
Abstract
Protein phosphatases play an essential role in cell signaling; however, they remain understudied compared with protein kinases, in part due to a lack of appropriate tools. In order to provide conditional control over phosphatase function, we developed two different approaches for rendering MKP3 (a dual-specific phosphatase, also termed DUSP6) activatable by light. Specifically, we expressed the protein with strategically placed light-removable protecting groups in cells with an expanded genetic code. This allowed for the acute perturbation of the Ras/MAPK signaling pathway upon photoactivation in live cells. In doing so, we confirmed that MKP3 does not act as a thresholding gate for growth factor stimulation of the extracellular signal-regulated kinase (ESRK) pathway.
In this commentary, we discuss the design principles considered in developing an optically controlled protein phosphatase, opportunities and limitations of the methodology, areas in need of improvement, and potential future applications.
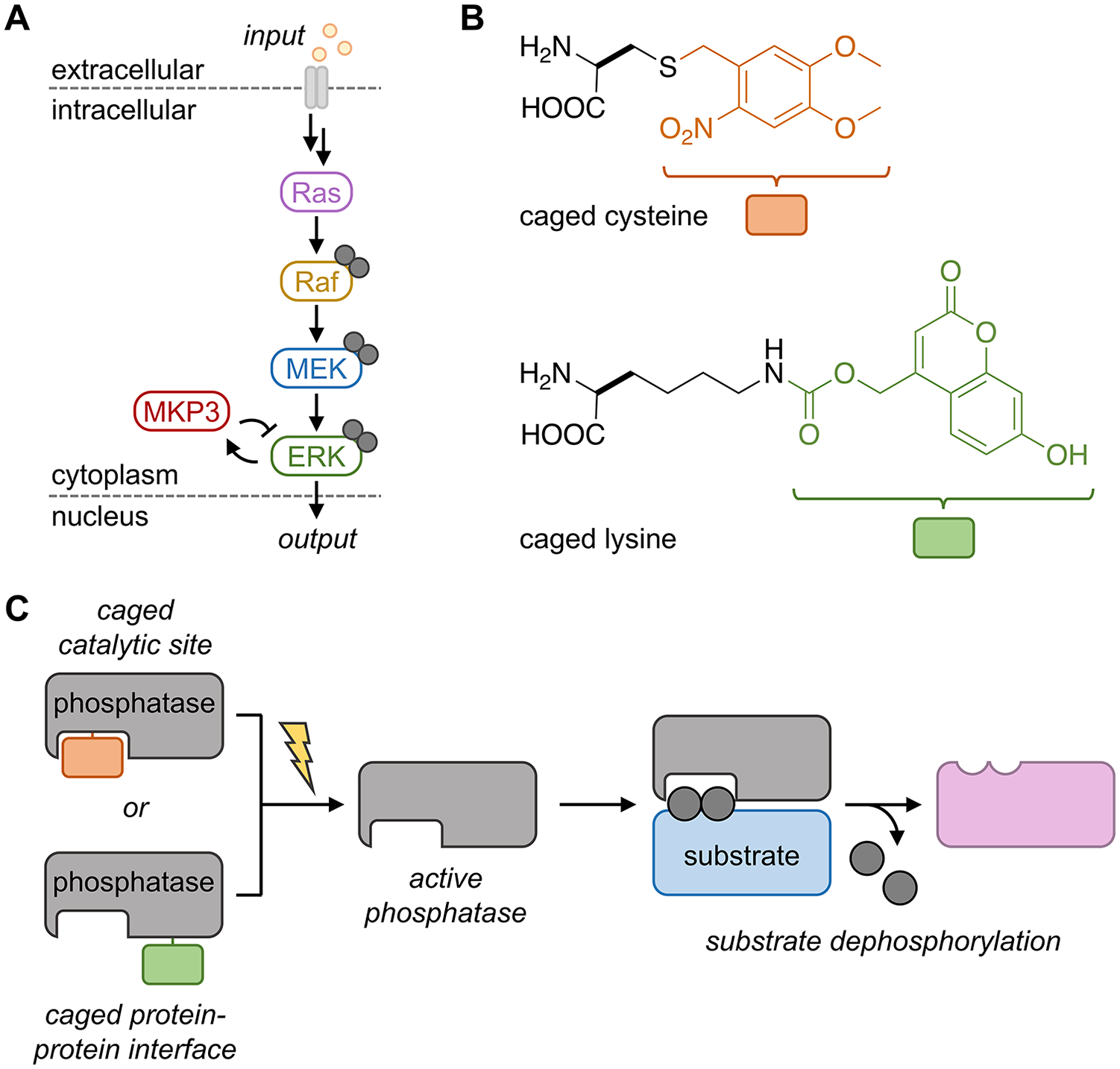
At the most basic level, signal transduction is the computing of extracellular stimuli, or inputs, and their conversion into a cellular response, an output. Protein kinases are likely the first enzyme class that comes to mind for their role in signal transduction, more specifically for their role in propagating phosphorylation throughout a pathway.1,2 However, protein phosphatases, arguably, play an equally important role in that they counteract kinases through the removal of a phosphate group. 3 Once thought to play an inferior role to kinases, the importance of phosphatases has become evident in recent years. Still, phosphatases remain understudied and poorly characterized compared with kinases. Traditional genetic tools, such as gene silencing or knockdown, have provided most of the knowledge that we have to date regarding protein phosphatase substrates and their various roles in disease. Two key factors contribute to the complexity and difficulty of studying protein phosphatases: (1) the detection of a negative signal (or removal of a phosphate group), which must first be installed by the appropriate kinase, and (2) the conserved, shallow active sites of phosphatases are not typically amenable to the design of specific inhibitors. 4 Despite these issues and due to their prevalence in disease, those up to the challenge have explored phosphatases as drug targets with some success. 5
Based on the difference in kinetics between cell signaling events (minute timescale) and traditional genetic tools (hour/day timescale), it became apparent that developing new tools for studying protein phosphatases might enable us to fill some of the existing knowledge gaps in signaling pathways. We envisioned that the development of an optogenetic protein phosphatase would afford acute activation of enzymatic activity in order to study the immediate effects of the engineered enzyme. Light is an excellent control element for cell signaling, as it can be readily adjusted in timing, location, wavelength, and amplitude. 6 To do this, we utilized a genetic code expansion technique in order to site-specifically introduce a photocaged amino acid at a defined site within our protein of interest. 7 A photocaged amino acid utilizes a photosensitive “protecting group” to mask the native amino acid side chain and render it inactive until irradiation with light cleaves the protecting group, thus generating the native amino acid residue—and thereby the native protein. In order to incorporate caged amino acids into a protein of interest, a tRNA/tRNA synthetase pair was engineered that recognizes the nonnatural amino acid and incorporates it into a growing polypeptide chain in response to an amber codon (TAG). This methodology has previously been applied for the generation of numerous caged protein kinases through incorporation of a caged lysine into the ATP binding pocket, thus blocking ATP binding and enzyme activity until photoactivation at a defined time point. 7
To adapt a similar strategy for protein phosphatases, we selected the dual-specific phosphatase 6 (DUSP6 or MKP3), which has high specificity for extracellular signal-regulated kinase (ESRK) over other mitogen-activated protein kinases (MAPKs), such as JNK or p38. Dysregulation of MKP3 expression (both overexpression and downregulation) has been implicated to play both oncogenic and tumor-suppressive roles in a range of cancers. Since the interaction between MKP3 and ESRK is well characterized, it provides an ideal starting point for proof-of-concept design. MKP3 functions to counteract the dual phosphorylation of ESRK (pESRK) by upstream kinases in the Ras/Raf/MEK/ESRK pathway (
(
MKP3 utilizes an essential cysteine to catalyze the dephosphorylation of pESRK through a direct nucleophilic attack onto the phosphate, transferring it from the target protein onto the thiol. Replacement of this catalytic residue with a caged cysteine (
Sequestration of ESRK by MKP3 is a result of the high-affinity (nanomolar range) interaction between the kinase interaction motif (KIM) of MKP3 and the docking groove of ESRK, and it is this interface through which kinase–phosphatase specificity is conferred. We hypothesized that a protein–protein interface could provide a second, complementary site for the caging of MKP3—and other phosphatases. Analysis of the crystal structure of ESRK2 bound to the KIM peptide of MKP3 (residues 60–76) revealed several potential residues to target for inhibiting the interaction. The MKP3 KIM peptide contains four basic amino acid sites that we hypothesized could be replaced with a caged lysine to sterically block the protein–protein interaction and abrogate electrostatic interaction, until light exposure removes the sterically demanding caging group and restores a positive charge to the residue. The optimal scenario would be replacement of K68 with a caged lysine in order to generate the native phosphatase upon light activation. Replacement of R64, R65, or R74 would require confirmation that an R-to-K mutation retained substrate recognition and enzymatic activity. Interestingly, we tested all four of these positions and found that caging of K68 and R74 was not sufficient to inhibit the phosphatase–kinase interaction in our cell-based reporter. We validated that individual replacement of R64 or R65 with lysine resulted in wild-type phosphatase activity; however, we were only able to incorporate a caged lysine into position 65. Through caging at K65, we demonstrated that caged lysine (
Two distinct, yet complementary, approaches were developed for rendering MKP3 light responsive and enabled acute perturbation of the ESRK pathway for further dissection of the role played by MKP3 (
When selecting residues to target at a protein–protein interface, it is advantageous to consider several different sites, as introduction of a small caging group may not be sufficient to abrogate interaction at all locations chosen (as seen in the case of our KIM-caging approach). One can also envision capitalizing on computational approaches to predict residues in order to minimize the experimental workload of site selection. 9 Modulation of protein phosphatase activity through reversible oxidation of cysteine residues provides an additional level of control of signal transduction; however, studying the effects of this reversibility is challenging with the current tools. 10 As an alternative to genetic code expansion, development of optically controlled phosphatases through optogenetic techniques (e.g., Dronpa or LOV domains) would provide the opportunity to reversibly control phosphatase activity; however, very careful design principles must be employed such that these large photoresponsive domains do not interfere with the endogenous functions of the selected phosphatase when fused to it.
We predict that these approaches can be combined with systems analyses in order to elucidate the downstream effects of acute phosphatase activation in a time-resolved manner, given the defined starting point of enzyme activation. Additionally, the defined starting point of phosphatase function in conjunction with reporter assays may enable screening for small-molecule inhibitors of signaling nodes in live cells. More broadly, we believe that the second approach opens the door for rendering a wide range of protein–protein interactions that rely on electrostatic interactions controllable with light, as we have demonstrated for nuclear transport. 11 Control of nonenzymatic functions via this strategy has the potential to expand our understanding of protein scaffolding, protein chaperoning, interactions involved in protein complex (dis)assembly, and others.
Footnotes
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: We would like to thank the National Science Foundation for support (CBET-1603930 to A.D. and a Graduate Research Fellowship to T.M.C.).