
Abstract
Drug discovery programs are moving increasingly toward phenotypic imaging assays to model disease-relevant pathways and phenotypes in vitro. These assays offer richer information than target-optimized assays by investigating multiple cellular pathways simultaneously and producing multiplexed readouts. However, extracting the desired information from complex image data poses significant challenges, preventing broad adoption of more sophisticated phenotypic assays. Deep learning-based image analysis can address these challenges by reducing the effort required to analyze large volumes of complex image data at a quality and speed adequate for routine phenotypic screening in pharmaceutical research. However, while general purpose deep learning frameworks are readily available, they are not readily applicable to images from automated microscopy. During the past 3 years, we have optimized deep learning networks for this type of data and validated the approach across diverse assays with several industry partners. From this work, we have extracted five essential design principles that we believe should guide deep learning-based analysis of high-content images and multiparameter data: (1) insightful data representation, (2) automation of training, (3) multilevel quality control, (4) knowledge embedding and transfer to new assays, and (5) enterprise integration. We report a new deep learning-based software that embodies these principles, Genedata Imagence, which allows screening scientists to reliably detect stable endpoints for primary drug response, assess toxicity and safety-relevant effects, and discover new phenotypes and compound classes. Furthermore, we show how the software retains expert knowledge from its training on a particular assay and successfully reapplies it to different, novel assays in an automated fashion.
Keywords
Introduction
Phenotypic assays have increasingly become a staple for biopharmaceutical R&D, with over half of first-in-class drugs having been estimated to originate from phenotypic instead of target-based screening campaigns. 1 Among phenotypic approaches, image-based high-content screening enables discovery in more biologically relevant, cellular model systems and leveraging of spatiotemporal information missing from biochemical target-based assays. 2 More recently, new approaches such as the cell painting assay have taken a multiplexed approach employing several morphological stains—instead of specific protein markers tied to a preselected biological response—in order to capture a broader, more unbiased profile of cellular phenotypes.3,4 In addition to the challenge of performing such assays, the analytical demands of identifying and classifying the resulting large volume of images with such high information content prove to be a major impediment to the routine application of multiplexed assays. In such situations, traditional computer vision analysis pipelines are complex and investment-intensive, creating an analytical bottleneck even when only a few phenotypes are present.
The recent introduction of deep learning-based methods presents a promising alternative solution capable of analyzing complex data at the quality and speed required for routine pharma research applications. 5 Image-based assays lend themselves to the application of deep learning thanks to recent progress in applying deep learning for general image recognition. As recently shown,6–10 deep learning-based image analysis reduces the effort needed to analyze large amounts of complex image data fast enough and with sufficient quality to use for pharmaceutical screening. It also seems to work very well on complex phenotypic data such as that from cell painting assays with their holistic capability to relate phenotypic effects of compounds with known mechanism of action (MoA) to compounds with an unknown MoA. Despite the utility of such assays, their widespread adoption remains hampered by the lack of algorithmic expertise among the biologists involved in assay development, 2 difficulty in recruiting such specialists to drug discovery, 11 and, until recently, the absence of an enterprise-appropriate software solution that easily integrates into discovery science workflows and allows iterative development and rapid assessment of screening assays for production use.
Here, we report an innovative deep learning application for automating the image analysis of phenotypic screens that enables their broad implementation, acceleration, and scaling. The application, Genedata Imagence, generates pharmacologically meaningful phenotypes via a single workflow from assay development to production screening (
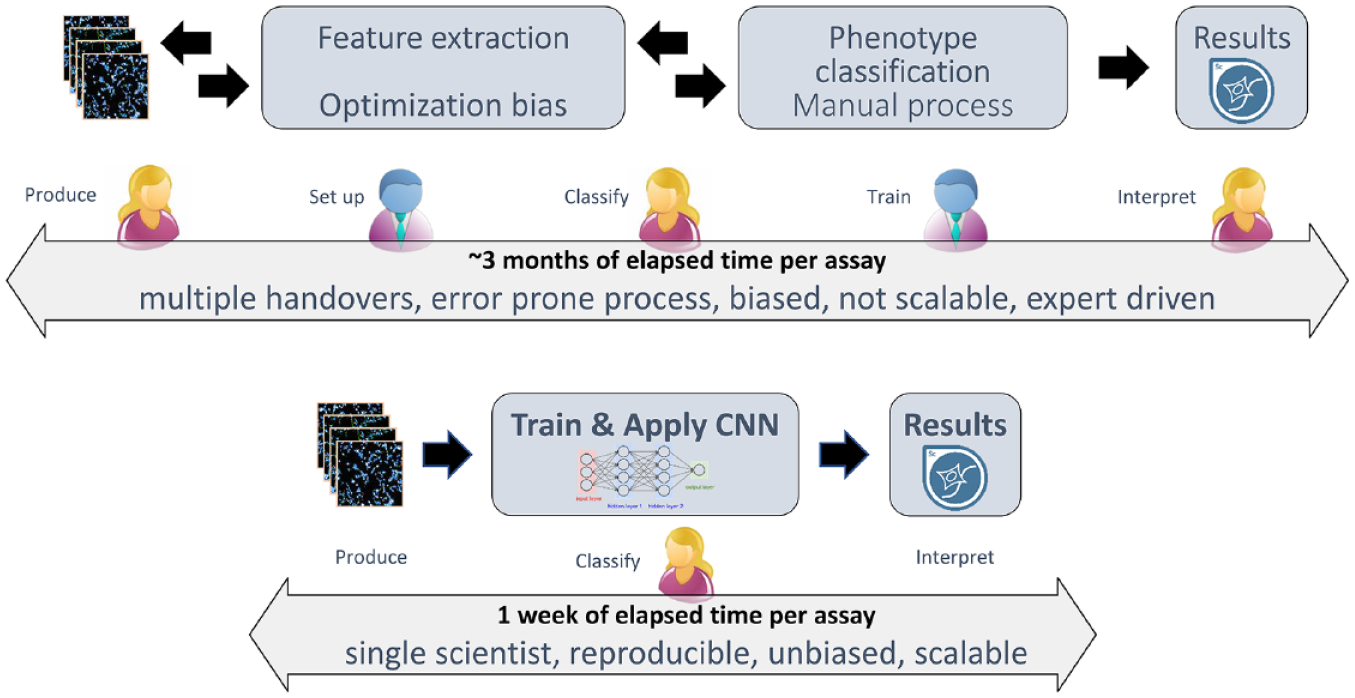
Comparison of classical HCS analysis workflow versus deep learning-based HCS workflow. In a classical HCS analysis workflow (top), establishing the analysis procedure is labor- and time-intensive. The work is usually split between distinct roles and people (assay biologists, yellow, and image analysis experts, blue) and involves several handovers. This workflow requires tight coordination and quality control to guarantee robust assay outcomes. In a deep learning-based HCS workflow (bottom), the same results can be generated by a single scientist in a fraction of the time. The scientist is responsible for training data generation and curation using the HCS images as reference, which is the only hands-on step in an otherwise automated workflow.
The application embodies five essential design principles, the implementation of which enables the everyday, routine production use of deep learning-based methods in discovery sciences: (1) insightful data representation, (2) automation of training, (3) multilevel quality control, (4) knowledge embedding and transfer to new assays, and (5) enterprise integration. Insightful data representation means the provision of human-interpretable representations of phenotypic space; automation of training includes both automated training set curation and network training to minimize hands-on time while maximizing the classification quality; quality control encompasses both the deep learning image analytical process and the pharmacological results derived from it, using typical screening visualizations and statistics to assess quality in a pharmacologically relevant context for drug discovery. We define knowledge transfer as the ability of deep learning-based solutions to embody learnings from previous training sessions toward the automated analysis of similar assays. Finally, enterprise integration refers to embedding this software and process with the infrastructure of global R&D organizations to use it across multiple sites, departments, and research groups.
Finally, we illustrate the benefits of applying such a solution to production-scale drug screens, on both a more conventional HCS receptor internalization assay and a more complex cell painting assay.
Materials and Methods
Software
Recently, we have reviewed deep learning applications in the life science and pharma domain and have assessed underlying development frameworks currently available in the public domain. 12 Our selection criteria encompassed agile software development and the ability to cope with a rapid algorithmic and intellectual turnover rate in this fast-moving field. From these, we selected TensorFlow and its family of high-level wrappers (Keras, TFlearn) for the de novo design of the classification network and its production deployment in Imagence. A distinct default pretrained convolutional neural network (CNN), which we term the “extraction CNN,” was tailored for an unsupervised feature extraction from high-content images.
Hardware
Algorithmic design and development were carried out on different workstations and servers with commodity hardware (Intel, Santa Clara, CA, XEON processor [10 cores], 128–512 GB RAM, 1–2 TB SSD hard drive and one high-end workstation graphic card [Nvidia, Santa Clara, CA, Titan X]). This setup is sufficient for processing image data from one 384-well microtiter plate (around 6500 images) to final results in approximately 1 h. On the same hardware, conventional image analysis using CellProfiler 13 takes around 1.3 h.
For production, a hardware setup with moderate state-of-the-art servers in a multiple graphics processing unit (GPU)-node configuration was applied (see
Image Data Set for Cell Painting Assay
Cell painting 8 is a morphological profiling assay that, instead of labeling molecular targets, multiplexes fluorescent dyes to reveal seven broadly relevant cellular components or organelles. Cell painting can be used to identify the phenotypic impact of chemical or genetic perturbations, grouping compounds and/or genes into functional pathways, or identify disease signatures. It can also be used to infer MoA through comparison of the phenotypes induced by compounds with unknown MoA with phenotypes of tool compounds of known MoA. We used the BBBC022 data set, which is publicly available as part of the Broad Image Data Base (https://data.broadinstitute.org/bbbc/BBBC022/). Typically, image analysis software like CellProfiler 13 is used to identify individual cells in the images and calculate ~1500 morphological features (various measures of size, shape, texture, and intensity) in order to produce a rich enough profile suitable for the detection of subtle phenotypes. The development of a robust image analysis algorithm generating such a rich profile can take more than a week. Using Genedata Imagence, this process can be shortened to a few hours.
Image Data Set for Receptor Internalization Assay
The neurotensin receptor 1 (NTR1) is a G-protein-coupled receptor. Upon activation the receptor is internalized into endosomes in a beta-arresting mediated process. The data shown in this publication stem from a screen for modulators of NTR1. 14 Briefly, the redistribution of β-arrestin-conjugated green fluorescent protein (GFP) was measured to assess the activation of NTR1.
Results and Discussion
Recent excitement around artificial intelligence stems from its capabilities in automated image classification, which outperform conventional computer vision methods by the introduction of deep neural networks with convolutional layers and appropriate training schemes. 15 In cell imaging, deep learning methods can replace the conventional approach of tedious and often highly biased manual selection of image analysis methods and pipelines to extract hundreds of cell features that are discriminative for a certain classification task. 16 For example, in a classical receptor internalization assay, the measurable translocation of the receptor upon signaling can be quantified by a set of manually engineered features specifying the relevant movement of a labeled receptor from the cell membrane to endosomes, for example, using a spot count measure within the cell body. These are steps that heretofore have required human expertise. The consequence of removing these steps is a significant reduction in data handovers and specialized intermediaries. Figure 1 contrasts a classical HCS analytical workflow with a deep learning-based HCS workflow and illustrates the advantages—including major time gains—of the latter.
A deep learning-based analysis holds the promises of increased speed and reliability at reduced complexity and dependency on image analysis experts. However, taking full advantage of these benefits necessitates a shift in how experts interact with their data, and requires their effective collaboration with artificial intelligence systems. To enable this shift and enable a more rapid adoption of deep learning-based workflows broadly across drug discovery, we have developed a set of five key design principles for an appropriate workflow and supporting software. In the next sections we present and discuss the rationale underlying these principles and show how these have been incorporated into Imagence, in the specific case of image-based phenotypic screening.
Automation of Training Data Generation and Insightful Data Representation
Applying deep learning to production-level high-content screens involves three main stages: (1) Generation of training data sets, a process that typically requires the assay biologist to manually classify images or—at minimum—curate an automatically proposed image set; for example, an experienced biologist might need to visually assess whether images represent a disease versus a nondisease state. (2) Training of the neural network on the training data set, which is an automated (hands-off) process. (3) Running images from a screening batch through the pretrained network. 3 In this process, the biologist spends time mostly on the first step, the training data generation. Therefore, once this process has been adopted, the greatest further efficiency returns can be gained from automating this process.
We therefore sought to develop a highly efficient image curation workflow that supports human decisions with the use of artificial intelligence and visual review tools (
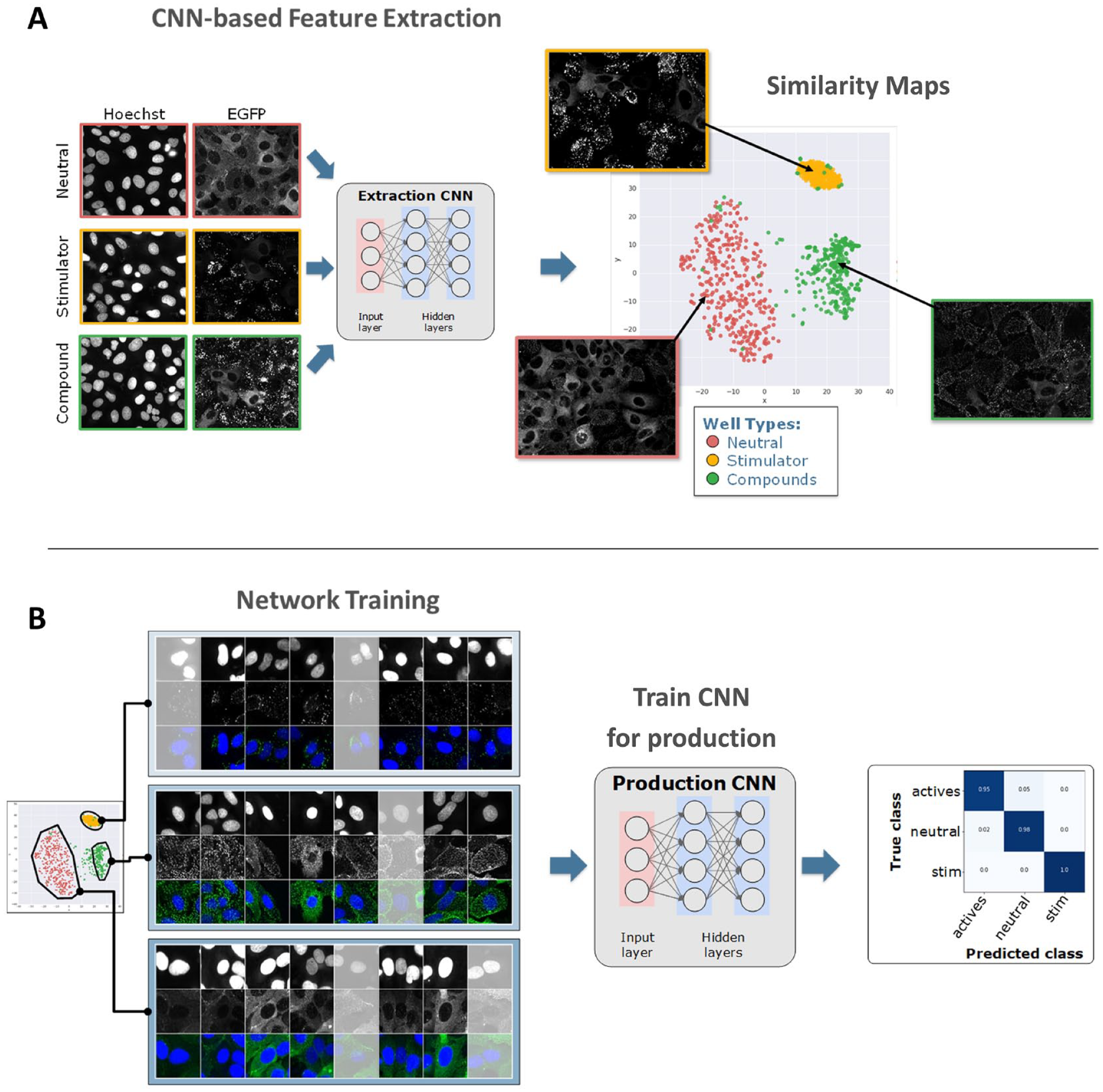
(
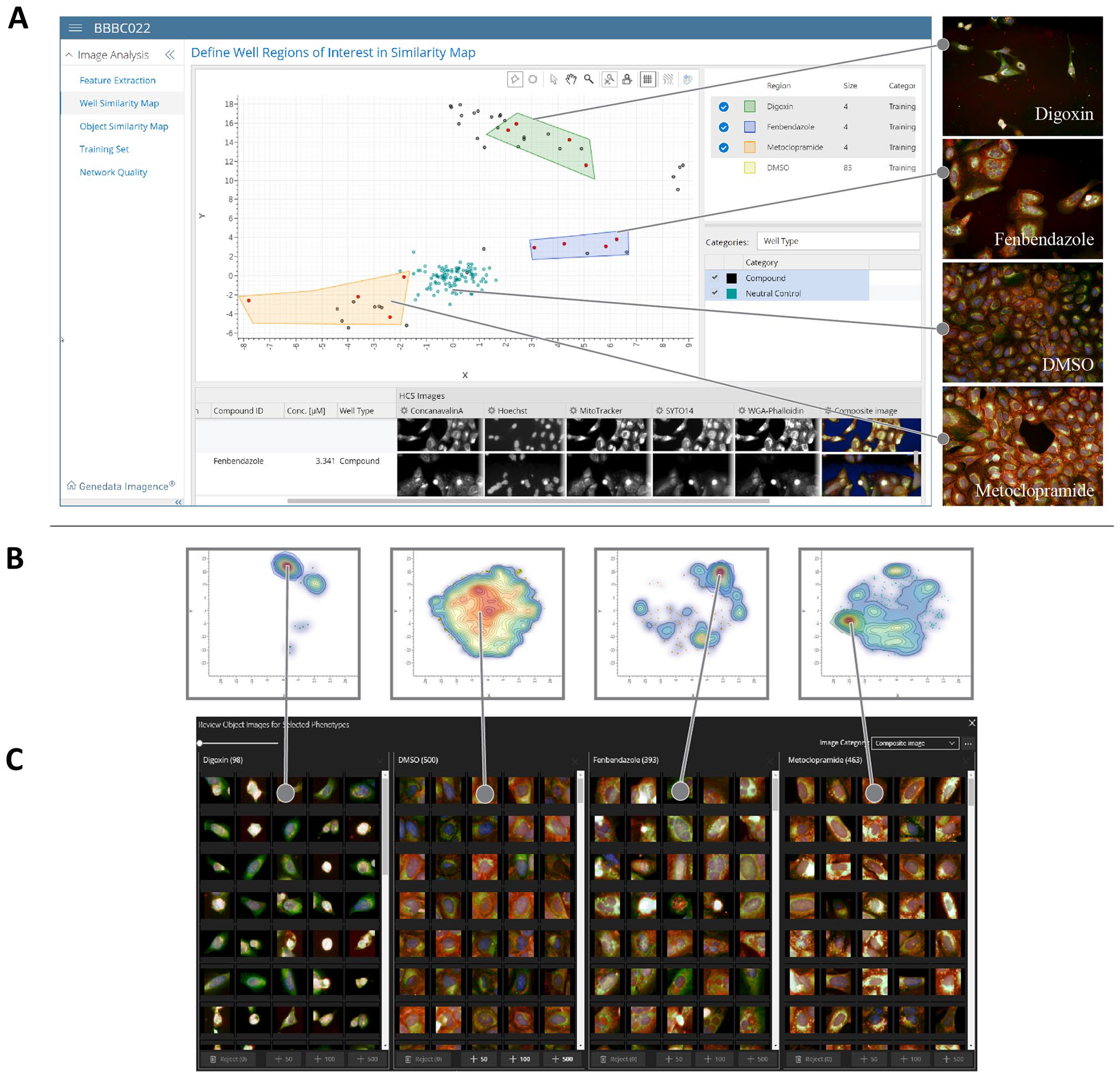
(
The class assignment finalized in this last curation step is used to train a classification or production network (
As an illustration of this workflow, we used a subset of the BBBC022 cell painting data set (
Quality Control: From Classification to Pharmacology
The desired quality of experimental results from a drug screening campaign mandates rigorous quality checks along the entire experimental and data analysis process. This begins with quality control of raw material (e.g., cell cultures) prior to experimentation and continues with postexperimental quality control of quantified responses (e.g., stability of a standard curve tested on each plate). For a machine learning application, a common form of statistical quality control is the holdout method, in which the data are split into different parts: a training set, a validation set, and a test set. The training and validation sets are used to train the network and quality is then assessed by the network’s classification accuracy on the test set. Best practice is to perform a variation of this procedure, where the data are split into multiple sets and the network is trained on each set while tested on the others, for a k-fold cross-validation.
17
The common visualization of such supervised learning quality is the confusion matrix where labeled holdout data are displayed in addition to the classifier outcomes (
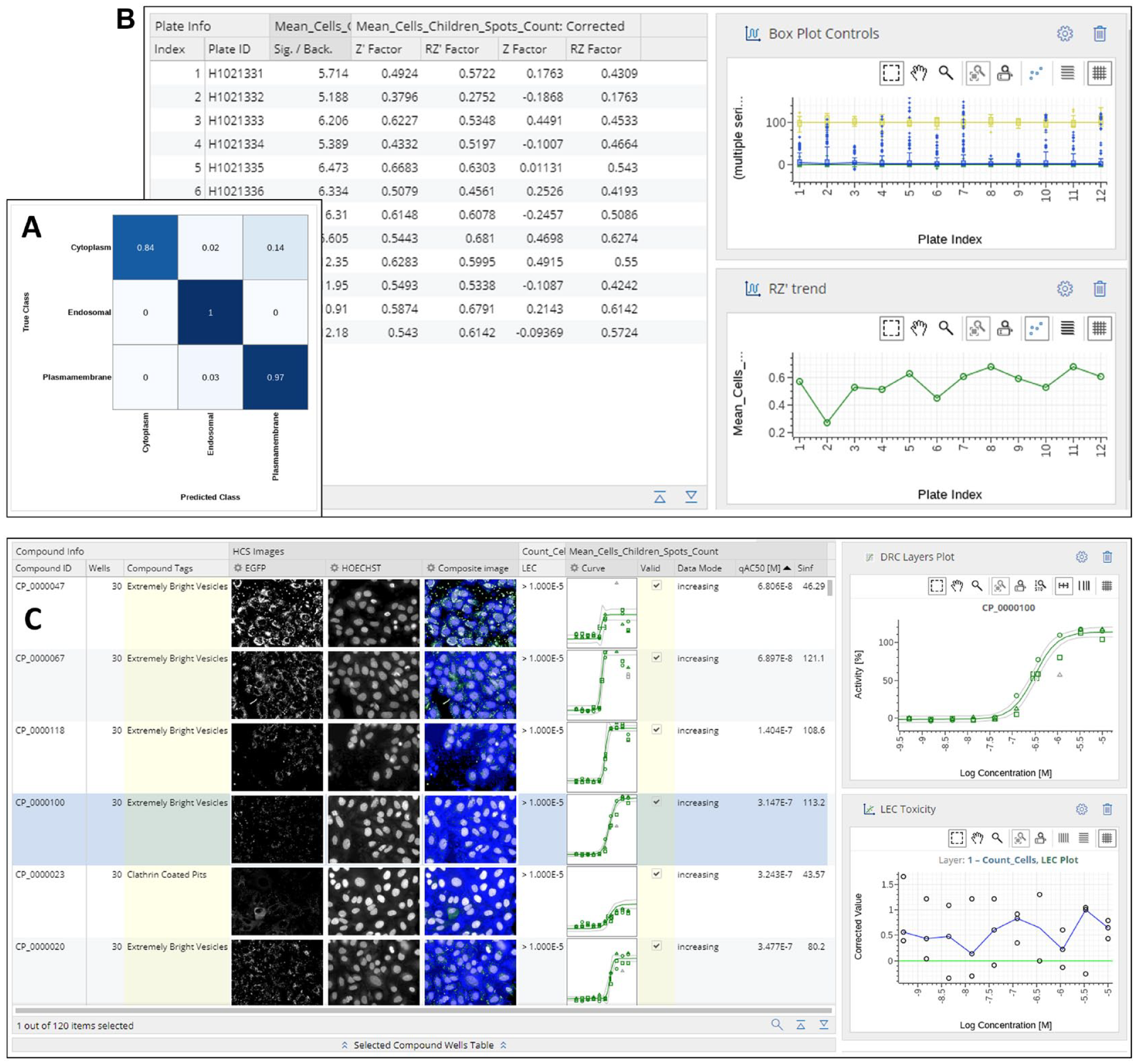
(
While these results serve as important controls for network quality, the abovementioned methods cannot be used to judge the adequacy of network training on accepted measures of pharmacology. Quite small differences in classification accuracy can have a huge impact on plate statistics and on many downstream parameters, such as the IC50 values, Hill slopes, or asymptotic plateaus in a concentration–response analysis.
3
This is even more important for drug screening, where the decision to move an assay from development to the production stage depends on clearing specific criteria, such as obtaining reliable and reproducible potency measurements for standard compounds or exceeding a given signal-to-background ratio or Z′-factor thresholds. In the workflow presented here, we fulfill this additional need by not only providing metrics on network quality (
Knowledge Transfer: Preservation and Reapplication of Analytical Knowledge
Generating training data with Genedata Imagence is an interactive, often explorative process. New assays may require more careful exploration, whereas for an established assay, curation may be much quicker if the human curator is very familiar with the expected results and visual appearance of phenotypes. However, even for an established assay, there are situations where conditions may be inconsistent and therefore difficult for a human curator to assess outcomes in a uniform, non-biased way, for example, in a screen performed on multiple cell lines, such as during panel screening, or a screen involving clinical materials such as patient-derived stem cells, where conditions are subject to greater variability. In order to reduce the time investment and introduction of potential bias by the human curator, we developed a workflow (“transfer learning workflow”) that preserves the knowledge of the human curator and reapplies this knowledge to new yet similar assays or conditions. The key element of this workflow is an incremental training data enrichment, which is used to retrain the network to adapt to a different domain (
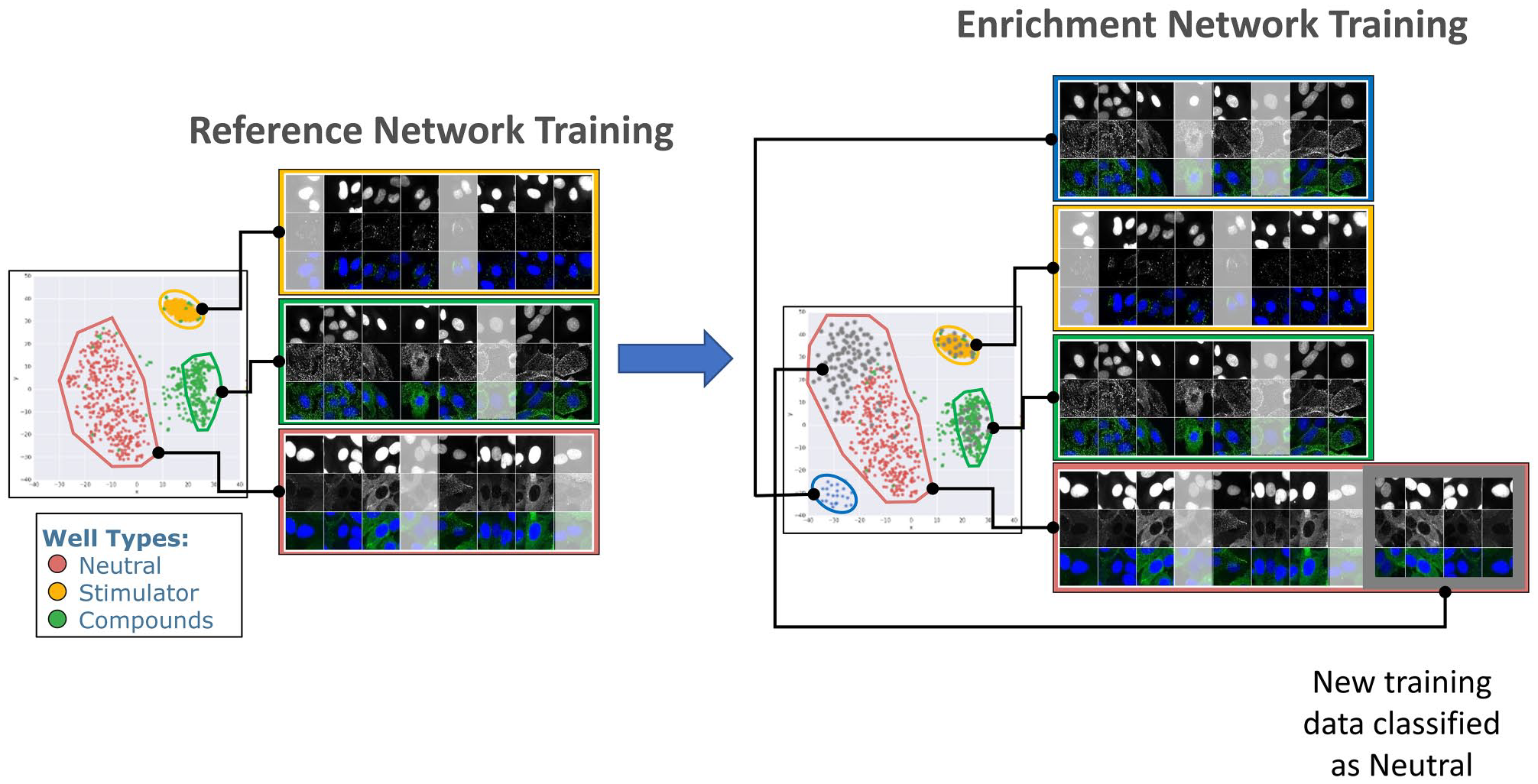
Transfer learning. Left: An initial reference network is trained on an established assay and clusters images into three classes: neutral, stimulator, and compounds. Right: Later, images from a second, related but distinct assay (gray points, images boxed in gray) are added to the initial training data set and the network is retrained. In this step, a new phenotype is discovered (blue dots), which was not present in the first data set and is used to form a fourth class. In addition, added data broaden the phenotypic space occupied by a previously identified neutral class (gray dots at the top left of the map). After this enrichment, data from the new assay can be related to the original assay, while the additional class serves for an additional endpoint for this assay.
Enterprise Integration
Due to the continued success of image-based screening technologies, enormous volumes of image data are often produced during image-based screens—a volume that is likely to grow exponentially, and with a growing interest in more physiologically relevant cellular models such as 3D organoids 19 and organs-on-a-chip, 20 image storage demands are only likely to further skyrocket. Compounded with this growth in image data volume, if more efficient CNN-based feature extraction and network training workflows such as those we have described above are used, there is the added complexity of managing and streamlining project data obtained across several labs or research groups.
The images generated in individual labs across an organization can be either stored and processed locally or transferred to centralized data centers with corresponding systems for image storage and analytics. An important consideration when deciding between these options is the potential expense versus speed of data transfer, as a typical requirement for image analysis systems is that they process the image data faster than they are acquired, in order to enable prompt reactions to experimental problems detectable only post-image analysis. To gain enough speed in a cost-efficient manner, we have implemented a high level of parallelization of both image transfer and computing, allowing the scaling of computational resources. Our architecture allows for a central server and one or more GPU nodes, with each GPU node in turn containing one or more GPUs (
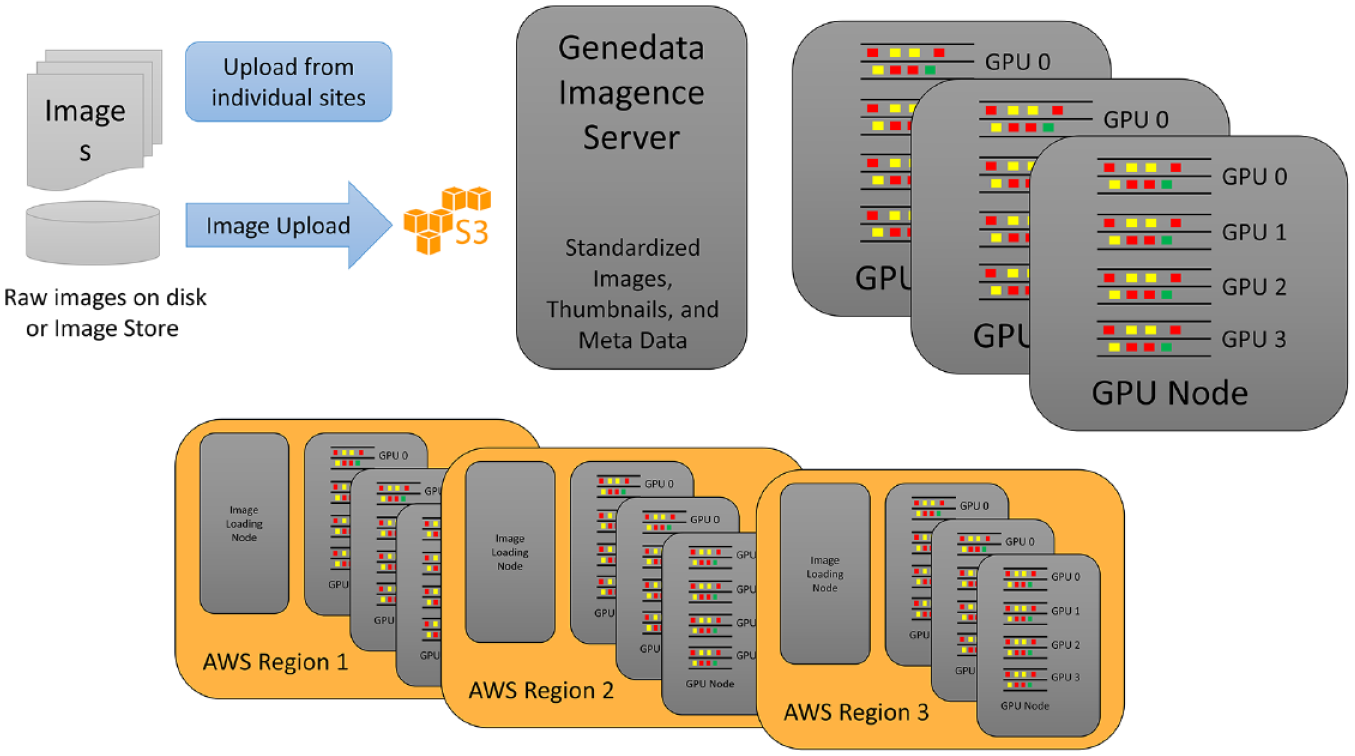
Infrastructure reflecting the individual and regional needs for an adaptive scaling of image transfer and computing. The main goal for this architecture is to enable two different scenarios. In the first, images are cheaply and efficiently transferred to central compute nodes that are mostly GPU driven (top). In this scenario, images are acquired by instruments and stored locally on the premises. They are transferred into a S3 bucket either by existing standard integrations (Cellomics, Thermo Fisher Scientific, Waltham, MA; HCS Connect, Molecular Devices, Danaher Corp., Washington, DC; MDC Store, PerkinElmer, Waltham, MT; Columbus, Yokogawa, Tokyo, Japan, CV7/8000 file-based image storage) or by custom uploaders. This S3 storage is close to the Genedata Imagence server. If, however, multiple geographic locations need to be served, it is less advantageous to transfer images over long distances; additional compute and image loading nodes can instead be configured for each geographic AWS, Amazon Inc., Seattle, WA, region, such that images are processed locally and network traffic is minimized (bottom). In both scenarios, the master Genedata server manages the deployment of the software on compute nodes, hosts the user interfaces, schedules jobs, and stores results.
Also important is to optimize the image upload process itself, including image annotation with standard metadata (e.g., file name, path, instrument, measurement date, and channel) and definition of further information like plate mapping and definition of wells containing experimental controls or a standard. In our solution, we have automated this process by constantly monitoring an image source such that immediately upon availability of a complete image set, it is uploaded and analyzed using the appropriate trained neural network. Results are then continually appended to an ongoing analysis session, allowing instantaneous real-time monitoring of the screening campaign. Business rules regarding plate and/or compound statistics can be applied automatically, which when violated trigger an email notification to the assay operator, alerting him of the assay issue and allowing him to react immediately.
A final key consideration when integrating an analysis workflow into daily operation is the sharing of validated results within and across organizations, such that they are available as quickly as possible to project teams, in an easily accessible and understandable form. Toward this end, our solution facilitates reporting to global data warehouses. We have also developed a framework of application programming interfaces supporting live access to any stage of the analysis session. Finally, our solution also allows the creation of Microsoft (Redmond, CA) Excel or Microsoft PowerPoint reports, for midterm sharing or presentational purposes.
To date, the approach we have described has been validated in more than 20 industry assays, including those involving full-production screening data sets (with hundreds of imaged microtiter plates).21,22 In these proof-of-principle projects, Imagence has generated excellent result quality, detecting biologically relevant effects with comparable or greater performance than classical methods, delivering at times superior plate and compound-level statistics. Our new approach has been shown to also be compatible with some of the most complex and analytically challenging assays now available in high-content analysis, including the cell painting assay. We view these initial successes as evidence that Imagence represents a step forward from the much discussed but abstract potential power of artificial intelligence to transform drug discovery, as a concrete realization of a real-world ready artificial intelligence-based analysis workflow—a view also recently articulated by Bio-IT World.23–26 Such a workflow is positioned to broaden the use of high-yield phenotypic assays in discovery screening and other life science applications; help discovery teams better exploit serendipitous findings; and lead to more rapid, relevant, production-level characterization of new molecules in drug discovery and development. Given these promising outcomes, we believe that the underlying principles of our solution might be adopted to also apply deep learning beyond image analysis across many domains of drug discovery, transitioning deep learning from a potentially powerful but still exotic approach to a mature, accessible, and integrated software enabler to the R&D of novel medicines.
Footnotes
Acknowledgements
We thank Dr. Oliver Duerr and Dr. Beate Sick for their technical help and assistance during the initial phase of this project. Additionally we would like to thank Dr. James Pilling and Dr. Dana Nojima for sharing industry relevant assay data and for being available for critical discussion during early phases of the project.
Declaration of Conflicting Interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: All authors are employed by Genedata AG (Switzerland) or Genedata Inc. (USA).
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The authors received financial support for the research, authorship, and/or publication of this article from the Commission for Technology and Innovation CTI, Switzerland.