
Abstract
Leucyl aminopeptidases (LAPs) are involved in multiple cellular functions, which, in the case of infectious diseases, includes participation in the pathogen-host cell interface and pathogenesis. Thus, LAPs are considered good candidate drug targets, and the major M17-LAP from Trypanosoma cruzi (LAPTc) in particular is a promising target for Chagas disease. To exploit LAPTc as a potential target, it is essential to develop potent and selective inhibitors. To achieve this, we report a high-throughput screening method for LAPTc. Two methods were developed and optimized: a Leu-7-amido-4-methylcoumarin–based fluorogenic assay and a RapidFire mass spectrometry (RapidFire MS)–based assay using the LSTVIVR peptide as substrate. Compared with a fluorescence assay, the major advantages of the RapidFire MS assay are a greater signal-to-noise ratio as well as decreased consumption of enzyme. RapidFire MS was validated with the broad-spectrum LAP inhibitors bestatin (IC50 = 0.35 μM) and arphamenine A (IC50 = 15.75 μM). We suggest that RapidFire MS is highly suitable for screening for specific LAPTc inhibitors.
Keywords
Introduction
Infection by parasitic pathogens remains a major human health burden. 1 Among these, Chagas disease is classified as a neglected tropical disease present in Latin and North America, Europe, and the Western Pacific. 2 The etiological agent is the kinetoplastid parasite Trypanosoma cruzi, responsible for 50,000 new cases and more than 10,000 deaths each year. 3
There are no effective vaccines for Chagas disease, and the current drugs employed in treatment are toxic and/or ineffective. Even with more than a century of research since the initial description by Carlos Chagas, only two nitroheterocyclic compounds, nifurtimox and benznidazole, have entered the clinic for treatment. 4 Both drugs have limited efficacy for treating the chronic stages of infection and have severe side effects. 5,6 In this context, new, effective, and safe drugs against T. cruzi are urgently required. 7
Proteases have multiple roles in cell functions and contribute with parasite physiology and virulence. 8 -10 Many are considered good drug targets, 11,12 among which are M17-leucyl aminopeptidases (LAPs; EC 3.4.11.1), enzymes that preferentially catalyze removal of an N-terminal leucine from peptides. 13 The process requires two divalent metal cations and a neutral/basic optimal pH. 14
The major M17-LAP from T. cruzi (LAPTc; UniProt: Q4DZJ3) is a 330-kDa homohexameric protein that has been proposed to be localized to vesicles in the parasite cytoplasm. 15 LAPTc could participate in the nutritional supply by degradation of peptides that are endocytosed by the parasite and delivery of leucine to the cytoplasm. Notably, the aminopeptidase inhibitor arphamenine A inhibits in vitro growth of T. brucei brucei, a parasite closely related to T. cruzi. 16 In addition, TbLAP1, the T. brucei ortholog of LAPTc, is involved in the late stages of segregation of kinetoplast DNA, as knockdown causes a delay in cytokinesis. 17 Furthermore, bestatin, a classic inhibitor of M1- and M17-aminopeptidases, 18 caused in situ inhibition of LAPTc in T. cruzi. 19 This latter evidence indicates that LAPTc is potentially druggable by bestatin and bestatin-like low-molecular-weight inhibitors.
M17-LAPs may also be exploitable in other parasites. The M17-LAP from Plasmodium falciparum plays an essential housekeeping function, as suggested by a specific bestatin-derived inhibitor. 13 The inhibition of Babesia bovis growth by bestatin has also been attributed to the inhibition of BbM17-LAP. 20 Finally, knockout of LAPTg from Toxoplasma gondii inhibits the ability of the parasite to invade cultured cells and also resulted in reduced replication and attenuated virulence in a murine model. 21 Hence, LAPs may also be of significant importance for the development of therapeutics in a wider context.
To identify highly specific LAPTc inhibitors in large-compound libraries, a high-throughput screening (HTS) assay is necessary. 22 Ideally, this assay needs to be sensitive, robust, and have a high signal-to-noise ratio. 23 Here we describe two biochemical assays for the identification of LAPTc inhibitors by HTS. The first is a fluorescence assay based on the fluorogenic substrate Leu-7-amido-4-methylcoumarin (Leu-AMC), and the second uses RapidFire mass spectrometry (RapidFire MS) with the peptide substrate LSTVIVR. We validated the selected method by assessing the half inhibitory concentration (IC50) of bestatin and arphamenine A toward LAPTc.
Materials and Methods
Substrates and Other Reagents
Superose 6 HR10/30 column was from Amersham Biosciences (Buckinghamshire, UK). DMSO, formic acid, NP-40, trifluoracetic acid (TFA), acetonitrile, and Leu-AMC were obtained from Sigma-Aldrich (St. Louis, MO). The LSTVIVR (786.953 Da) and STVIVR* (683.794 Da) peptides were supplied by Cambridge Research Biochemicals (Billingham, UK) and dissolved in water.
Protein Expression and Purification of Recombinant LAPTc
LAPTc was produced in recombinant form in Escherichia coli according to Izquierdo et al. 24 As a second purification step, gel filtration chromatography was performed using a Superose 6 HR10/30 column with 50 mM Tris-HCl buffer, pH 8.0, 300 mM NaCl. The flow rate was 1 mL/min, the fraction size was 2 mL, and runs were monitored at 280 nm. Protein samples were quantified by the Bradford method. Sodium dodecyl sulfate–polyacrylamide gel electrophoresis was performed with NuPAGE 4% to 12% Bis-Tris Gels (Invitrogen, Paisley, UK). Purified LAPTc was stored at –80 °C in the same buffer.
Kinetic Assay with the Fluorogenic Substrate Leu-7-Amido-4-Methylcoumarin
LAP enzymatic activity was assessed by a continuous kinetic method using 1 mM (one value of apparent Michaelis-Menten’s constant -appKM-) Leu-AMC fluorogenic substrate solubilized in DMSO. 25 The fluorescence due to the release of AMC (excitation: 380 nm, emission: 460 nm) was monitored over a 30-min period using a microplate spectrofluorometer (PHERAstar FSX, BMG LABTECH, Aylesbury, UK). Kinetic assays were carried out with concentrations of LAPTc that were linearly related to the initial velocities, at 25 °C, in 96- or 384-well black plates (200, 100, and 50 µL final volume). The activity buffer was 50 mM Tris-HCl pH 7.5. The final concentration of DMSO was 1%, 10%, and 20% (v/v). Only the linear portions of progress curves, corresponding to substrate consumption lower than 5%, were used to measure the reaction rates. Slopes with R 2 < 0.98 were not considered. All assays were performed in triplicate.
Determination of the enzyme concentration range linearly related with the initial velocity
Three enzyme concentrations (0.036, 0.09, and 0.18 μM) were tested, and initial velocities were determined in 96-well black plates with a final volume of 200 μL.
Determination of apparent KM for LAPTc
Assays were performed with 0.036 μM LAPTc, in 96-well black plates and 200 µL final volume, at 11 concentrations of Leu-AMC substrate prepared by serial dilutions in the 9.375 to 9600 μM range. The experiment was performed at 1% and 20% DMSO. The appKM was calculated by fitting the Michaelis-Menten rectangular hyperbola function 26 to the experimental data using the software OriginPro 8 SR0 (version 8.0724 [B724]; OriginLab Corporation, http://www.OriginLab.com).
Determination of the bestatin effect
Assays were performed by preincubating 0.18 μM LAPTc with 1 mM bestatin for 15 min before adding substrate, in 384-well black plates, for a final volume of 50 µL and 20% DMSO.
Kinetic Assay with the LSTVIVR Peptide Substrate by RapidFire MS
LAP enzymatic activity was assessed by a discontinuous kinetic method. Basically, this method consisted of mixing enzyme and substrate, allowing the reaction, and further quantifying the product by MS. For this, the reaction was loaded into the mass spectrometer after a liquid chromatography step. The mass spectrometer contains a triple quadrupole; the precursor selection occurs in the first, the fragmentation occurs in the second, and fragment selection occurs in the third quadrupole. Peak areas were integrated, and area ratios of product to the internal standard were used for quantitation.
The assay was performed in 384-well clear F-bottom polypropylene plates with a final reaction volume of 15 μL. The reaction mixture contains 7.5 µL LAPTc in 50 mM Tris-HCl, pH 7.5, 0.005% NP-40, plus 7.5 µL LSTVIVR peptide substrate in the same buffer. The reaction was performed at room temperature for 40 min and then stopped with 85 µL 1% formic acid containing 0.15 μg/mL STVIVR* internal standard.
RapidFire MS was performed using a RapidFire 365 system (Agilent, Santa Clara, CA) coupled with a triple quadrupole mass spectrometer 6740 (Agilent). The samples were loaded onto a C18 cartridge (Agilent) using 0.1% TFA in deionized water at flow rate of 1.5 mL/min and eluted to the mass spectrometer using 0.1% TFA in acetonitrile/deionized water (90/10, v/v) at a flow rate of 1.25 mL/min. The sipper was washed to minimize carryover with deionized water followed by acetonitrile. Aspiration time, load/wash time, elution time, and reequilibration time were set to 600, 3000, 5000, and 500 ms, respectively, with a cycle time of approximately 10 s. The triple-quadrupole mass spectrometer with electrospray ion source was operated in positive multiple reaction monitoring (MRM) mode. The detailed setting for the mass spectrometer parameters was as follows: capillary voltage, 3000 V; gas temperature, 350 °C; gas flow, 7 L/min; nebulizer, 40 psi; sheath gas temperature, 300 °C’ sheath gas flow, 11 L/min; and nozzle voltage, 1500 V. The MRM transitions (Q1 and Q3) for LSTVIVR peptide as a reaction product and LSTVIVR* peptide as an internal standard were set as 337.8/486.3 and 342.8/491.3, respectively. The mass resolution window for both parental and daughter ions was set at as a unit (0.7 Da). The dwell time, fragmentor, and collision energy for each transition were 50 ms, 175 V, and 10 eV, respectively. Peak areas were integrated, and area ratios of the LSTVIVR peptide to the internal standard LSTVIVR* peptide were used for quantitation.
Determination of the enzyme concentration range linearly related with the initial velocity at different incubation times
LAPTc was tested at 0, 1.56, and 3.13 nM with 2 mM (∼12 appKM) LSTVIVR peptide substrate. Different incubation times were tested (0, 5, 10, 15, and 20 min).
Determination of apparent KM for LAPTc
LAPTc was tested at 3 nM with 10 LSTVIVR peptide substrate concentrations, prepared by serial dilutions in water, spanning the range of 3.906 to 2000 μM. The incubation time was 20 min.
Determination of the incubation time range linearly related with the initial velocity
LAPTc was tested at 3 nM with 150 μM (∼1 appKM) LSTVIVR peptide substrate at 0, 10, 20, 30, 40, 50, and 60 min incubation.
Determination of the LAPTc tolerance to DMSO
LAPTc was tested at 3 nM with 150 μM (∼1 appKM) LSTVIVR peptide substrate at 40 min incubation in the presence of 0%, 1%, 2%, 3%, and 4% DMSO.
Dose-response study for bestatin and arphamenine A
LAPTc was tested at 3 nM with 150 μM (∼1 appKM) LSTVIVR peptide substrate at 40 min incubation. Before addition of substrate, LAPTc was preincubated with the inhibitors or DMSO for 15 min. Bestatin and arphamenine A were prepared in 10 concentrations in DMSO by serial dilutions in the ranges of 0.005 to 100 μM and 1.76 to 900 μM, respectively. A control without inhibitor (the same volume of DMSO, 0% inhibitory effect) and another without enzyme and inhibitor (100% inhibitory effect) were prepared.
The IC50 values were calculated by the nonlinear fit of the Hill function to the experimental data, using OriginPro 8 SR0 software (version 8.0724 (B724); OriginLab Corporation, http://www.OriginLab.com) with default parameters.
Statistical Calculations
Blank Median = Median (Blank) (nonexcluded BLANK Raw Values)
High Control (Ctrl) Median = Median (Ctrl) (nonexcluded control raw values).
When the raw value for the Blank was lower than the raw value for the Ctrl, the following formulas were used:
Robust S/B = Median (Ctrl)/Median (Blank)
Robust Z′ Factor = 1 – (3 × (1.4826 × MAD(Ctrl)) + 3 × (1.4826 × MAD(Blank)))/Median(Ctrl) – Median(Blank)
where MAD is the median absolute deviation.
When the raw value for Ctrl was lower than the raw value for the Blank, the following formulas were used:
Robust S/B = Median (Blank)/Median (Ctrl)
Robust Z′ factor = 1 – (3 × (1.4826 × MAD(Blank)) + 3 × (1.4826 × MAD(Ctrl)))/Median(Blank) – Median(Ctrl)
Results
Isolation and Purification of LAPTc
LAPTc was expressed in Escherichia coli (1575 bp synthetic gene cloned in the plasmidic vector pET-19b between the NdeI and XhoI restriction sites, fused to an N-terminal tag of 10 histidines) and purified by immobilized metallic ion affinity chromatography according to Izquierdo et al.
24
Here, to improve purity, we introduced a second purification step based on gel filtration chromatography. After this chromatography, the protein was obtained in sufficient amount (25.36 mg from 600 mL of culture) and with a purity of 92% (
Fluorescence Assay Development
First, we determined the enzyme concentration range that was linearly correlated with the initial velocity. Assays were performed in 96-well black plates and 200 µL final volume. LAPTc has maximal activity at pH 9.0, 50 °C, and 4 mM Co2+. 24 However, the conditions tested here were pH 7.5, 25 °C, and no cation, to have an assay close to physiological conditions in which inhibition is relevant and favors the stability of the compound set. As the result of this first experiment, an enzyme concentration range of 0.036 to 0.18 μM was found to be linearly correlated with the initial velocity ( Fig. 1A ).
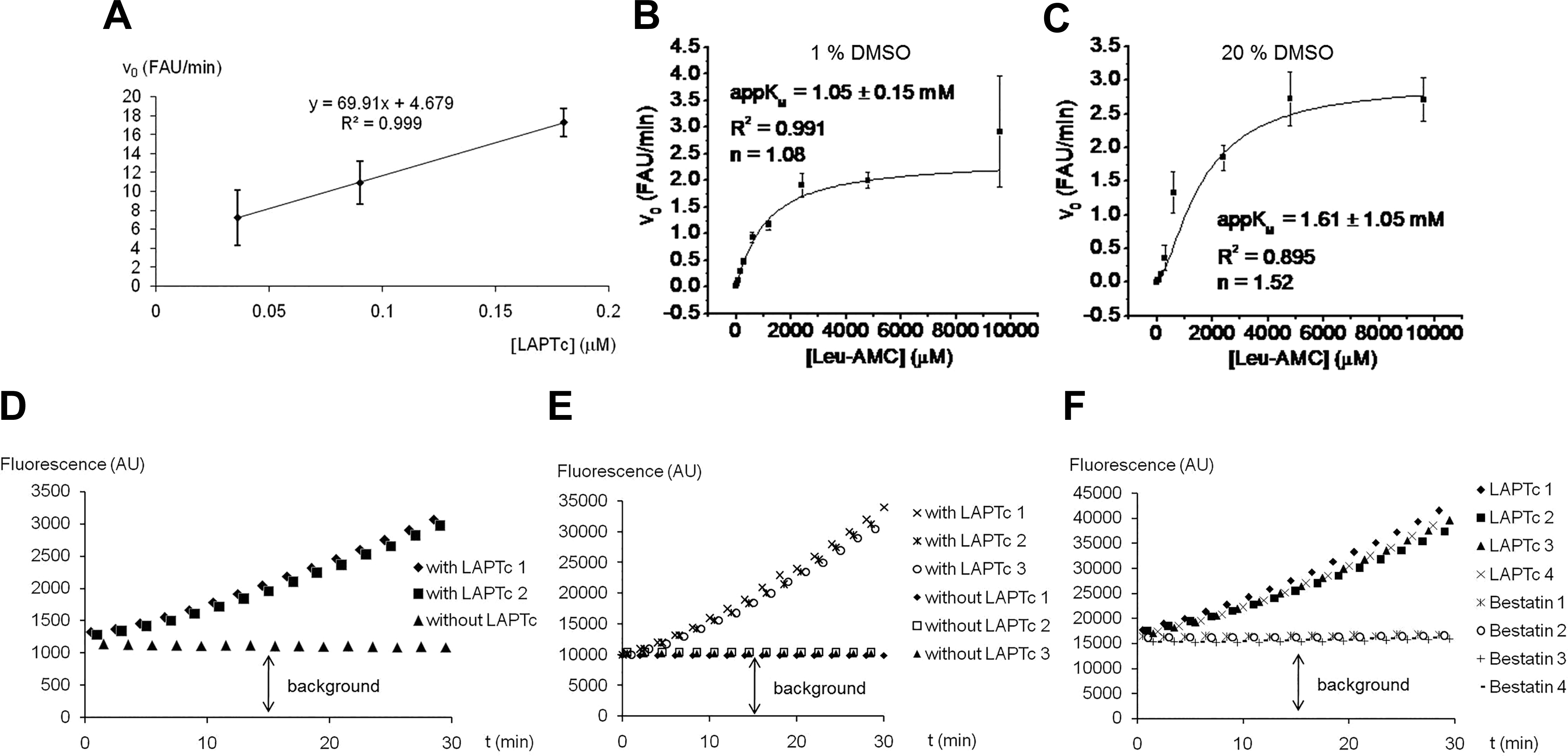
Development of the fluorescence assay based on the Leu-7-amido-4-methylcoumarin substrate. (
Second, the appKM value of LAPTc with the Leu-AMC substrate was determined. The KM is expressed as an apparent value because the enzyme is not 100% homogeneous. For HTS biochemical assays, it is desirable to reduce the reaction volume as much as possible, and here we tested 200, 100, and 50 μL. For this reason, we determined appKM in the presence of 1% DMSO (the substrate is dissolved in this solvent and represents 1% final volume in the 200 µL assay) and 20% DMSO (when the assay volume is reduced to 50 μL, it is not accurate to dispense 0.5 µL of substrate and inhibitor; then, both volumes should be increased to 5 μL, each representing 10% final volume). LAPTc is known to be a DMSO-tolerant enzyme, as it retains activity in the presence of 20% DMSO. At this organic solvent concentration, the affinity for the Leu-AMC substrate is, however, only slightly diminished with respect to 1% DMSO, with appKM increased only from 1.05 to 1.61 mM ( Fig. 1B, C ). A 1 mM substrate concentration was selected for the remaining experiments.
Despite clearly reporting LAP activity, a high background signal was observed ( Fig. 1D ) and was also present in 384-well plates with a 100 µL reaction volume and 10% DMSO, as well as 50 μL, 20% DMSO, and 1 mM bestatin ( Fig. 1E, F ). Although the assay could detect inhibitors such as bestatin ( Fig. 1F ), the low signal-to-noise ratio suggested that the assay was not adequate for identification of LAP inhibitors by HTS.
RapidFire MS Assay Development
With the poor performance of the fluorescence-based assay, we turned to RapidFire MS. This assay was performed without added exogenous divalent metallic cation, like the fluorescence assay. The enzyme concentration range linearly correlating with initial velocity was determined for different incubation times. As is shown in Figure 2A , below 3.13 nM LAPTc, a linear relationship with initial velocity was obtained for all incubation times. Therefore, 3 nM LAPTc was selected for the next steps.
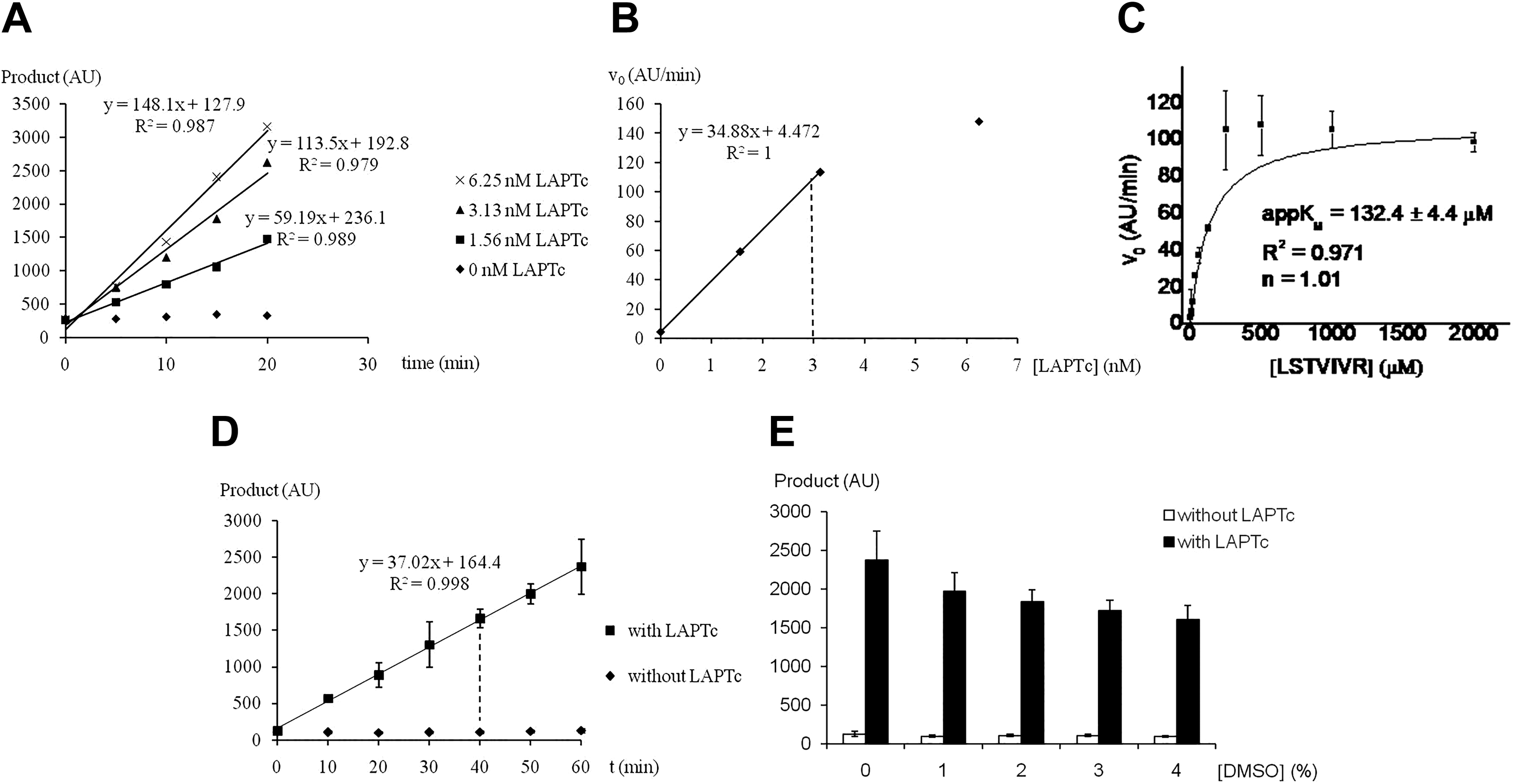
Development of the RapidFire mass spectrometry assay with the LSTVIVR peptide substrate. (
Next, the appKM value against the LSTVIVR peptide substrate was determined as 132 μM ( Fig. 2B ). Hence, 150 μM substrate was used in the next experiments. Incubation time was linearly related with the initial velocity up to 60 min ( Fig. 2C ) and a 40 min incubation time was selected. When different DMSO concentrations were tested, the DMSO-tolerant property of LAPTc was corroborated. At 4% DMSO, the enzyme retained 67.8% of activity ( Fig. 2D ).
To test assay robustness and signal-to-noise ratio, a mock screen without inhibitors was performed. Two 384-well plates were used, and the last column in both cases lacked the enzyme (
Finally, a dose-response study for known aminopeptidase inhibitors bestatin and arphamenine A against LAPTc was performed ( Fig. 3 ). The IC50 values for bestatin and arphamenine A are 0.35 μM and 15.75 μM, respectively.
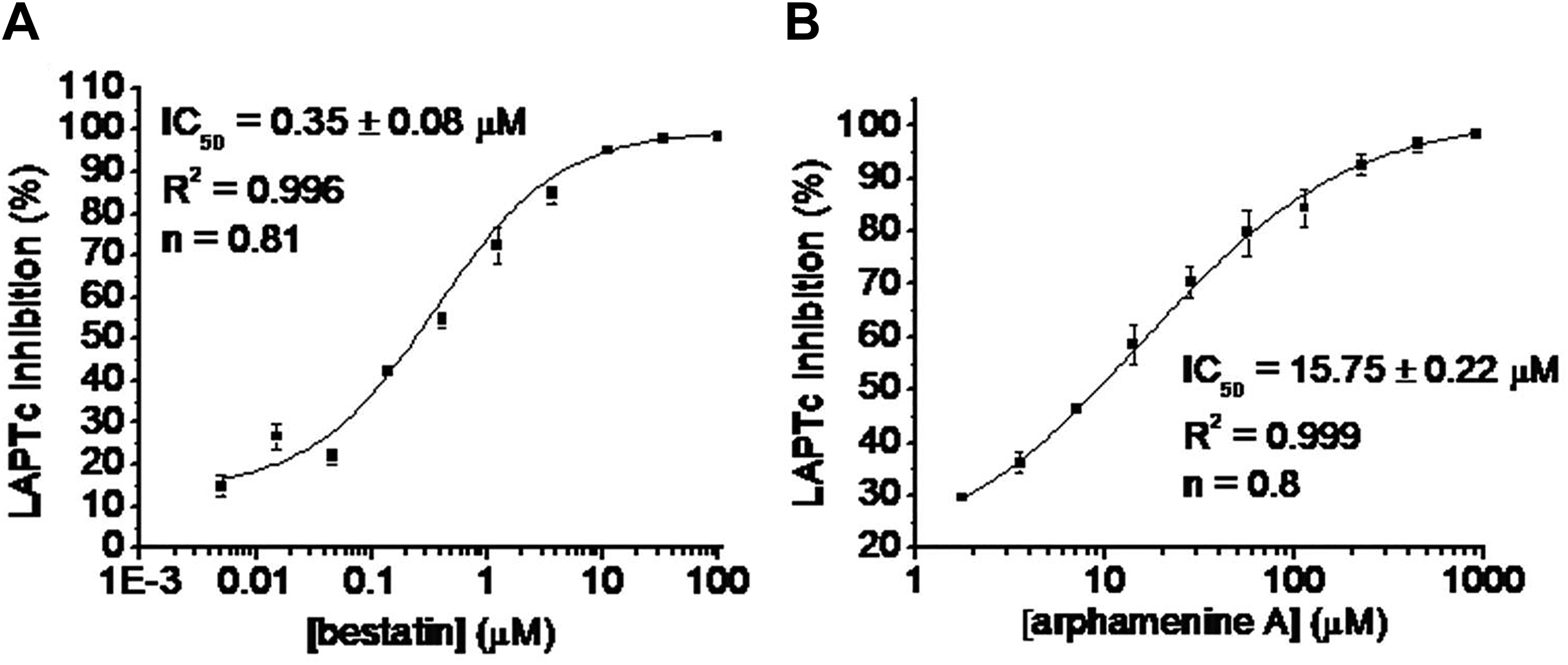
Dose-response study for bestatin (
In Table 1 , both kinetic methods tested in this work for LAPTc activity and its inhibition are compared in their main parameters.
Comparison Between the Fluorescence Assay and the RapidFire MS Assay.

Discussion
Leucine is an important amino acid for trypanosomatids. It has been proposed that leucine catabolism produces mevalonate, a metabolite necessary for the ergosterol biosynthesis, which is a cholesterol-related essential parasite lipid. 27,28 Because many parasites, including trypanosomatids, have no or limited ability to synthesize essential amino acids, including leucine, it is possible that LAPs are important for the provision of this amino acid. This contribution with leucine production, at least in part, could explain why M17-LAPs are critical for parasite development in the mammalian host. 29,30
In this work, we developed and optimized two kinetic assays for identification of LAPTc inhibitors by HTS. This is necessary to detect potent inhibitors of this M17-LAP in libraries of thousands of synthetic compounds, as the first step in the target-based drug discovery process.
In our previous work, we purified LAPTc in a single step by IMAC.
24
To improve purity, we introduced an additional gel filtration step and obtained LAPTc protein in a sufficient amount and purity for the development of kinetic assays, one of which proved to be suitable for HTS (
M17-LAPs are usually assayed at alkaline pH and elevated temperature,
29,31
-34
and the T. cruzi enzyme displays maximal activity at pH 9.0 and 50 °C.
24
Although these conditions are incompatible with HTS, LAPTc retained sufficient activity at pH 7.5 and room temperature for HTS assay development (
Figs. 1A
,
2A, and 2C
). In our fluorescence assay, the lowest concentration showing activity was 36 nM (
Fig. 1A
), whereas 3 nM LAPTc was sufficient for RapidFire MS (
Fig. 2A
; and
The drastic difference in the appKM value for LAPTc between Leu-p-nitroanilide (Leu-pNA; 74 μM
24
) and Leu-AMC substrates (1.05 mM;
Fig. 1B
), at 1% DMSO could be due to structural and size differences between both artificial substrates. The natural peptide substrate LSTVIVR has an appKM value also in the submillimolar order (132 μM;
Fig. 2B
). On the other hand, the increasing of DMSO concentration to 20% could produce conformational change in the protein (or simply interfere with substrate binding), diminishing the affinity to the Leu-AMC substrate (appKM is increased to 1.61 mM;
Fig. 1C
). However, LAPTc, as other enzymes,
35
shows a high DMSO tolerance (
Figs. 1C, 2D
; and
The fluorescence method has a high background under conditions tested ( Fig. 1D–F ). Although this background could theoretically be subtracted in assays with several replicates, in HTS assays that use single-point measurements of thousands of compounds, background subtraction substantially increases the probability of losing active molecules. For this reason, this method was rejected.
The peptide LSTVIVR was selected as the substrate in the RapidFire MS assay. The selection of the first two residues of this sequence was based on the work of Trochine et al., 19 who found LS dipeptide accumulation after treatment of T. cruzi epimastigotes with bestatin. The C-terminal arginine was chosen because this amino acid is amenable to isotope labeling, 36 which is used as an internal standard to quantify reaction products by RapidFire MS.
The RapidFire MS–based assay we devised meets all requirements for HTS methodologies
23
: it is robust (with Z′ values of 0.80 and 0.61;
RapidFire MS successfully detected the inhibitory activity in bestatin and arphamenine A in a dose-dependent manner ( Fig. 3 ). For bestatin, we have reported an IC50 of 6.62 μM and a noncompetitive inhibition mode toward LAPTc using Leu-pNA substrate. 24 The IC50 from RapidFire MS (0.35 μM; Fig. 3A ) is different from the Leu-pNA, 24 because the IC50 and inhibition constant (Ki) depend on the substrate and method used. 38 In addition, to the best of our knowledge, this is the first report to indicate that LAPTc can be inhibited by arphamenine A ( Fig. 3B ). This is a promising result, because arphamenine A inhibits the in vitro growth of T. brucei, a parasite closely related to T. cruzi. 16
In conclusion, as part of a campaign to exploit LAPs for therapeutic intervention, we developed a novel RapidFire MS assay for LAPTc. This technology was used by Leveridge et al. 39 in HTS to identify LRRK2 kinase inhibitors for the treatment of Parkinson’s disease and more recently to identify an arginase II inhibitor, 35 but its use for aminopeptidases has not been reported. We propose that this methodology is suitable for identifying LAPTc inhibitors by HTS.
Supplemental Material
Supplemental_Material_for_HTS_LAPTc_by_Izquierdo_et_al - Development of a High-Throughput Screening Assay to Identify Inhibitors of the Major M17-Leucyl Aminopeptidase from Trypanosoma cruzi Using RapidFire Mass Spectrometry
Supplemental_Material_for_HTS_LAPTc_by_Izquierdo_et_al for Development of a High-Throughput Screening Assay to Identify Inhibitors of the Major M17-Leucyl Aminopeptidase from Trypanosoma cruzi Using RapidFire Mass Spectrometry by Maikel Izquierdo, De Lin, Sandra O’Neill, Martin Zoltner, Lauren Webster, Anthony Hope, David W. Gray, Mark C. Field and Jorge González-Bacerio in SLAS Discovery
Footnotes
Acknowledgments
J.G.-B. and M.I. are grateful to Susan Farrell (training manager, Wellcome Centre for Anti-Infectives Research, University of Dundee) for the opportunity to participate in the WCAIR training scheme. We also thank her for help with the logistics of their stay in Dundee and making them feel so welcome. Equally, we thank Alain Pierre-Petit (Dundee) for help with the second step of protein purification and members of the Drug Discovery Unit compound management team.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article: This work was supported by Wellcome Trust Centre awards (203134/Z/16/Z, 204697/Z/16/), the second to M.C.F.; the International Foundation for Sciences (grant F/4730-2) to J.G.-B.; and the project assigned to J.G.B. and associated with the Cuban National Program of Basic Sciences.
Supplemental material is available online with this article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.