
Abstract
Using mass spectrometry to detect enzymatic activity offers several advantages over fluorescence-based methods. Automation of sample handling and analysis using platforms such as the RapidFire (Agilent Technologies, Lexington, MA) has made these assays amenable to medium-throughput screening (of the order of 100,000 wells). However, true high-throughput screens (HTS) of large compound collections (>1 million) are still considered too time-consuming to be feasible. Here we propose a simple multiplexing strategy that can be used to increase the throughput of RapidFire, making it viable for HTS. The method relies on the ability to analyze pooled samples from several reactions simultaneously and to deconvolute their origin using “mass-tagged” substrates. Using the JmjD2d H3K9me3 demethylase as a model system, we demonstrate the practicality of this method to achieve a 4-fold increase in throughput. This was achieved without any loss of assay quality. This multiplex strategy could easily be scaled to give even greater reductions in analysis time.
Keywords
Introduction
Recently, there has been much debate on the value of high-throughput screening (HTS) of large, diverse compound libraries in research and development (R&D) as a means of identifying lead molecules. Some argue that HTS has dramatically reduced the cost and time taken to find molecules 1 and that screening collections of millions of diverse compounds is a successful method for identifying leads for more than 60% of targets. 2 Others believe that HTS is a “brute-force” approach and moves away from classic and more productive drug discovery approaches.3,4 This has led some companies to abandon large diversity screens in favor of knowledge-based, focused screening efforts. 5 Despite this ongoing debate, there is also widespread agreement that the focus of much R&D effort is now on hard-to-treat, rare diseases, often exploring new areas of biology and novel target classes that may offer greater commercial and therapeutic value in terms of differentiation in the marketplace.3,5 For these less established protein targets, knowledge-based screening is very difficult, since the knowledge with which to design focused compound sets simply does not exist. In these cases, diversity screening at large scale becomes invaluable in identifying starting points.
One such novel area of biology is epigenetics. Covalent modification of histones by epigenetic enzymes such as histone acetyl transferases, histone deacetylases (HDACs), and histone demethylases (HDMs) is crucial for transcriptional regulation. 6 Disruption of these processes has been linked to numerous disease states, particularly in the areas of oncology and inflammation.7,8 The best understood of these enzymes are the HDACs, with compounds such as Vorinostat already marketed. The HDMs, the largest class of which is the Fe2+, O2 and α-ketoglutarate–dependent JmjC domain–containing (Jumonji) histone lysine demethylases, 9 on the other hand, are less established and clinically unproven as tractable drug targets. Diverse screens to identify chemotypes for use as tools in cellular mechanistic studies are therefore essential to improve understanding of these enzymes. This has recently been demonstrated by the disclosure of GSK-J1, a selective KDM6 inhibitor with anti-inflammatory activities derived from the output of a high-throughput screen. 10
A number of fluorescent assay formats are available for HTS of JmjC demethylases. Assays that generate a fluorescent signal by coupling through formaldehyde, a product of the demethylase reaction, have previously been described. These assays either use formaldehyde dehydrogenase linked to NADH detection or detection of formaldehyde via acetoacetanilide. 11 Both formats, however, are likely to suffer from interference from autofluorescent compounds due to the blue spectral region of the detection signal. Indeed, a screen of JmjD2e, a JmjC family demethylase, using the NADH detection method yielded a very high hit rate >10%, of which the majority were shown to be fluorescent false positives. 12 Amplified Luminescent Proximity Homogeneous assay (AlphaScreen) formats have also been reported for screening of JmjC demethylases 13 but are prone to interference from compounds that scavenge singlet oxygen. Although these formats are easily miniaturized and suitable for HTS, the high potential false-positive rates can lead to a lack of confidence in the output, and stringent deconvolution strategies are required to identify true positives.
Mass spectrometry offers an attractive alternative to fluorescent assays for screening of JmjC family demethylases, because relative methylation levels can be monitored directly based on mass, 14 without the risk of fluorescence interference. We have previously reported on the application of RapidFire (Agilent Technologies, Lexington, MA) high-throughput mass spectrometry to screening of ~100,000 compounds against JmjD2c. 15 However, despite the throughput advances of the RapidFire system relative to liquid chromatography–mass spectrometry (sample times of ~8 s/well),16,17 the system is still not adequate for large-scale HTS in a reasonable time frame. One could enable HTS by RapidFire with access to multiple systems in parallel; however, it is unlikely that capital investment in a single technology at this scale would occur within the pharmaceutical industry today. Another option to increase throughput for RapidFire screening is to pool multiple test compounds in a single well. This is an approach that has been used successfully by a number of groups. 18 The additional time taken to deconvolute the output of a pooled screen can be minimized through the use of so-called orthogonal pooling, whereby compounds are present in multiple pools, and statistical hits are derived from compounds that flag as active in more than one pool. 19 This approach does, however, require complex information technology solutions and compound management logistics, which are not always available and would take significant time and investment to implement, making this solution less than ideal. In addition, the act of pooling compounds serves to reduce the effective screening concentration, which is not helpful when screening novel targets that are likely to have low hit rates. Finally, the screening set must be distributed into pools that do not combine components that have chemical reactivity with each other, a nontrivial optimization exercise.
Here we describe an alternative, straightforward, and scalable method for increasing throughput on the RapidFire system, using JmjD2d as a model system. In the approach described here, a panel of histone H3–derived peptides, each differing by just one or two amino acids, and trimethylated at lysine 9 was generated. The peptides are all substrates of JmjD2d and bind to the enzyme with equal affinity but differ sufficiently in their mass, so they can be differentially detected by mass spectrometry. A RapidFire assay was established with analytical methods for all peptides in parallel, such that multiple compound plates could be assayed against JmjD2d with different substrates and then pooled for analysis (see
Materials and Methods
Materials
Oxidized 3-acetyl pyridine adenine dinucleotide (APAD+), reduced 3-acetyl pyridine adenine dinucleotide (APADH), Pseudomonas putida formaldehyde dehydrogenase (FDH), bovine serum albumin (BSA), ascorbic acid, and α-ketoglutarate were all purchased from Sigma-Aldrich Ltd. (Gillingham, Dorset, UK). All buffer components were from Sigma-Aldrich Ltd. unless otherwise stated. Assay buffer was 50 mM Hepes, 100 mM NaCl (pH 7.0). RapidFire assays were performed in 384-well V-base polypropylene plates, whereas FDH assays were performed in 384-well low-volume black plates (both Greiner Bio-one, Stonehouse, UK). Iron (II) sulfate (Fe2+) and solvents for mass spectrometry were from Fisher Scientific (Loughborough, UK). JmjD2d was cloned, expressed, and isolated in house (GSK, Stevenage, UK). Trimethylated peptides were supplied by Cambridge Research Biochemicals (Cleveland, UK).
Methods
H3K9me3 peptide synthesis
Peptides were supplied by Cambridge Research Biochemicals. In detail, the protected peptides were assembled on a solid-phase synthesizer using either preloaded Wang or a RINK amide resin and used standard Fmoc synthesis protocols. The crude peptide was obtained after cleavage from the resin with a mixture of trifluoroacetic acid (TFA), triisopropylsilane, and water (95:2.5:2.5) for 3 h at room temperature and was then purified using a C18 reverse-phase column using a 0.1% TFA-buffered water/acetonitrile gradient. The resulting fractions, which were >95% pure by analytical high-performance liquid chromatography (HPLC) and giving the correct molecular weight (mw) (by matrix-assisted laser desorption-ionization time-of-flight [MALDI-TOF] mass spectroscopy), were analyzed, pooled, and freeze dried. The final material was analyzed by HPLC and MALDI-TOF mass spectroscopy.
The trimethyl lysine was incorporated into the sequence during assembly as Fmoc-Lys(TriMe)-OH. This was made by reaction of a suitably protected α-amino lysine derivative with an excess of methyl chloride or bromide to form the quaternary salt.
FDH coupled assay
This assay couples the formaldehyde product of JmjD2d to FDH, resulting in the oxidation of formaldehyde to formate as APAD+ is reduced to APADH. The reactions were monitored following an increase in fluorescence intensity (λex = 360 nm, λem = 485 nm) associated with the formation of APADH under initial rate conditions. Assays contained 50 µM ascorbate, 50 µM Fe2+, 200 µM α-ketoglutarate, varied concentrations of trimethylated peptide substrate, 1 µM JmjD2d, 1 mM APAD+, and 0.004 U/µL FDH in a total reaction volume of 15 µL. Enzymatic rates in arbitrary fluorescence units per unit time were converted to quantity of product formed per unit time using an APADH standard curve. Measurements were made using a Tecan Ultra Evolution instrument (Tecan Group Ltd, Seestrasse, Switzerland).
RapidFire mass spectrometry assay
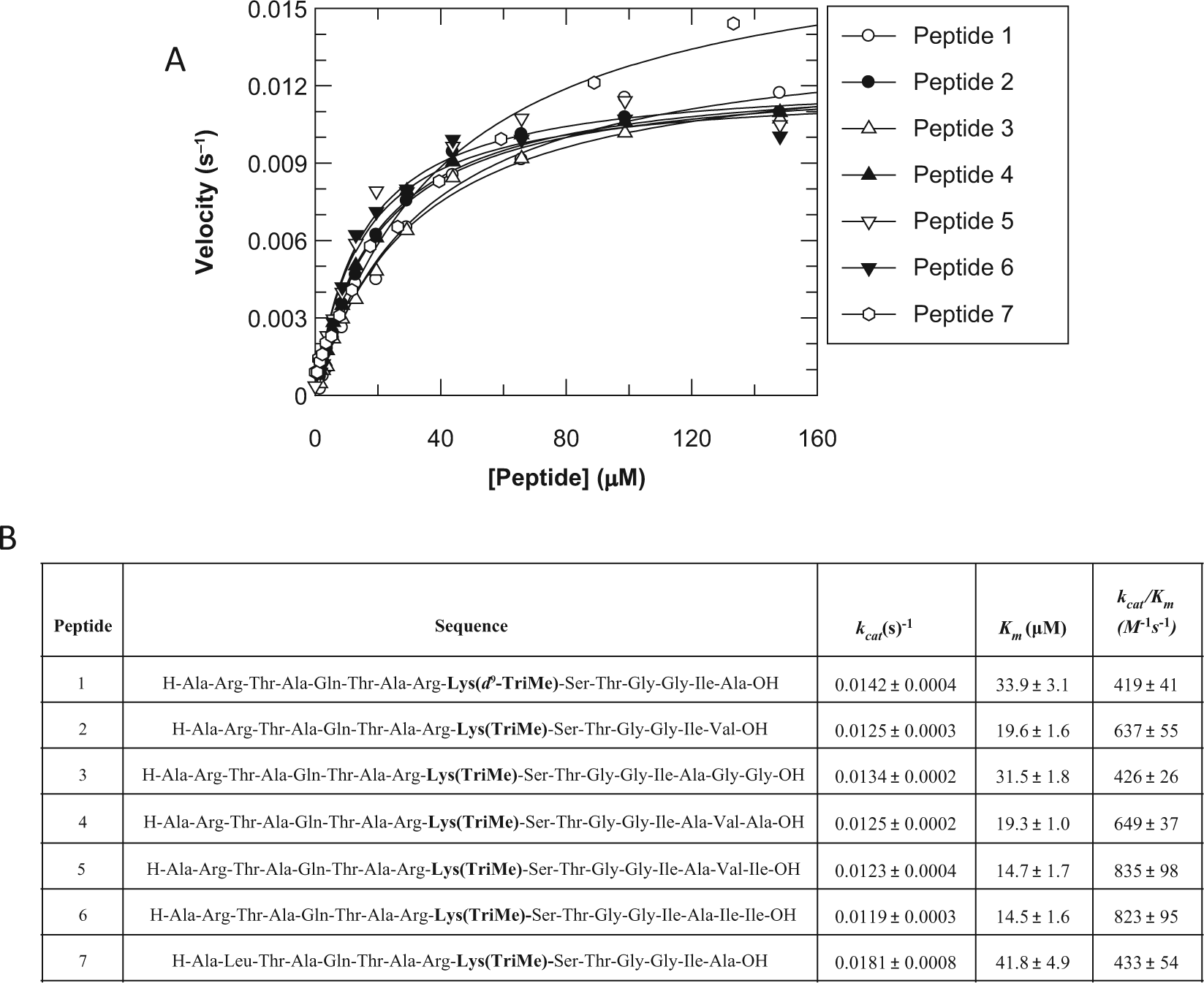
This assay monitors demethylation of a histone H3 peptide containing trimethylated K9 by recombinant JmjD2d enzyme. Assays were performed either with peptide 1 ( Fig. 1 ) as the substrate for all plates tested (referred to herein as the single-peptide assay) or, in the case of the multiplexed assay, with separate plates each screened with one of the multiplex peptides 2 to 7 ( Fig. 1 ) as the substrate and pooled for analysis. Four different peptides were used in the final multiplex assay format; where more than four compound plates were tested, they were divided into batches split between the four peptides.
(
Compound plates were prepared by transferring 100 nL of test compound in DMSO into 384-well V-base polypropylene plates using an Echo 555 acoustic dispenser (Labcyte, Sunnyvale, CA). For dose-response experiments, compounds were prepared in a 3-fold, 11-point serial dilution from a 100-µM final assay concentration. For single-concentration testing, compounds were screened at either 10 µM or 100 µM. In addition, 100 nL DMSO was added to columns 6 and 18 of the plates for use as controls.
Initially, 5 µL of an enzyme solution containing 1.5 µM JmjD2d and 0.5 mg/mL BSA in assay buffer was added to plates, to give final concentrations of 750 nM JmjD2d and 0.25 mg/mL BSA. Plates were centrifuged at 1000 rpm for 1 min and then incubated on the bench for 10 min at room temperature. To initiate the reaction, 5 µL of a substrate solution containing 100 µM ascorbic acid, 20 µM α-ketoglutarate, 100 µM Fe2+, and 50 µM of the relevant H3K9me3 peptide, all diluted in assay buffer, was added to all wells. This gave final assay concentrations of 50 µM ascorbic acid, 10 µM α-ketoglutarate, 50 µM Fe2+, and 25 µM peptide. Plates were centrifuged at 1000 rpm for 1 min before being incubated on the bench at room temperature for a further 9 min. All additions were performed using a Multidrop Combi (Thermo Fisher Scientific, Waltham, MA).
In the case of the single-peptide assay, 30 µL of 0.5% TFA was then added to all wells to stop the reaction and plates transferred directly to the RapidFire for analysis. In the case of the multiplexed assay, 5 µL of 1% TFA was added to all wells to stop the reaction. Then, 10 µL from each of the four plates containing a different peptide substrate was pooled into a fresh 384-well V-bottom polypropylene plate using the Biomek FX (Beckman Coulter, Wycombe, UK), giving a final volume of 40 µL. This plate was transferred to the RapidFire for analysis. For assays with greater than four plates, plates were divided between the four peptides, and one pooled plate was generated per four compound plates. The total analysis time for four compound plates was 3 h and 28 min in the single-peptide assay and 52 min in the multiplex assay.
Once transferred onto the RapidFire system, 10 µL of sample was aspirated directly from quenched assay plates and loaded onto the micro-scale C4 solid-phase extraction (SPE) cartridge (Agilent Technologies) to remove buffer salts with HPLC-grade water containing 0.09% formic acid and 0.01% TFA in a 3-s wash cycle. Analytes were then coeluted into the mass spectrometer in a 3-s elution cycle using 80% acetonitrile containing 0.09% formic acid and 0.01% TFA. TFA can result in suppression of the mass spectrometry signal but was included since it was found to enhance retention on the SPE cartridge and improve peak shape at low concentration. H3K9me3 peptide substrate and H3K9me2 peptide product were monitored on a Sciex API triple quadrupole (QqQ) mass spectrometer (Applied Biosystems, Concord, Ontario, Canada) in the positive electrospray ionization mode. The peptides used for this study were particularly resistant to collisionally induced fragmentation within the mass spectrometer
15
; hence, a quasi–multiple-reaction monitoring (MRM) method was developed for each peptide according to
Data Analysis
For RapidFire high-throughput mass spectrometry (HT-MS) assays, peaks were integrated using the RapidFire integrator software and percent conversion from H3K9me3 substrate to H3K9me2 product was calculated according to equation (1), where IA is the integrated peak area for the respective analyte.
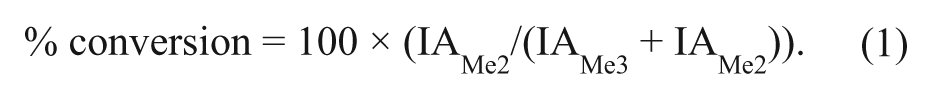
Dose-response curves were analyzed within ActivityBase software (ID Business Solutions Ltd, Surrey, UK), with data fitted to equation (2), using the raw data derived from equation (1):
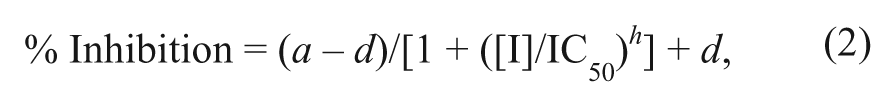
where a is the uninhibited value, d is the fully inhibited value, [I] is the inhibitor concentration, IC50 is [I] that gives ½ × (a – d), and h is the Hill slope.
For peptide Km determinations, data were fitted to the nonlinear curve-fitting programs of GraFit version 5.0.12 (Erithacus Software Ltd, Surrey, UK). Saturation curves were fitted to equation (3):
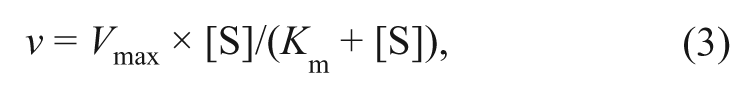
where Vmax is the maximum velocity (apparent kcat), [S] is the substrate concentration, and Km is the Michaelis constant.
For multiplexed assays, the data file exported from the mass spectrometer was first processed via a Microsoft Excel (Microsoft, Redmond, WA) macro to separate the data for each peptide substrate into a single file that was then processed via ActivityBase as above.
Results and Discussion
RapidFire HT-MS is an attractive assay format for the study of novel enzyme targets such as the JmjC family demethylases. It offers a direct, label-free readout of product formation with reduced risk of interference compared with alternatives and no requirement for antibody generation.16,17 However, the capacity of this system renders it a medium-throughput rather than a high-throughput technology. Fluorescent-based methods are therefore still employed for diversity screening of these targets. Although an HT-MS–based detection system provides the opportunity for multiplexing, this generally has been demonstrated only to increase the number of proteins that can be studied at any one time, rather than increasing the number of individual compounds screened.
Here we describe a novel method of multiplexing to increase compound throughput. A panel of peptide substrates for JmjD2d was designed to enable parallel assaying of four compound plates with four distinct peptides and subsequent pooling for RapidFire analysis. Using this approach, the throughput of the assay was increased 4-fold, offering the possibility of large focused screens or even high-throughput diversity screens to be run on this system.
Peptide Design and Selection
In a previous screen of JmjD2c using the RapidFire, an H3K9me3 peptide substrate was synthesized that gave optimal retention on a C4 SPE cartridge and excellent sensitivity in a QqQ mass spectrometer. 15 This peptide corresponds to the first 15 amino acid residues of histone H3, with K4 and K14 substituted by alanine and isoleucine, respectively, to reduce basicity and increase sensitivity. This peptide (peptide 1, Fig. 1 ) was also shown to bind JmjD2d. To enable the JmjD2d multiplexed assay described here, a panel of six additional peptides was generated, with sequences almost identical to peptide 1, but with an amino acid change at one or two residues to alter the molecular mass for differential detection by mass spectrometry. The additional peptides (peptides 2–7, Fig. 1 ) retain the reduction in overall molecular weight and basicity of peptide 1.
To enable any combination of peptides 1 to 7 to be used as substrates and the data pooled, the kinetic parameters of each peptide must remain unchanged despite the change in sequence. The FDH coupled assay was used to generate Km and kcat values for each peptide as this assay enables real-time monitoring of the reaction time course. The Km values were all within 2- to 3-fold of peptide 1, and the kcat were all ~0.01 s−1 ( Fig. 1B ). Changing the peptide sequence by one or two residues has, therefore, not altered the catalytic properties of the peptidic substrates beyond what is deemed acceptable for batch-to-batch variation. Assay conditions were, therefore, kept identical for all peptides for pharmacology testing.
Another important aspect of multiplexing peptides is the specificity with which the substrate can be detected on the mass spectrometer. As multiple peptides were analyzed simultaneously from a pooled sample, there had to be minimal crosstalk between mass ranges for each substrate and product. Initially, a broad set of seven peptides was designed with some overlap of mass between substrates and products—for example, the H3K9me3 mass m/z for peptide 5 overlaps with the H3K9me2 m/z for peptide 6. The set was then narrowed down through a series of optimization experiments to a final set of four, with no overlap in m/z for either the H3K9me2 or H3K9me3 masses (
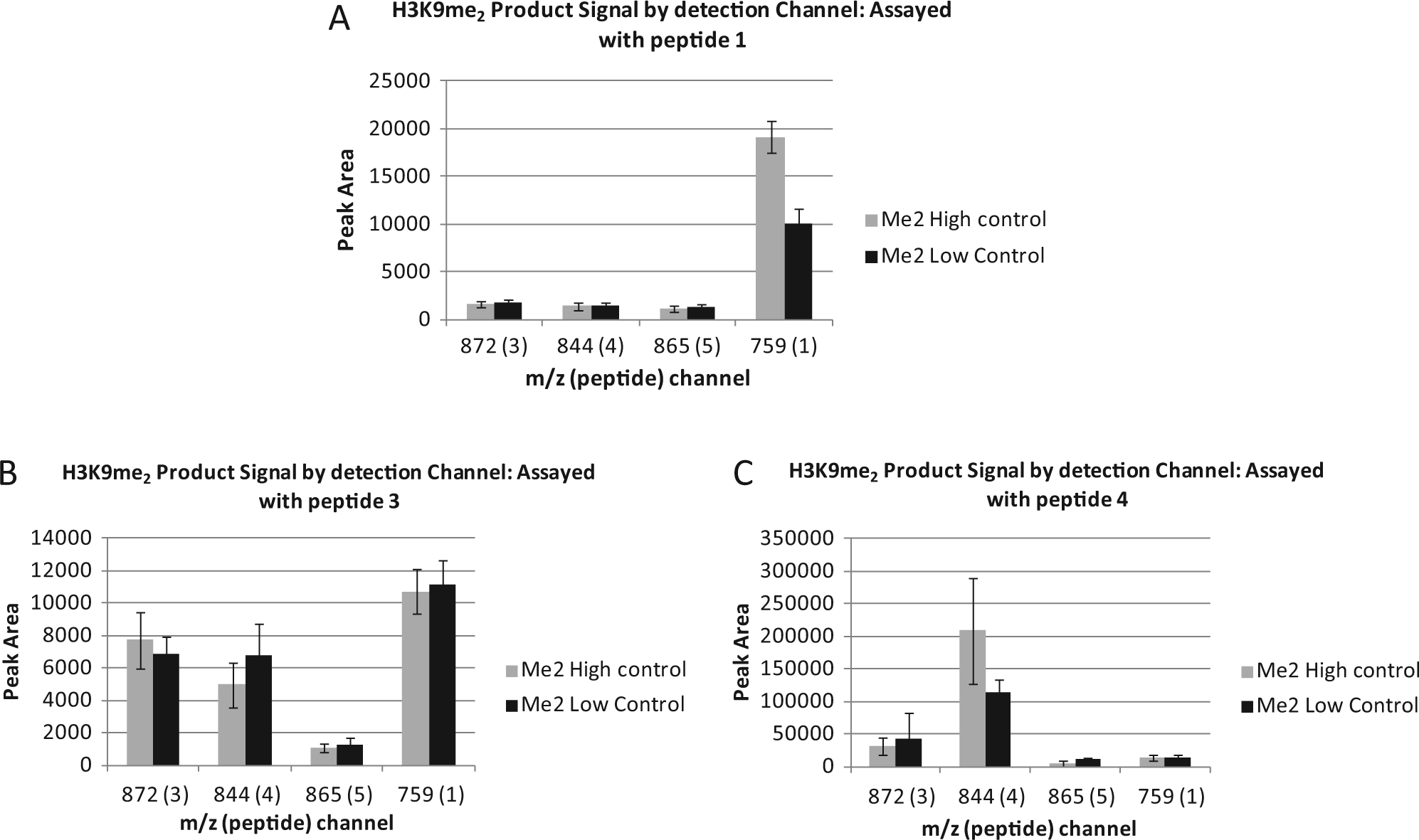
Analysis of crosstalk of H3K9me2 product signal between peptide m/z ratios. Peptides 1 (
Multiplexed Assay Development
Having determined that the kinetics of each multiplex peptide was comparable to that of peptide 1, the final assay conditions for both single-peptide and multiplexed assays were established. Both assays were run under identical peptide and α-ketoglutarate substrate concentrations, as well as equal concentrations of the cofactors ascorbic acid and Fe2+. One challenge when pooling samples is the effect of dilution on the detection of the analytes of interest. Peptide concentration could not be increased to overcome this dilution, as concentrations at ~Km for the substrates were required to ensure comparable pharmacology. An alternative approach was a reduction in volume of TFA added to stop each reaction, such that the final pooled volume was identical to that of the single-peptide assay (40 µL final volume). A reaction time course with each multiplex peptide was therefore performed, using a reduced volume of 5 µL of 1% TFA (compared with 30 µL of 0.5% TFA in the single-peptide assay) to stop the reaction. The results demonstrated that 5 µL of 1% TFA was sufficient to stop the reaction, and this volume and concentration were used for all subsequent multiplex assays (data not shown). Once pooled, this gave a total volume of 40 µL per well, identical to the single-peptide assay, to minimize the dilution effect.
Using these final conditions, the assays were compared for signal window and Z′. Under assay conditions previously established for the single-peptide assay (i.e., 500 nM JmjD2d), the multiplexed assay gave slightly poorer Z′ and lower signal to background (data not shown). This could be due to the small dilution effect still present when pooling samples, despite efforts to minimize this. In addition, the reduced quality could be due to lower signal and higher variability on the mass spectrometer when eight MRM transitions are monitored simultaneously, corresponding to product and substrate masses for all four multiplex peptides, as opposed to just two transitions in the single-peptide assay. This is because the dwell time for each individual mass transition must be reduced when multiple transitions are monitored, resulting in reduced peak definition. Finally, it is possible that the additional pipetting steps required for the multiplex assay add an additional degree of variability. To improve the robustness, the JmjD2d concentration was increased from 500 to 750 nM for both assays to ensure that the multiplex data were of sufficient quality for compound testing. This effectively increased the theoretical tight binding limit of the assay from 250 to 375 nM IC50. Although this may be of concern for structure-activity relationship (SAR) screening, it is less of a concern for hit identification and HTS, where assay robustness is more important. To reduce the protein requirement in the future, one could further optimize the peptide substrates such that they are no longer resistant to collisionally induced fragmentation and hence enable a true MRM transition method, rather than the quasi-MRM methods employed here. This approach has been successfully applied to H3K27 demethylases 21 and gave a much reduced assay background.
Compound Testing
To ensure consistent pharmacology of inhibitors within the multiplexed and single-peptide assays, a set of 110 compounds with varying potency against JmjD2d was assayed side by side. Figure 3 shows the correlation of pIC50 for this set. Potency and rank order are unaffected by the multiplex step, with a correlation coefficient of 0.98 between formats.
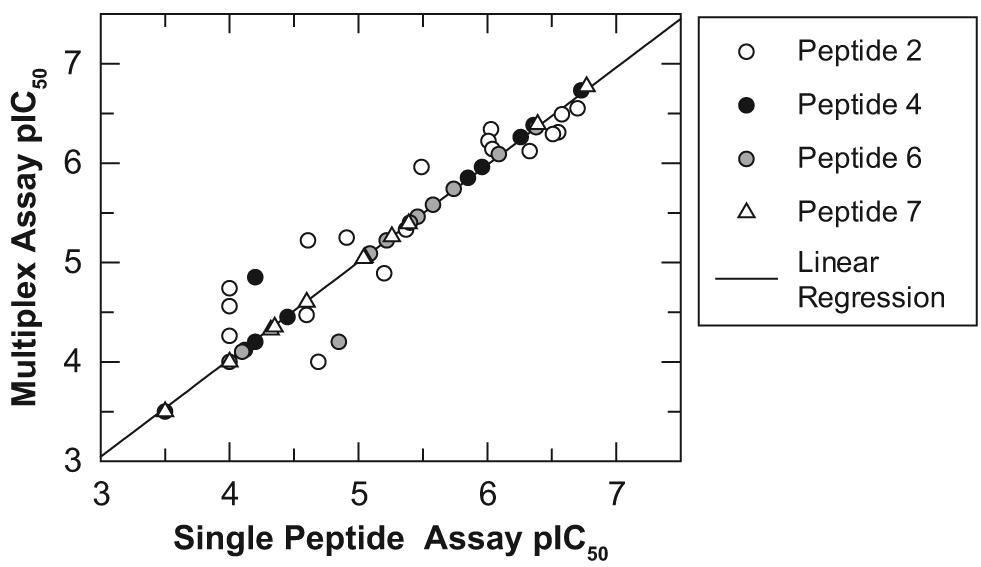
Correlation of pIC50 values for the JmjD2d RapidFire assay run with either a single substrate or a multiplex of four substrates. Four assay plates containing a range of JmjD2d inhibitors were assayed with peptide 1 added as the substrate to all four plates (referred to as single-peptide assay) or peptides 2, 4, 6, and 7 (legend shows which compound was assayed with which peptide in the multiplex) each added to one of the four plates and pooled in a multiplex. The correlation coefficient is 0.98. All assay conditions were as described in the Materials and Methods.
Although multiplexing could be useful to increase throughput and save machine time for weekly SAR compound profiling to generate IC50 data, the primary aim of this study was to determine whether the multiplexed assay could increase throughput for diversity screening of compounds at single concentrations. A set of 1408 diverse chemotypes (four complete 384-well plates, allowing for control wells) was therefore cherry picked from the GSK HTS collection and screened in both the multiplexed and single-peptide assays. Both assays performed well, with the multiplexed assay giving a mean Z′ of 0.72 and the single-peptide assay a mean Z′ of 0.85. However, the hit rate of this diverse compound set was extremely low, with just one hit identified in both assays above a robust cutoff of 25% inhibition at a 10-µM compound concentration (data not shown). This set was therefore not suitable to determine whether the same hits would be identified using the multiplexed or the single-peptide method. The data generated from this set do, however, give confidence that the multiplex approach is viable for single-shot screening, as the assay was robust and no false positives were identified. In addition, this extremely low hit rate strengthens the argument for high-throughput, large-scale diversity screening as a means to identify leads for novel targets such as JmjD2d, suggesting that screening a focused subset rather than a full HTS collection could result in one missing the few active compounds that may reside within that collection. Designing specific knowledge-based sets is also challenging for such targets, when little is known about their biology and few tool compounds exist. Establishing methods to increase throughput of technologies like RapidFire, such that they could be used to run large-scale HTS campaigns, is therefore essential to help drive chemistry efforts for these novel targets.
Since the small diversity set contained too few actives to validate the multiplexed assay, a second experiment was performed using DMSO plates spiked with known inhibitors of JmjC family demethylases. Compounds such as 2,4-PDA 20 and members of the 8-hydroxyquinoline series 12 were included at either 10 or 100 µM. Compounds were spread across four plates and spiked in 14 random locations. Figure 4 shows the correlation of activity of this spiked set between the multiplexed and single-peptide assays. Of the 14 spiked wells, 13 were identified as hits in the multiplexed assay, whereas all 14 were identified as hits in the single-peptide assay. The compound that failed to be identified in the multiplexed assay was a weak JmjD2d inhibitor (IC50 > 10 µM). This compound gave 15% inhibition at 10 µM in both assays, but since the robust cutoff (calculated from the mean response of the samples plus 3 standard deviations) was 18% in the multiplexed assay compared with 14% in the single-peptide assay, this compound was not flagged as a hit. Despite this slightly increased variability when screening in a multiplex and the risk of missing very weak compounds, a robust cutoff of 18% would still be considered acceptable for hit identification. Overall, the identification of spiked compounds was excellent in both assay formats, giving a correlation coefficient of 0.90.
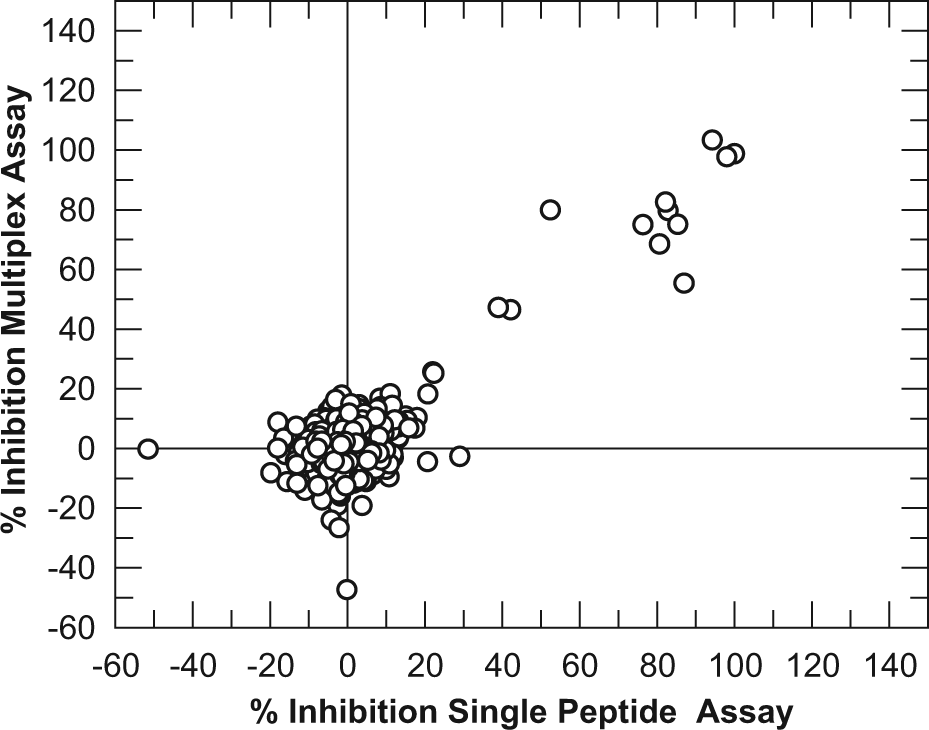
Correlation of % inhibition data for the JmjD2d RapidFire assay run with either a single substrate or a multiplex of four substrates. Four 384-well assay plates were spiked in random well locations with 100 nL of either 10-mM or 1-mM solutions of one of three known JmjD2d inhibitors. All other wells contained 100 nL DMSO. Plates were then assayed as per the Materials and Methods, with either peptide 1 added as the substrate to all four plates (referred to as single-peptide assay) or peptides 2, 4, 6, and 7 each added to one of the four plates and pooled in a multiplex (referred to as multiplex assay).
Actives from the spiking experiment were followed up to dose-response testing to confirm IC50 values. Figure 5 shows that the IC50 for 2,4-PDA in the multiplexed assay and the single-peptide assay are very consistent, at 0.5 and 0.3 µM, respectively. Overall, the data suggest that the multiplex approach is suited to identifying genuine inhibitors of JmjD2d from a diverse compound set screened at a single concentration.
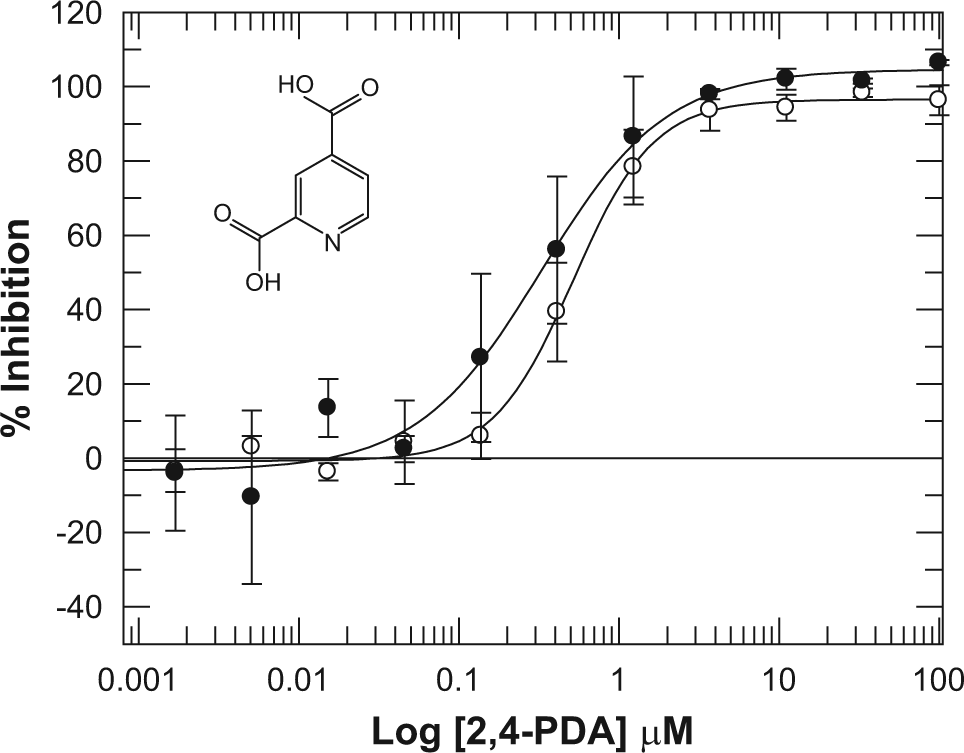
Dose-response curves for 2,4-pyridinedicarboxylic acid (2,4-PDA) in the JmjD2d RapidFire assay run with either a single substrate (●) or in a multiplex of four substrates (○). All conditions were as described in the Materials and Methods.
In conclusion, this study has demonstrated that RapidFire HT-MS assays lend themselves well to multiplexed analysis, and such methods could be used to improve compound throughput for focused and diversity screening. In this example, overall throughput was improved by 4-fold. Using two RapidFire instruments in this way, weekly capacity of a screening laboratory could be as high as 280,000 compounds relative to just 70,000 in a standard assay, enabling a high-throughput screen of 1 million compounds to be completed in fewer than 4 working weeks. Although in this example, four peptides were combined in a multiplex, as a proof of concept, one could envisage combinations of greater numbers of peptides to further increase capacity. The upper limit on peptide numbers would be determined by balancing assay robustness, dilution effects, and the maximum number of MRM transitions that can be simultaneously monitored.
This method of multiplexing could be easily adopted for any RapidFire assay that uses a peptide substrate, including numerous other epigenetic enzymes. The approach maximizes the quantity of data that can be generated from a single instrument, ensuring the most cost-effective and efficient use of equipment. Of course, the single-target, single-compound concentration multiplex approach described here does not preclude the additional application of multiplexing to assay multiple proteins simultaneously for panels of related proteins such as the cytochrome p450 enzymes. 22 In addition, there is the possibility of assaying different compound concentrations with different peptides and pooling to generate a dose-response curve in a single well, as has been reported by other groups. 23 All of these methods of multiplexing serve to increase throughput in different ways and address challenges at different stages of the lead discovery process.
There are of course alternative approaches to achieve the same outcome. The use of tandem mass tagging has been described for up to six-plex analysis, for example. 24 The method described here offers the advantage of lower cost than these alternative techniques, although assay development times are longer due to the substrate optimization required. There is also the possibility of adding nonpeptidic functional groups to the peptide core to generate mass differences while retaining the original core peptide 1 substrate. This may lower the risk of altering the kinetic properties of the substrate. However, the current method offers the advantage that the chemical properties of the various peptides are unchanged, and hence identical RapidFire and mass spectrometry methods can be applied to all with little additional optimization. These different approaches in reality could be used in combination and will suit different targets and assay types.
In summary, there is currently a paradigm shift toward screening with information-rich, physiologically relevant label-free assay techniques, such as mass spectrometry, to enable assays for more novel targets. These emerging technologies offer many advantages but share common throughput limitations that, until recently, have precluded their suitability for HTS. Advances in methods to increase the throughput of these techniques, such as the approach described here, are vital to enable diversity screening of these novel targets, identify tool molecules, and build understanding of their biology and therapeutic potential.
Footnotes
Acknowledgements
We thank Stuart Baddeley, Steve Rees, and Penny Smee for intellectual input and for supporting the project. We also thank Sue Westaway, Jack Brown, Phil Humphreys, and other members of the Epinova DPU for providing test compounds, chemistry input, and program leadership.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.