buffer components, such as sodium chloride, additionally attenuate signal intensity, mainly induced by sodium adduct formation. For this purpose, we established an on-target washing procedure with 10 mM NH4H2PO4/0.1% TFA to reduce adduct formation, which significantly increased the MALDI-MS signal (area internal standard peptide:
Assessment of the MALDI-TOF Assay
With the optimized assay buffer, we first verified the linearity of the substrate and product signals in an assay-relevant concentration range, in order to ensure quantitative measure of the enzyme activity using the MALDI-TOF readout. As shown in Figure 3A , both peptide species show a linear response up to 5 µM, enabling direct correlation of the signal intensity to the enzyme activity. The dephosphorylated product peptide exhibited a higher sensitivity than the substrate peptide, represented by a lower limit of detection (defined as the lowest concentration with precision better than 20% and a signal to noise ratio of >3) of 17 nM (1.7 fmol on the target) for the product and 53 nM (5.3 fmol on the target) for the substrate peptide ( Fig. 3A ). This is in line with previous knowledge that phosphate groups reduce the ionization efficiency of peptides. 29
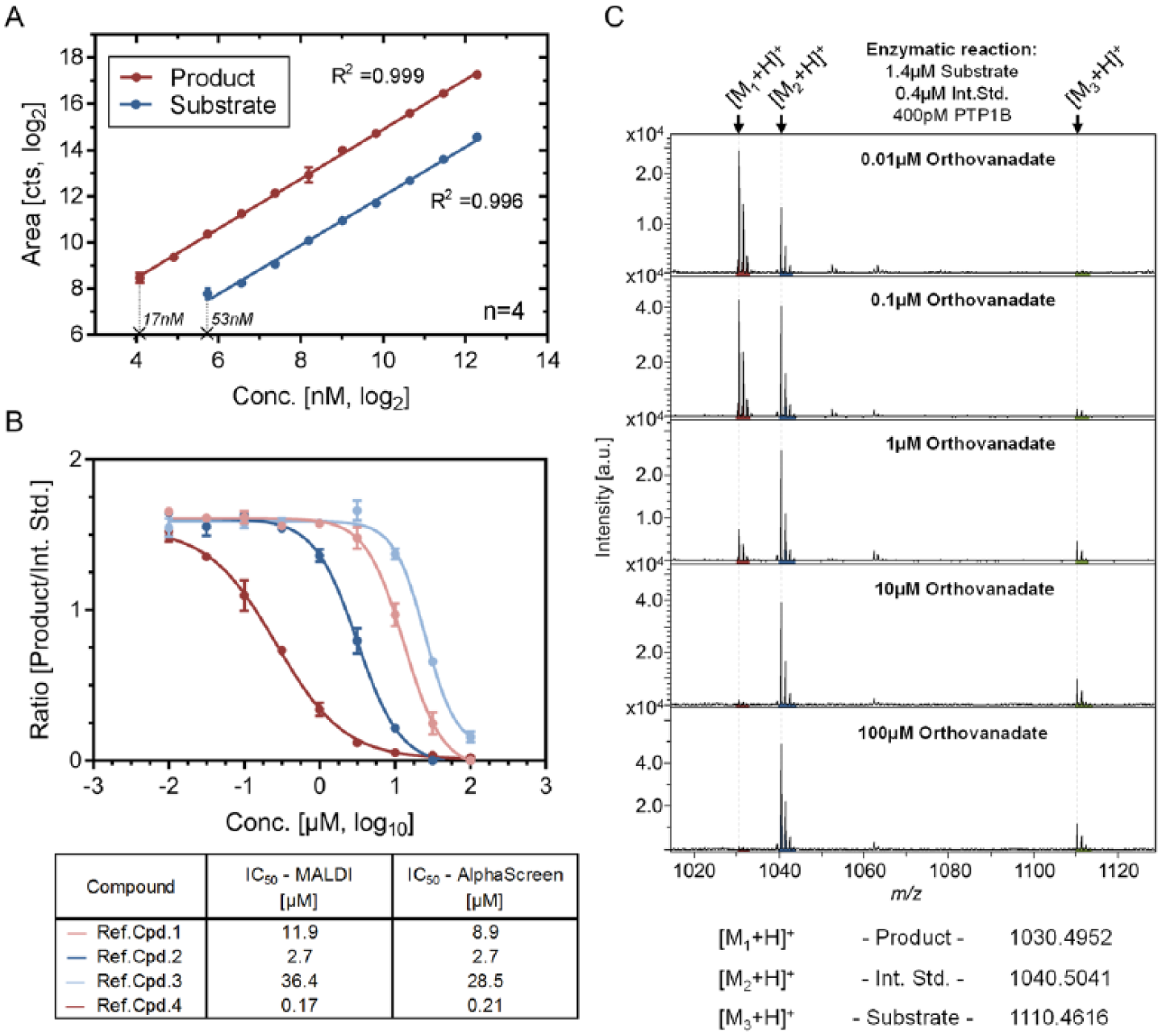
Verification of MALDI-TOF suitability for compound profiling of the PTP1B assay. (
Subsequently, we performed compound profiling assays to confirm the suitability of the MALDI-TOF readout to track enzymatic activity in the PTP1B assay. For this purpose, four reference compounds were used to determine the half-maximal inhibitory concentration (IC50), as illustrated via dose–response curves in Figure 3B . Representative mass spectra, illustrated in Figure 3C , depict the prevalent enzyme inhibition by incubation with ascending concentrations of the well-known protein tyrosine phosphatase inhibitor orthovandate. 30 Generated IC50 values were used to assess the consistency of the MALDI-TOF readout. For this purpose, previously generated AlphaScreen data for the tested compounds were cross-checked and confirmed the validity of the obtained results, suggesting comparable biochemical pharmacology of the MALDI-TOF and AlphaScreen assays ( Fig. 3B ).
Automated Sample Spotting Using CyBio Well Vario
A major challenge for MALDI-TOF-based HTS assays is the respective integration into the HTS environment, which requires sophisticated liquid handling solutions. For this purpose, we selected and applied the CyBio Well vario liquid handling system for MALDI target preparation due to its unique performance characteristics, which include (1) the transfer of 384- and 1536-well formats within one step; (2) the unique possibility to wash the spotted samples on target, enabling the removal of salt interferences; (3) simultaneous drying in a vacuum chamber, providing extremely homogenous spot shapes; (4) a perfectly uniform grid of spots over the target plate, enabling reliable automated analysis; and (5) highly efficient scheduling of individual tasks within the spotting procedure (spotting, cleaning of capillary tips, drying, and on-target wash) to keep up with the MALDI-TOF analysis speed. A limitation of the CyBio Well vario is the necessity for washing steps between the spotting of assay plates in order to prevent carryover. Our optimized washing protocol included three consecutive washing steps with 70% isopropanol in 0.1% TFA, leading to no observable product/internal standard signal in three concatenated spotting steps without repetitive sample aspiration. Hence, this washing procedure was adequate to prevent carryover of the analytes in our assay. The time restraints of this washing procedure vanished with the precise interlacing of different liquid handling steps within the MALDI target plate preparation.
Compared with alternative liquid handling equipment previously reported in this context (Echo 55012 and Mosquito13,14), the CyBio Well vario liquid handling system offers the implementation of automatic and parallelized washing steps in 384- or 1536-well format in a controlled fashion. Moreover, the CyBio Well vario includes simultaneous deposition of all sample spots, reduced consumable consumption compared with the Mosquito, and the ability to spot any matrix/assay buffer composition without prearrangements, such as instrument calibrations, which are necessary for the Echo. One limiting factor of CyBio Well vario applicability is the potential carryover by the ceramic tips, which requires additional washing steps. For the presented assays, no carryover was observed as described above. However, this has to be reviewed carefully for every upcoming project in order to exclude alterations induced by the spotting procedure. Altogether, we demonstrate the suitability of the CyBio Well vario liquid handler for MALDI target preparation and the integration of MALDI-TOF into the HTS environment.
Competitiveness to the AlphaScreen Technology
In order to assess performance characteristics of the developed MALDI-TOF assay, we determined IC50 values for 103 compounds and compared them with the ones obtained by the AlphaScreen technology. All generated IC50 values are listed in Supplemental Table S1 . Scatterplots of the IC50 values generated with AlphaScreen and MALDI-TOF, respectively, are illustrated in Figure 4A . The observed correlation coefficient (R = 0.95) emphasizes high accordance between the two independent technologies. Of note, compounds without inhibitory activity in the tested concentration range were removed from the plots in Figure 4 , reducing the number of plotted substances (n < 103). The obtained correlation is in line with those of previous studies that examined the comparability of MALDI-TOF and label-based assays (IC50 correlations of R = 0.83, 14 R = 0.95, 14 and R = 0.9612 for 105, 23, and 13 tested compounds, respectively). In addition, we demonstrate the equal robustness of the MALDI-TOF and AlphaScreen assays by comparison of Z′ values (Z′AlphaScreen: 0.86/0.76; Z′MALDI-TOF: 0.87/0.86—two independent replicates). This together enables robust and accurate analysis of IC50 values with both assay technologies and demonstrates the suitability of the MALDI-TOF-based MS readout to complement label-based HTS readouts. In addition, our data confirm the structural class representatives that were previously identified with AlphaScreen, offering confirmation via an orthogonal analytical technology.
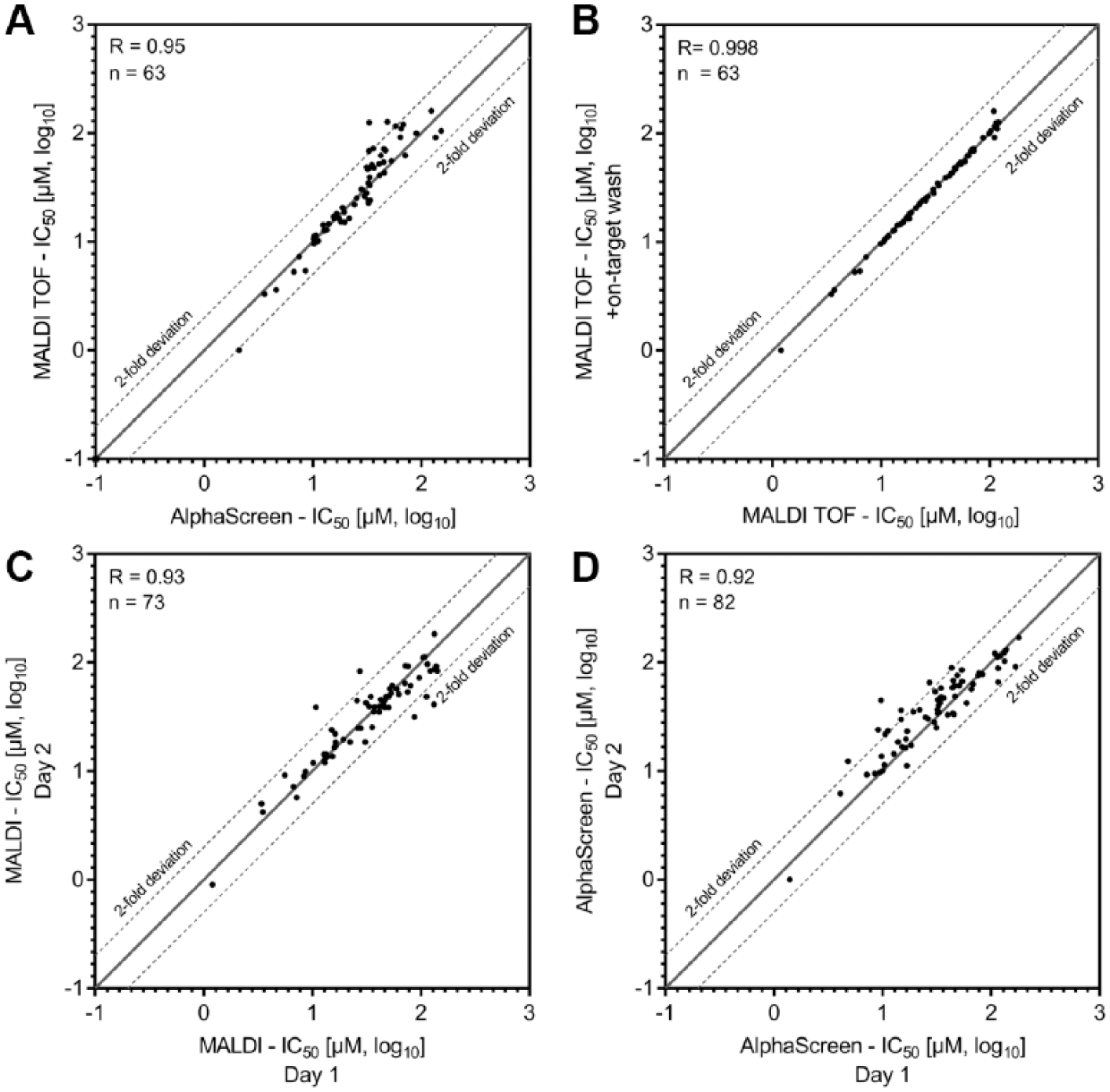
Scatterplots to assess the consistency of MALDI-TOF-generated IC50 values. (
In order to verify the capability of on-target washing, we compared IC50 values generated with and without the cleanup, respectively. A high correlation coefficient of 0.998 ( Fig. 4B ) between the two datasets demonstrates that no bias is raised by the additional processing step. This, combined with the significantly increased signal intensity by on-target washing (approximately 2.5-fold signal increase), emphasizes the merits of the procedure.
Finally, interday reproducibility was verified by high correlation of the MALDI-TOF assay (R = 0.93) performed on different days ( Fig. 4C ). This is comparable to the correlation coefficient of IC50 values generated by AlphaScreen in the same manner (R = 0.92) ( Fig. 4D ). This interday repeatability guarantees comparable results of data generated on different days, a prerequisite for the application of MALDI-TOF in HTS campaigns.
Compound-Dependent Enzyme Modification
It has been previously shown that many compound classes inhibit PTP1B activity by covalent modification or oxidation of the catalytic site (Cys215). 31 In order to identify compounds with this (typically undesired) mode of action, we established a secondary assay using MALDI-TOF. This assay comprised an elevated PTP1B concentration, compared with the drug discovery assay (500-fold), to compensate sensitivity limitations. Following compound incubation, the protein was tryptically digested and the peptide surrounding the catalytic cysteine (Cys215; called CatCysTide) was analyzed, as shown in Figure 5A . This enables the identification of compound-dependent modification of the CatCysTide (PTP1B200–221), and moreover, by MS/MS experiments we can even provide evidence that the modified amino acid is the catalytic cysteine, as outlined in Supplemental Figure S3 .
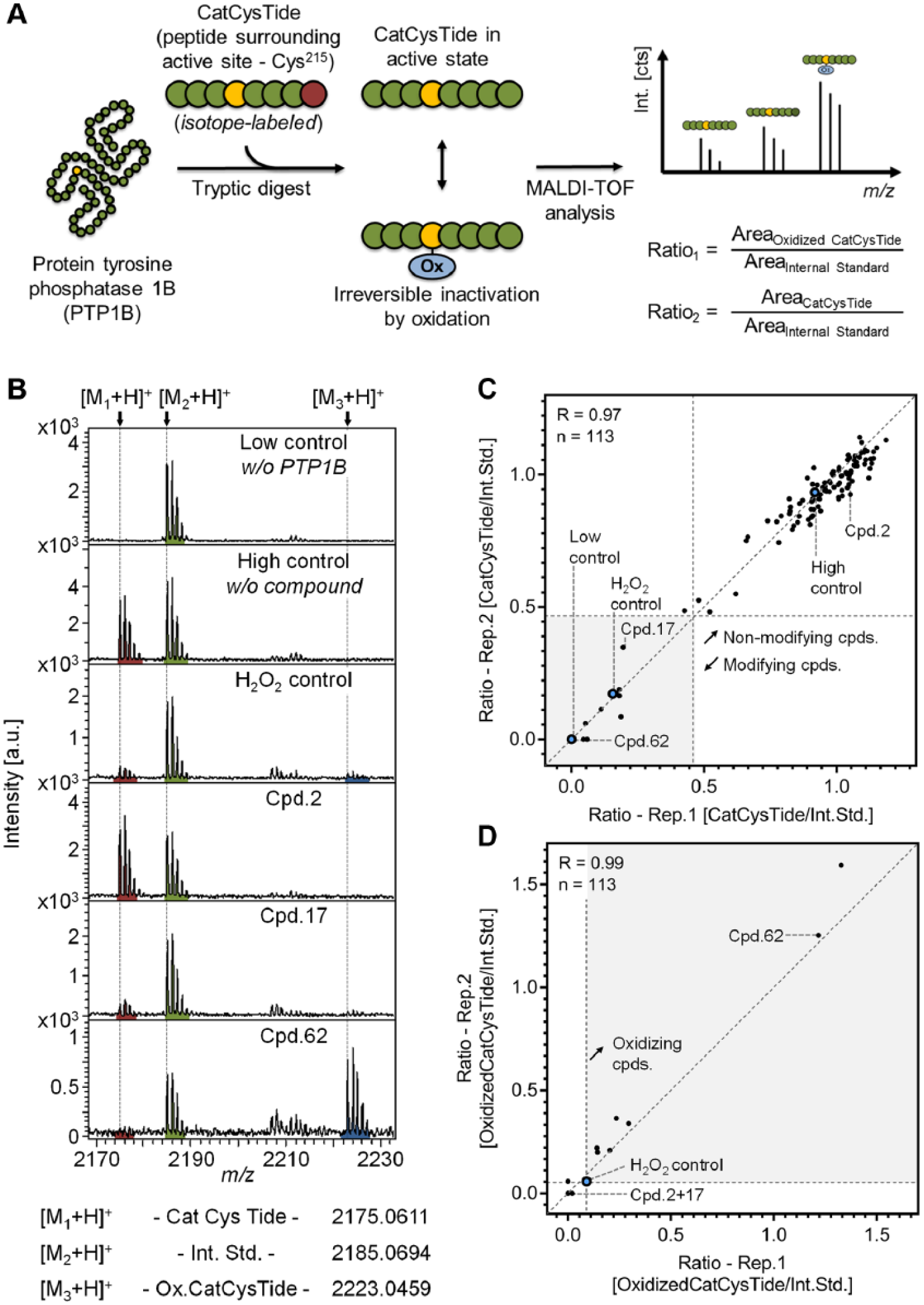
Characterization of compound-dependent PTP1B modification. (
For accurate quantification of the CatCysTide, we spiked in an isotope-labeled internal standard peptide, enabling us to monitor compound-dependent vanishing of the CatCysTide signal. Furthermore, we tracked the sulfonic acid generation at the catalytic cysteine ( Suppl. Fig. S4 ) by measuring the threefold oxidized version (+47.98 Da) of the CatCysTide, as only the hyperoxidized form of cysteine is irreversibly modified. 32 Analyzing these three features enables the identification of CatCysTide-modifying compounds and reveals if this is induced by irreversible oxidation. As presented in Figure 5B , we applied three controls for this experiment: (1) low control lacking PTP1B, (2) high control without compound, and (3) oxidation control incubated with H2O2. This oxidation control is composed of a vanished CatCysTide signal while the peak of the threefold oxidized peptide appears (improved visibility in Suppl. Fig. S5A,B ).
Additionally, three compounds were selected and illustrated in
Figure 5B
representing three potential compound characteristics that have to be differentiated within this assay. Compound 2 represents a nonmodifying compound being characterized by the high intensity of the CatCysTide signal at a level comparable to that of the high control. On the contrary, compounds 17 and 62 eradicate the CatCysTide signal, which is a result of its covalent modification induced by compound incubation. A raising signal at m/z 2223.05, which is the threefold oxidized form of the CatCysTide, enables differentiation between nonoxidizing (no. 17) and oxidizing (no. 62) compounds. In this context, 113 compounds were analyzed. To facilitate data interpretation, the ratio of CatCysTide to internal standard peptide is plotted for two independent replicates in
Figure 5C
. This reveals a strong cluster of nonmodifying compounds in close proximity to the high control. Moreover, high correlation between the two independent biological replicates (R = 0.99) confirms high repeatability of this assay. In order to identify compounds that modify PTP1B at the CatCysTide, we defined a threshold for the observed ratio to be less than half of the high control mean ratio. This revealed 11 modifying compounds in the set of 113 (
In order to exclude vanished CatCysTide signal raised by hampered enzymatic digestion of PTP1B induced by compound-dependent inhibition of trypsin, we additionally tracked another PTP1B peptide ( 13 SGSWAAIYQDIR 24 ). Analysis of this PTP1B peptide being distant from the catalytic cysteine provides a measure for successful tryptic digestion of PTP1B. Analysis of its abundance ( Suppl. Fig. S5 ) demonstrates that none of the tested compounds inhibit trypsin efficacy and confirms validity of the identified modifying compounds. Due to this finding, all unveiled modifying compounds were subsequently excluded.
This depicted secondary assay deciphering the mode of action of small-molecule inhibitors demonstrates the assets of MALDI-TOF for the follow-up of primary screens. As demonstrated, MALDI-TOF offers the valuable possibility to design tailored assays to cut down the list of potential drug candidates prior to expensive lead optimization processes.
Conclusion
Conclusively, we established MALDI-TOF as an in-line reader for drug discovery research. We demonstrate successful integration into the HTS environment using the liquid handling system CyBio Well vario, offering unique advantages for this approach. MALDI-TOF is capable of producing compound profiling data of high quality, comparable with the well-established AlphaScreen technology, while providing significant advantages, such as simultaneous detection of physiologically relevant assay components, together with the less error-prone readout. Due to this, follow-up screening concepts (counterscreens) to identify false-positive primary hits can be omitted or at least reduced for the MS-based HTS readout. Together, MALDI-TOF has the potential to reduce time and costs for the implementation of HTS campaigns. Further, the developed secondary assay for the identification of compound-dependent enzyme modification at the catalytic center of PTP1B emphasizes the versatility of MALDI-TOF as an integrated drug discovery readout. Overall, the presented data highlight MALDI-TOF as a viable and versatile technology for drug discovery research per se.
Footnotes
Acknowledgements
We thank Margit Bauer for providing the enzyme PTP1B and Romina Schnegotzki for assistance during assay optimization. Further, we thank Dirk Gottschling, Peter Haebel, Dieter Wiedenmayer, Dennis Fiegen, Markus Zeeb, and Christofer Tautermann for being part of the project team. We thank Meike Hamester, Franz J. Mayer, Miriam Denkert, and Astrid Erdmann from Bruker Daltonics (Bremen, Germany) as well as Adrian Siemers and Swen Tyrasa from Analytik Jena (Jena, Germany) for their valuable input and helpful discussion.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article. Everything was completed as work-for-hire for the employer Boehringer Ingelheim Pharma GmbH & Co. KG.
Supplementary material is available online with this article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.