
Abstract
To expedite G-protein-coupled receptor (GPCR) drug screening studies, cell lines amenable to transfection (e.g. CHO cells) have been widely used as cellular models. These cells can be frozen in a ready-to-use format, allowing screening of a single batch of cells and validation of the cellular material prior to the screening run. A common method used to deliver frozen cells to screening programs is to γ-irradiate the cells, abrogating cell division after thawing and ensuring consistency in the number of cells analyzed per well. With the recognition that signaling proteins such as ERK and Akt are important markers of GPCR activation, along with the availability of suitable assays for their measurement, these outputs have become important for GPCR screening programs. Here we show that several γ-irradiated and frozen CHO-K1 cell lines expressing transfected GPCRs, initially optimized for performing cAMP or AequoScreen calcium flux assays, can be used for the measurement of GPCR-mediated ERK and Akt phosphorylation. Furthermore, CHO-K1 cells transfected with NOP or GAL1 receptors show pharmacology for a number of agonists and antagonists that is consistent with non-irradiated cultured lines. These data indicate that γ-irradiated CHO-K1 cells can be reliably used for the measurement of GPCR-mediated kinase signaling outputs.
Introduction
G-protein-coupled receptors (GPCRs) are a large family of cell surface receptors that are involved in many cellular processes. GPCRs have proven very amenable for therapeutic intervention, and GPCR-targeted compounds represent a large proportion of Food and Drug Administration (FDA)–approved drugs. 1 However, still only around 80 GPCRs identified as potential therapeutic candidates are currently addressed by drugs in the clinic. 2 Therefore, a large number of potential targets remain for pharmaceutical intervention, and the area remains one with much potential.
For drug discovery screening, some GPCR-mediated signaling pathways have been well established. In particular, the mobilization of intracellular Ca2+ reserves, or the formation of cAMP, is widely used. There are several well-established cell-based screening technologies for the measurement of both of these analytes, along with well-characterized native and recombinant cellular systems. However, the measurement of calcium or cAMP alone can be insufficient to monitor all of the signaling events arising from a particular receptor.1,3 Allosteric modulation, ligand-induced G-protein-coupling selectivity, receptor hetero-oligomerization, and G-protein-independent signaling, among other mechanisms, have all emerged to add complexity to the drug discovery process.1,3 Therefore, the need for interrogation of a broader range of GPCR signaling pathways, such as intracellular kinase cascades, has gained increasing attention.
The activation of G-proteins and β-arrestin by GPCRs leads to the activation of many kinase cascades, including protein kinase A (PKA), protein kinase C (PKC), or extracellular regulated kinase (ERK)–associated signaling, and the regulation of these cascades represents an important part of the mode of action of GPCRs.4-7 In addition, the importance of GPCR-mediated Akt signaling is also becoming better understood, with evidence for an important role in the regulation of action of drugs targeting psychiatric disorders.8-12
From a drug discovery perspective, the ability to screen for specific kinase targets in a cell-based setting has been hampered by the availability of suitable assays with robust characteristics for handling, variation, and reproducibility. However, in recent years, this limitation has been addressed with the commercial availability of high-throughput screening (HTS)–suitable homogeneous technologies, such as AlphaScreen SureFire assays (PerkinElmer, Waltham, MA) and, for some targets, homogeneous time-resolved fluorescence (HTRF) assays, as well as the emergence of high-content analysis. Similarly, from a cellular perspective, generating consistent cellular material to support an HTS-scale program can be challenging. For this reason, transfected and frozen model cell lines such as Chinese hamster ovary (CHO) cells for cell-based screening are widely used within the drug discovery community, despite limitations associated with the use of transfected cellular models for GPCR analysis. 13 Separating cell production from the actual screening campaign increases flexibility but also improves the data consistency as the cellular material can be controlled and validated before running the functional assay.
One of the methods used to deliver frozen cells as a consumable to the final user is to γ-irradiate the cells. Irradiation inhibits cell division after thawing, ensuring consistency in the number of cells analyzed per well, across multiple plate batches. Although the use of this cellular material has been well established for performing cAMP assays or calcium flux assays, less is known about the effect of irradiation on signal transduction through intracellular kinase signaling cascades. Here we test several commercially available CHO cell lines carrying transfected GPCRs, as well as their γ-irradiated frozen counterparts—generated and optimized for performing cAMP assays or calcium flux assays—and show these cellular materials can also be used for the measurement of GPCR-mediated ERK and Akt phosphorylation. Furthermore, γ-irradiated frozen cells show virtually indistinguishable pharmacological characteristics in response to a number of receptor agonists and antagonists when compared with nonirradiated cultured lines. These data indicate that γ-irradiated frozen CHO cells are reliable for use when measuring GPCR-mediated mitogen-activated protein kinase (MAPK) and phosphoinositide 3-kinase (PI3-K) signaling outputs.
Materials and Methods
Materials
All γ-irradiated frozen and nonirradiated CHO cell lines, ProxiPlate-384 Plus microplates, and AlphaScreen SureFire ERK (p-Thr202/Tyr204) and Akt (p-Thr308) phosphorylation assays were from PerkinElmer (Waltham, MA). Trypsin-free cell dissociation solution was from Sigma-Aldrich (St. Louis, MO). Adrenomedullin, M32, M35, and galanin (human) were from Bachem AG (Bubendorf, Switzerland). Galanin 2-29 (rat), J113397, M40, nociceptin, and UFP-101 were from Tocris Bioscience (Ellisville, MO). Human CCL19 (MIP-3β) was from R&D Systems (Minneapolis, MN). All cell culture media and additives were from Life Technologies (Carlsbad, CA). IgG-free bovine serum albumin (BSA) was from Jackson Immunoresearch Laboratories (West Grove, PA). TC-treated 96-well plates and growth flasks were from Thermo Fisher Scientific (Rochester, NY).
Cell Culture
When fresh cells were used for comparison, they were cultured according to the protocol given in the technical datasheet of the corresponding cell lines. Briefly, cells were cultured at 37 °C/5% CO2 in Ham’s F12 medium, containing 10% fetal bovine serum (FBS) and 1× penicillin-streptomycin (Pen-Strep). The cells were passaged every 2 to 3 days, when 70% to 80% confluent.
Cellular Assays
Cultured Cells
Cultured cells were harvested from flasks with trypsin-free cell dissociation solution and seeded at 40 000 cells/well in 96-well tissue culture microplates, in fresh Ham’s F12 medium, containing 10% FBS and 1× Pen-Strep. Generally, the cells were cultured for approximately 6 h at 37 °C/5% CO2. The medium was then aspirated and replaced by serum-free culture medium, and the plates were incubated overnight in a cell culture incubator (37 °C, 5% CO2; serum starvation step). The following day, the serum-free culture medium was removed by aspiration, and the cells were treated with various concentrations of agonist diluted in serum-free culture medium, containing 0.1% IgG-free BSA.
Irradiated and Frozen Cells
Frozen cells were rapidly thawed, diluted in 5 mL culture medium, and centrifuged (3000 rpm for 3 min) to remove DMSO, and the cell pellet was resuspended to the concentration of 200 000 cells/mL in Ham’s F12 medium, containing 10% FBS and 1× Pen-Strep. Cell suspension (200 µL) was dispensed into the wells of a 96-well tissue culture microplate, yielding 40 000 cells/well. Generally, the cells were cultured for approximately 6 h at 37 °C/5% CO2. The medium was then aspirated and replaced by serum-free culture medium, and the plates were incubated overnight in a cell culture incubator (37 °C, 5% CO2; serum starvation step). The following day, the serum-free culture medium was removed by aspiration, and the cells were treated with various concentrations of agonist diluted in serum-free culture medium, containing 0.1% IgG-free BSA.
Antagonist Assays
For antagonist treatments, cells were cultured in microtiter plates as described above, except that prior to agonist treatment, the cells were treated with 50 µL of antagonist and diluted in serum-free culture medium containing 0.1% BSA for 15 min—a time period anticipated to allow maximal binding of the antagonist to the receptor prior to introduction of the agonist. The cells were then stimulated with the addition of 50 µL agonist, yielding a final volume of 100 µL in each well.
AlphaScreen SureFire ERK and Akt Phosphorylation Assays
The AlphaScreen SureFire assays are homogeneous sandwich immunoassays that use the AlphaScreen proximity-based detection system. Signal is generated only when both antibodies—one binding a constant region on the analyte and the other binding an inducible phosphoepitope—are bound to the analyte. When this occurs, the AlphaScreen Donor and Acceptor beads, each of which specifically binds only one of the antibodies in the reaction, are brought into close proximity, allowing signal transfer from the Donor to the Acceptor bead.
Briefly, after treatment, the medium was removed from the cells by aspiration, and the cells were lysed in 50 µL of 1× AlphaScreen SureFire lysis buffer, with shaking for 10 min at room temperature. An aliquot of cellular lysate (4 µL) was transferred to a ProxiPlate-384 Plus assay plate and assayed for either phospho-ERK1/2 (phospho-Thr202/Tyr204) or phospho-Akt (phospho-Thr308) according to the respective AlphaScreen SureFire kit’s instructions. At the completion of the assay, signal in the wells was measured using an EnVision Alpha plate reader (PerkinElmer).
Data Handling
Raw data from lysates generated from at least three individual wells of cells were averaged, and the standard deviation was determined. Dose–response and dose inhibition curves were analyzed and fitted using sigmoidal variable-slope models, using GraphPad Prism software (GraphPad Software, La Jolla, CA).
Results
Optimization of Cellular Assay Parameters
As a first step in determining if the kinase signaling mechanisms remained intact after irradiation and freezing, we looked at the key assay parameters of cell seeding density and time course of stimulation to test and optimize signaling output prior to comparison with nonirradiated cells.
To determine the number of cells to seed per well in 96-well tissue culture microplates required to elicit an easily measureable ERK phosphorylation response upon agonist treatment using γ-irradiated frozen cells, two cell lines—namely, CHO cells transfected with either CCR7 (CHO-CCR7) or galanin receptor 1 (CHO-GAL1)—were examined. The cells were seeded at between 10 000 and 40 000 cells per well and subsequently stimulated with agonists expected to elicit a maximal response for each respective receptor. The amplitude of the ERK phosphorylation response, generated using a high agonist concentration relative to the basal signal from unstimulated cells, was determined (data not shown). From these results, which were in agreement with our previous experience of optimal cell density for nonirradiated CHO cells, 14 we selected 40 000 cells per well as the cell seeding density for further experiments.
From previous work with cultured recombinant CHO cells, a 5- to 15-min agonist treatment at room temperature was often an optimal stimulation time for GPCR-stimulated ERK phosphorylation. 14 To determine if this remained true when working with γ-irradiated frozen cells and to determine an optimal time point for Akt phosphorylation, CHO cells transfected with GAL1 receptor, nociceptin opioid receptor (CHO-NOP), or the adrenomedullin receptor 2 (CRLR + RAMP3; CHO-AM2) were treated with their respective agonist for varying lengths of time ( Fig. 1 ). Agonist-mediated ERK phosphorylation was observed in all three cell lines, with the phosphorylation peak at around 10 to 15 min. Akt phosphorylation at Thr308 was observed in CHO-GAL1 and CHO-NOP cells, with a slightly faster responding time course compared to ERK. However, no Akt response was observed in CHO-AM2 cells. From these experiments, a 10-min agonist treatment time was determined as the standard stimulation time for performing ERK and Akt phosphorylation measurements. Based on these results, a consistent workflow for comparison of fresh cultured cells versus irradiated frozen cells was defined.
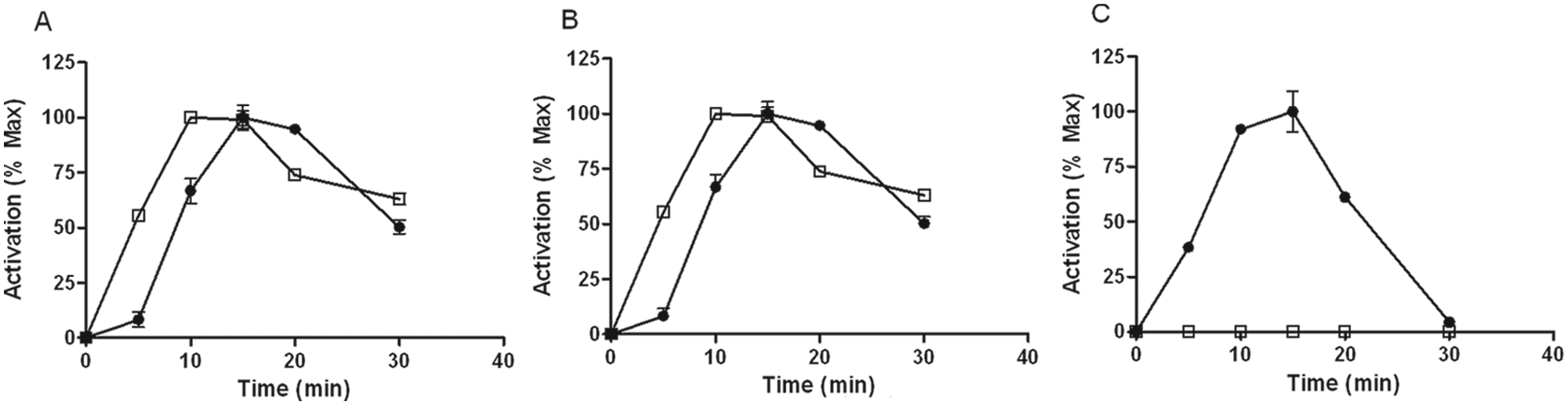
Time course of kinase pathway activation in various transfected Chinese hamster ovary (CHO) cells. γ-Irradiated frozen: (A) CHO-GAL1, (B) CHO-NOP, or (C) CHO-AM2 cells were thawed and seeded in 96-well plates for 6 h and then serum-starved overnight. The next day, the cells were stimulated with 100 nM galanin, 10 nM nociceptin, or 1 µM adrenomedullin (1–52), respectively, for varying lengths of time. The cells were subsequently lysed and analyzed for either (●) phosphorylated ERK or (□) phosphorylated Akt (p-Thr308). Signal is representative of n = 4 wells of cells and expressed as a percentage of the maximum signal, after subtraction from the signal obtained in unstimulated cells.
Comparison of Akt and ERK Signaling in Irradiated Frozen versus Nonirradiated Cells
To examine the reliability of agonist-mediated ERK and Akt phosphorylation in γ-irradiated frozen CHO cells in detail, we compared two γ-irradiated frozen CHO cells, CHO-GAL1 and CHO-NOP, with their cultured counterparts and determined their response to several agonists and antagonists.
Irradiated versus Nonirradiated CHO-GAL1 Cells
For characterization of the response of irradiated frozen versus freshly cultured CHO-GAL1 cells, we looked at several peptides known to bind to the GAL1 receptor—namely, rat galanin (2–29), M32, M35, and M40. These ligands have all been shown to bind the GAL1 receptor with low nanomolar affinities.15-18 Here we found that all compounds tested in this study behaved as agonists for the GAL1 receptor as measured for both ERK ( Fig. 2 ) and Akt phosphorylation ( Fig. 3 ), with comparable low-nanomolar EC50 for all treatments observed for both ERK at Akt phosphorylation (summarized in Table 1 ). Although no significant differences in EC50 were observed between irradiated frozen versus nonirradiated CHO-GAL1 cells, the peak signals were either similar or slightly higher in irradiated cells compared to cultured cells. The ERK phosphorylation response over basal in nonirradiated cells ranged from 11.2 to 33.6 (Z′ = 0.51–0.93) in these experiments, depending on the agonist used, whereas the response in the irradiated cells was slightly higher, ranging from 22.2 to 47.7 (Z′ = 0.68–0.96). The Akt phosphorylation response over basal in nonirradiated cells ranged from 3.8 to 4.8 (Z′ = 0.35–0.79), whereas the response in the irradiated cells ranged from 4.4 to 6.7 (Z′ = 0.59–0.91).
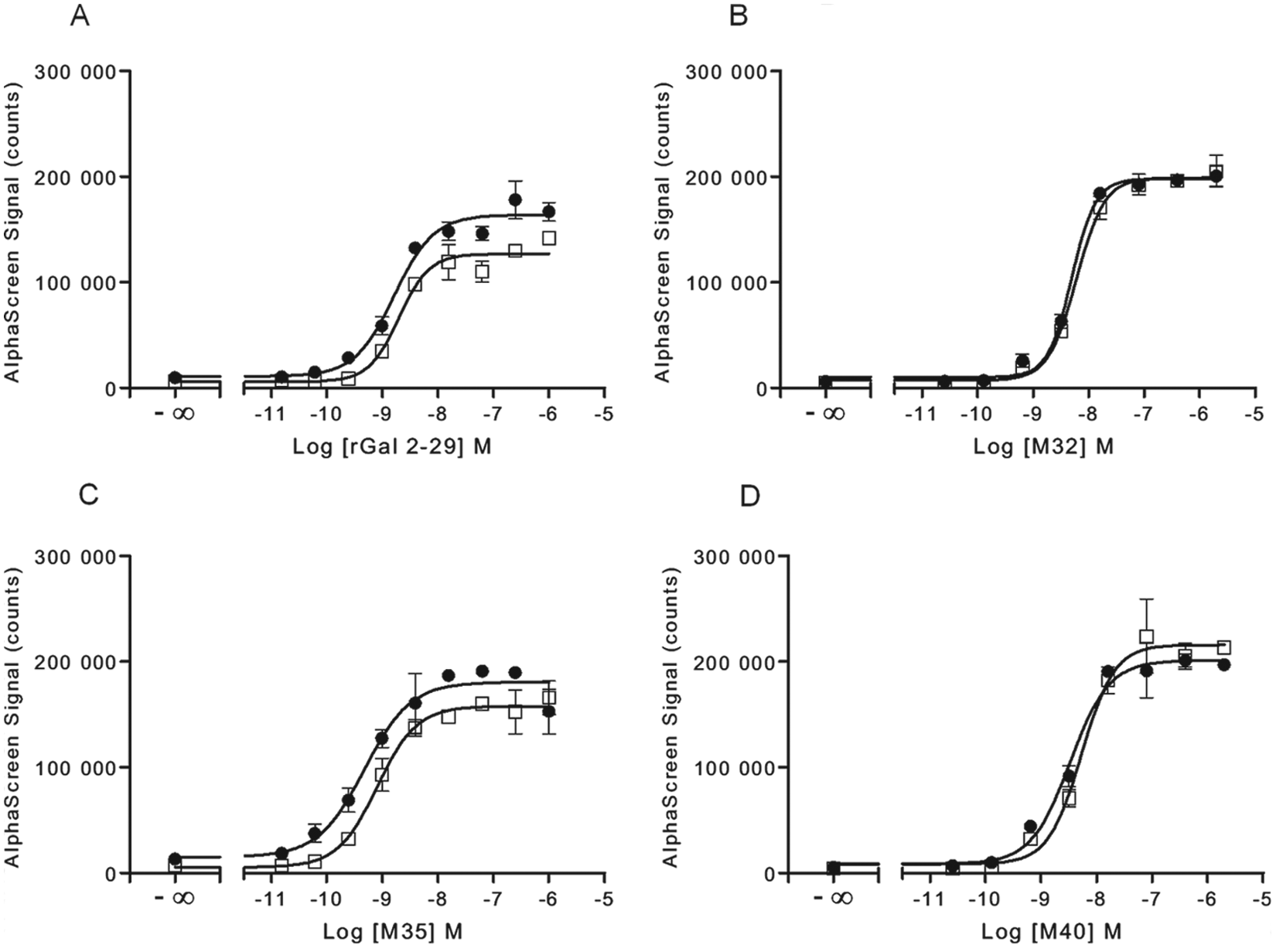
Comparison of ERK phosphorylation in cultured and γ-irradiated frozen CHO-GAL1 cells. Either (●) fresh cultured or (□) irradiated frozen CHO-GAL1 cells were seeded into 96-well microplates for 6 h in medium containing 10% fetal bovine serum (FBS) and then serum-starved overnight. The next day, cells were stimulated with the indicated agonists for 10 min and lysed. A portion of lysate (4 µL) was transferred to an assay plate and analyzed for ERK phosphorylation, using the standard AlphaScreen SureFire protocol. Results are the mean ± SD of six separate wells of cells.
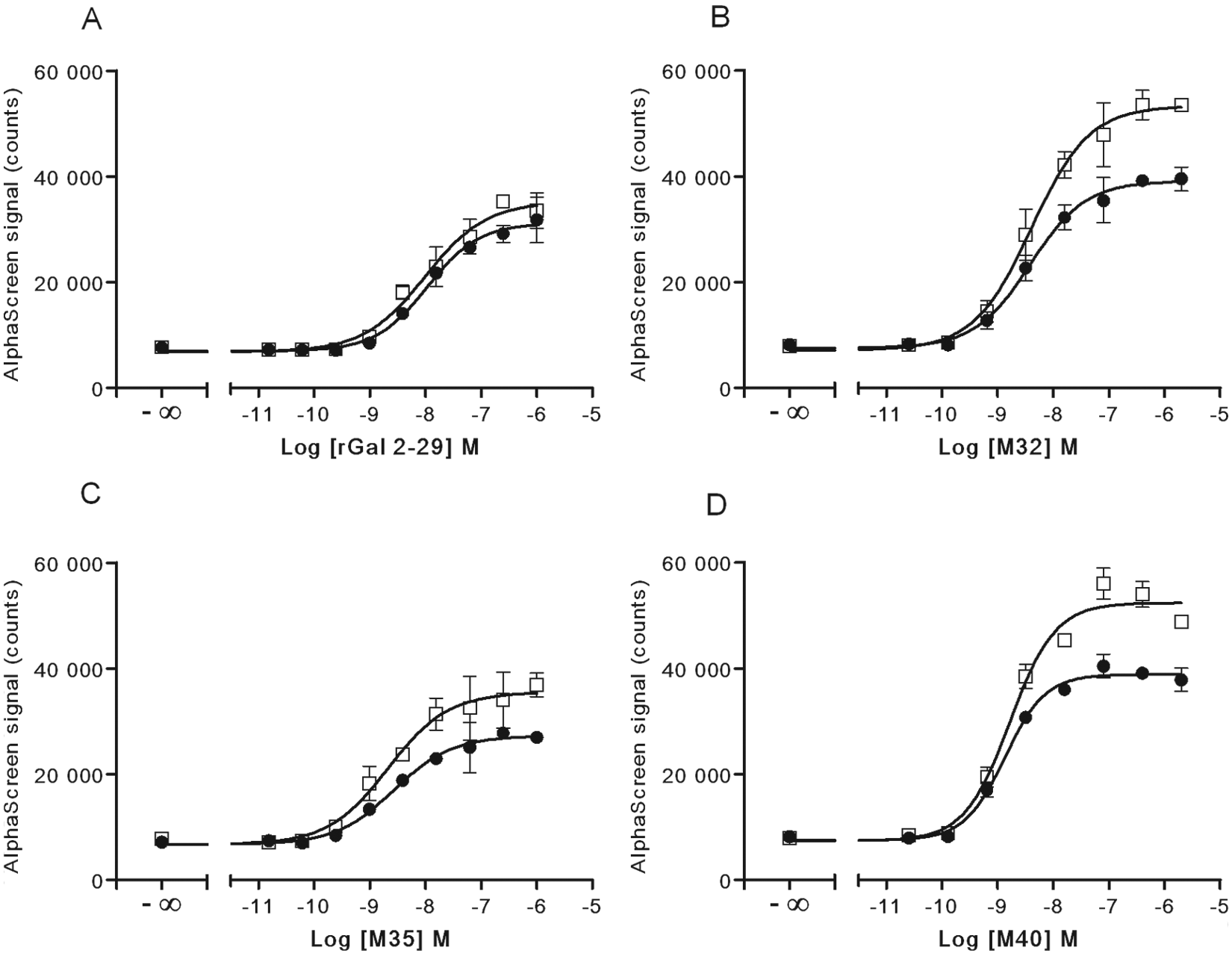
Comparison of Akt phosphorylation in cultured and γ-irradiated frozen CHO-GAL1 cells. Either (●) fresh cultured or (□) irradiated frozen CHO-GAL1 cells were seeded into 96-well microplates for 6 h in medium containing 10% fetal bovine serum (FBS) and then serum-starved overnight. The next day, cells were stimulated with the indicated agonists for 10 min and lysed. A portion of lysate (4 µL) was transferred to an assay plate and analyzed for Akt phosphorylation, using the standard AlphaScreen SureFire protocol. Results are the mean ± SD of six separate wells of cells.
Comparison of ERK and Akt Phosphorylation Response in Cultured versus Irradiated Frozen Cells
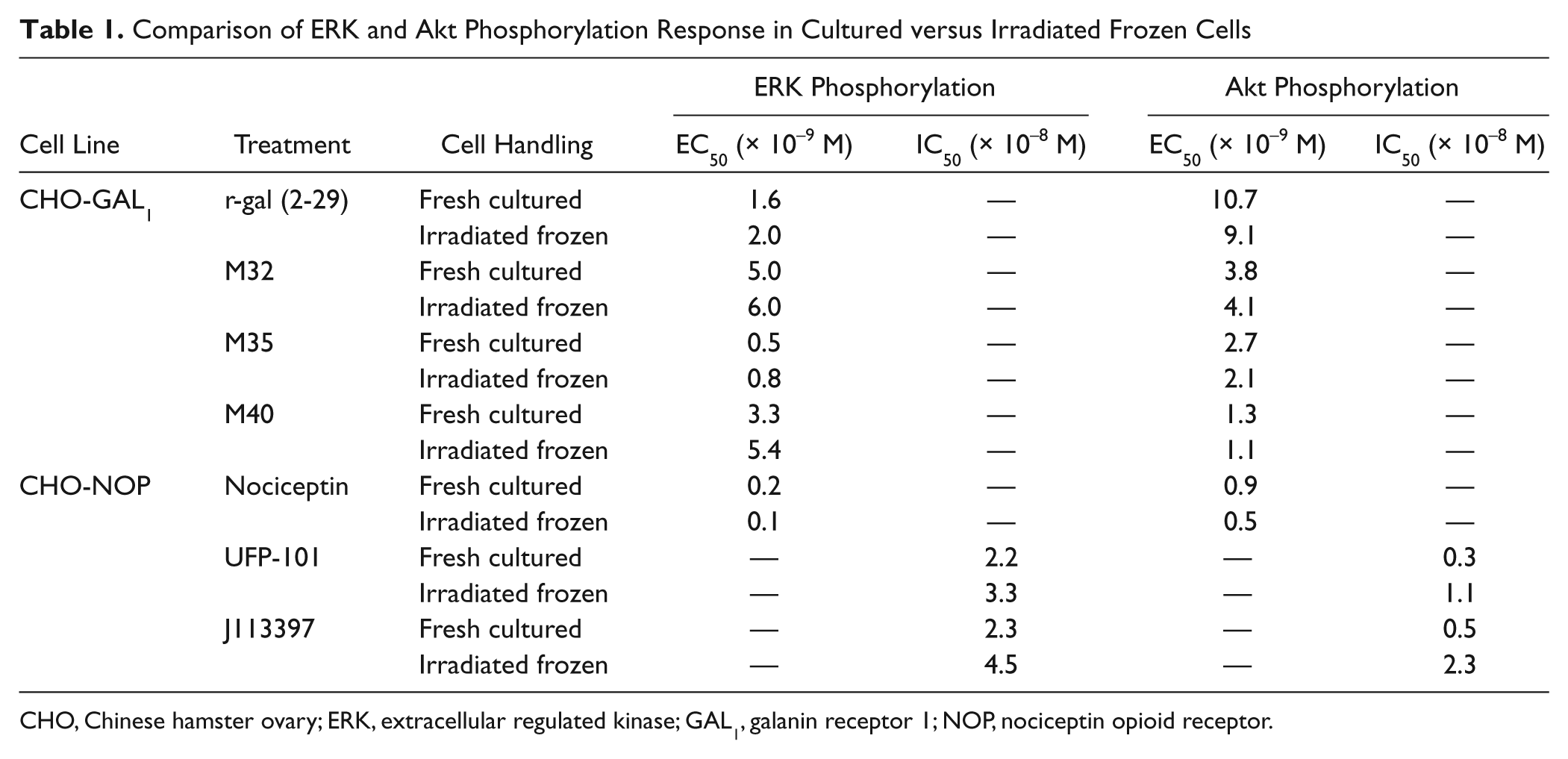
CHO, Chinese hamster ovary; ERK, extracellular regulated kinase; GAL1, galanin receptor 1; NOP, nociceptin opioid receptor.
Irradiated versus Nonirradiated CHO-NOP Cells
For characterization of the response of irradiated frozen versus freshly cultured CHO-NOP cells, we examined a known NOP receptor agonist, nociceptin, which has been shown to inhibit forskolin-induced accumulation of cyclic AMP in CHO-NOP cells with an EC50 in the picomolar range. 19 Here we found that nociceptin stimulated both ERK and Akt phosphorylation ( Fig. 4 ), with an EC50 in the picomolar range for ERK and Akt phosphorylation for both irradiated frozen and nonirradiated CHO-NOP cells ( Table 1 ). Here, the ERK phosphorylation response over basal in nonirradiated cells was 3.0 for both nonirradiated and irradiated cells (Z′ = 0.79 and 0.74, respectively), limited by a much higher basal phosphorylation than was observed in the CHO-GAL1 cells. The Akt phosphorylation response for nonirradiated and irradiated cells was 4.6 (Z′ = 0.66) and 5.2 (Z′ = 0.70), respectively.
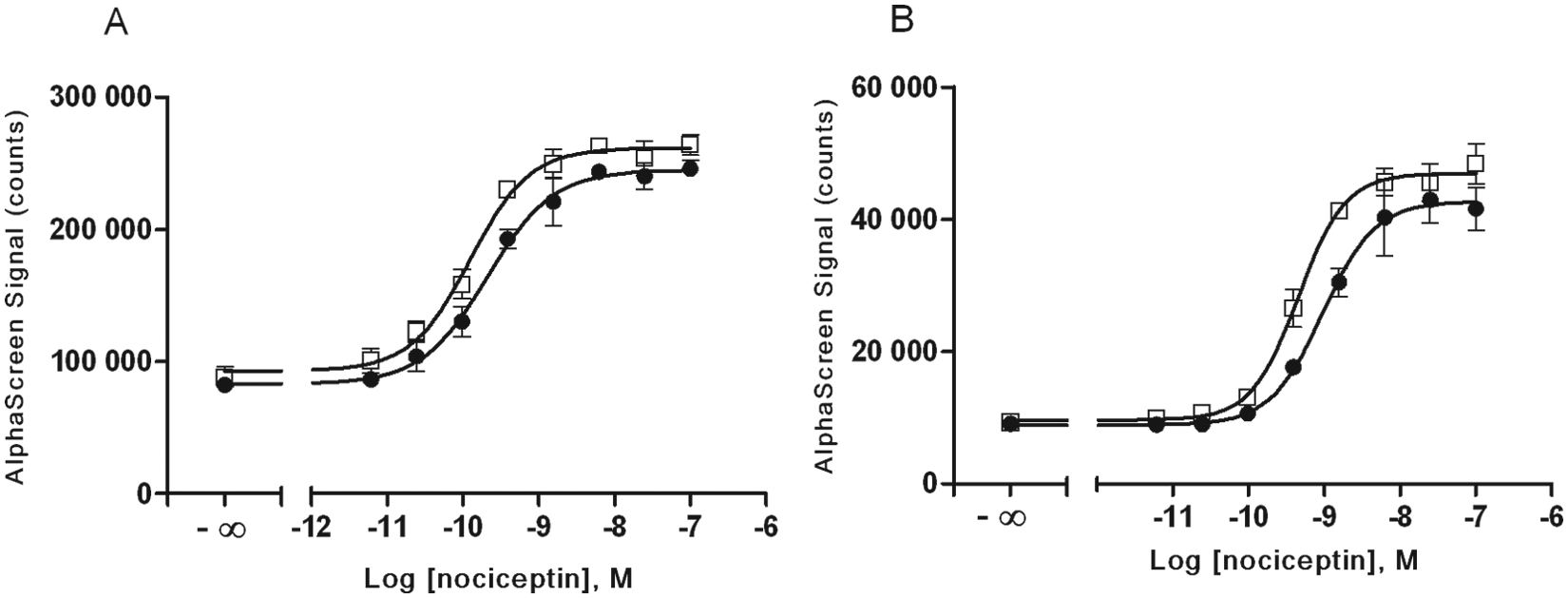
Comparison of agonist-mediated ERK and Akt phosphorylation in cultured and γ-irradiated frozen CHO-NOP cells. Either (●) fresh cultured or (□) irradiated frozen CHO-NOP cells were seeded into 96-well microplates for 6 h in medium containing 10% fetal bovine serum (FBS) and then serum-starved overnight. The next day, cells were stimulated with nociceptin for 10 min and lysed. A portion of lysate (4 µL) was transferred to an assay plate and analyzed for either (A) ERK phosphorylation or (B) Akt (p-Thr308) phosphorylation, using the standard AlphaScreen SureFire protocol. Results are the mean ± SD of six separate wells of cells.
We also examined NOP receptor antagonists, J113397 and UFP-101. Again, both the irradiated frozen and nonirradiated CHO-NOP cells exhibited similar behavior in response to the antagonists tested ( Fig. 5 ), with observed IC50 values in the low nanomolar range for both ERK and Akt phosphorylation ( Table 1 ).
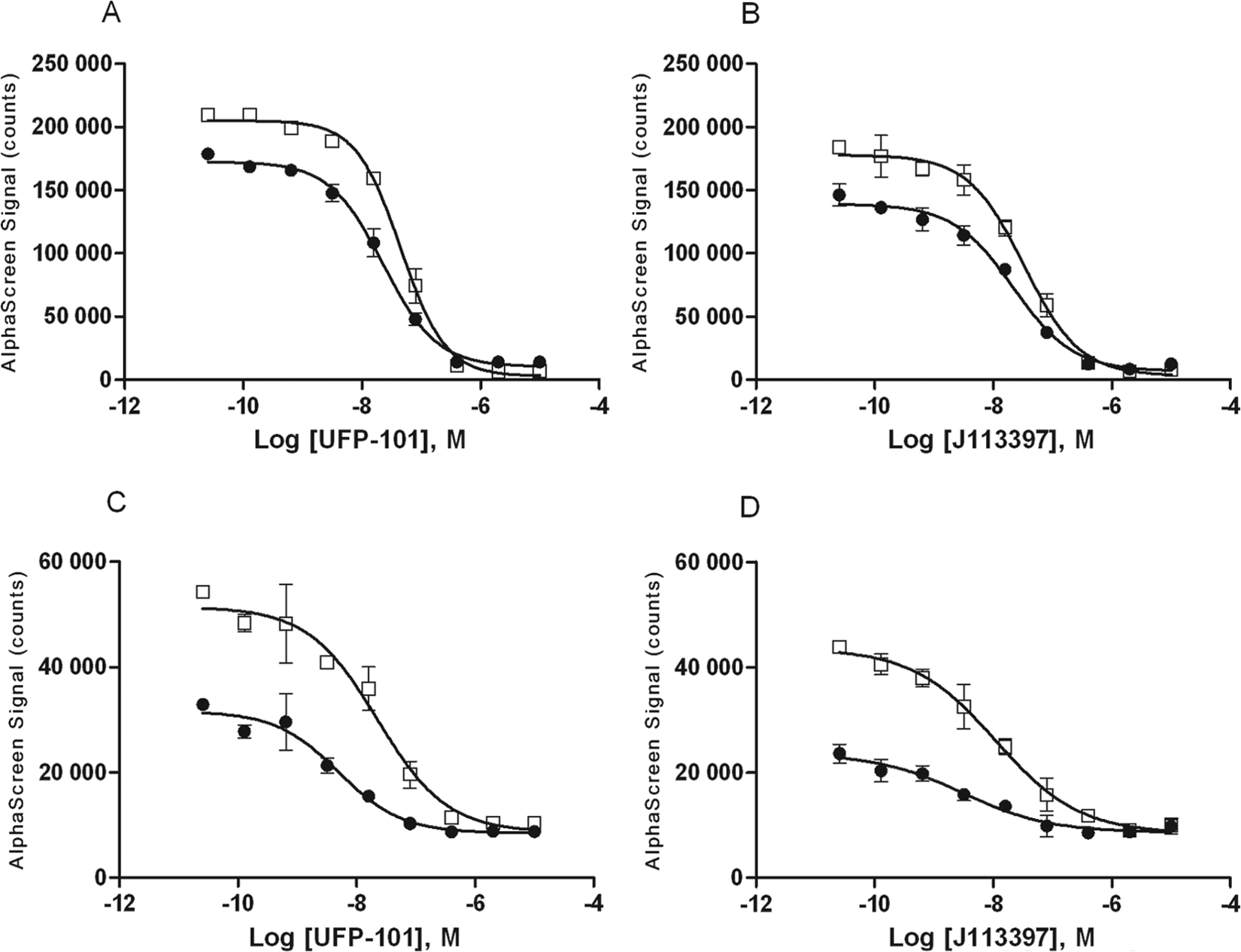
Comparison of inhibition of agonist-mediated ERK and Akt phosphorylation in cultured and γ-irradiated frozen CHO-NOP cells. Either (●) fresh cultured or (□) irradiated frozen CHO-NOP cells were seeded into 96-well microplates for 6 h in medium containing 10% fetal bovine serum (FBS) and then serum-starved overnight. The next day cells were treated with NOP inhibitors UFP-101 or J113397 for 15 min, then stimulated with the nociceptin for 10 min and lysed. A portion of lysate (4 µL) was transferred to an assay plate and analyzed for either (A, B) ERK phosphorylation or (C, D) Akt (p-Thr308) phosphorylation, using the standard AlphaScreen SureFire protocol. Results are the mean ± SD of three separate wells of cells.
Discussion
The use of γ-irradiated and frozen model transfected cell lines has become widely accepted for drug discovery programs and has been well established for performing cAMP assays or calcium flux assays. However, less is known about the effect of irradiation on signal transduction through intracellular kinase signaling cascades. Here we have tested several γ-irradiated and frozen CHO cell lines for the measurement of GPCR-mediated ERK and Akt phosphorylation.
Initially, a protocol was developed that involved seeding the cells at a near-confluent density of 40 000 cells per well and allowing the cells to adhere to the plate for approximately 24 h prior to agonist treatment. We have found for many cells that allowing 18 to 24 h for adhesion to the plate is required for full MAPK signaling (data not shown). Furthermore, there are reports that ionizing radiation can trigger or modulate signaling in cells, including calcium, PKC, JNK, PI3K/Akt, Rho, and Raf-MEK-ERK pathways.20-22 However, the kinetics of the activation of kinases by ionizing radiation was reported to be rapid and transient, 20 followed by a return to basal levels within a few hours. Consequently, using the protocol determined here, where cells are in culture for around 24 h after thawing before treatment and lysis, no interference caused by a γ-irradiation response was expected. Indeed, following this protocol, we did not observe any increases in basal phosphorylation levels when comparing cultured and γ-irradiated cells for any of our experiments.
Interestingly, during optimization experiments, no Akt response was observed in CHO-AM2 cells. GAL1 and NOP are known to couple through Gαi in transfected CHO cells,23,24 whereas the AM2 (CRLR + RAMP3) receptor couples through Gα q and Gαs proteins. 25 This coupling was confirmed for these experiments by pretreating cells with pertussis toxin, which completely abrogated agonist-mediated ERK and Akt signal in CHO-GAL1 and CHO-NOP cells but not did not abrogate ERK signal in CHO-AM2 cells (data not shown). As specific Gαi-coupled receptors tested here (GAL1, CCR7, and NOP) showed a consistent Akt response, the lack of response in CHO-AM2 cells suggests that the Akt response may be mediated by Gαi proteins in CHO cells.
We next moved to explicitly examine whether the irradiated frozen cells showed similar receptor-mediated activation of MAPK and Akt signaling, when compared to cultured cells carrying the same receptors. For this, we used irradiated frozen CHO-GAL1 and CHO-NOP cells that were prepared from the same cell lines that were used for culturing. In this testing, the γ-irradiated frozen cells show virtually indistinguishable pharmacological characteristics in response to a number of receptor agonists and antagonists when compared with nonirradiated cultured lines.
The CHO-GAL1 cells were treated with a range of peptides known to elicit a signaling response at the GAL1 receptor, including rat galanin, and the chimeric peptides M32, M35, and M40. It is to be noted that although M32, M35, and M40 are sometimes described as antagonists to the GAL1 receptor, 26 we found them to have agonistic properties in the phospho-ERK and phospho-Akt assays ( Figs. 2 and 3 ). This is in agreement with our previous observation that M35 and M40 behave as full agonists in a calcium flux assay performed on the same receptor (data not shown) and with the agonistic properties of the same two peptides at both GAL1 and GAL2 receptors. 27 We also examined NOP receptor antagonists J113397 and UFP-101. J113397 has been shown to inhibit forskolin-stimulated accumulation of cyclic AMP in CHO-NOP cells. 28 UFP-101 has been shown to inhibit nociceptin binding at the NOP receptor and subsequent cAMP accumulation in CHO-NOP cells with a similar potency. 29
No significant differences in EC50 or IC50 were observed between either irradiated frozen versus nonirradiated CHO-GAL1 or CHO-NOP cells for any of the agonists or antagonists tested here ( Table 1 ), indicating that the irradiation and freezing processing of these cells had no effect on subsequent receptor-mediated ERK or Akt signaling pharmacology for these cells. However, despite initial seeding at the same density, the observed peak response to agonists was generally higher in irradiated cells, a phenomenon particularly noticeable for Akt ( Figs. 2 - 5 ). An irradiation-mediated accumulation of signaling molecules, including signaling proteins, was recently described in irradiated prokaryotic cells. 30 Our data indicate this could also occur in irradiated CHO cells, but this would need further testing to confirm. In any case, this caused no apparent discrepancy in receptor-mediated signaling pharmacology, manifesting simply as an increased signal window when using irradiated frozen cells for most experiments.
In summary, we have examined whether division-arrested CHO-K1 cells can also be used to measure GPCR-mediated ERK and Akt signaling. Here, when irradiated and frozen cells were compared to cells in culture, they behaved in a manner similar to the nonirradiated cultured cells in response to a number of different agonists and antagonists. Although ERK was activated in each of the GPCRs tested here, Akt provided an alternative method to record GPCR activation and appeared to be activated by the Gαi-coupled receptors tested here, but not by the AM2 receptor, which couples via Gαq or Gαs. These data show that γ-irradiation of cells is compatible with subsequent assaying of GPCR-mediated activation of MAPK and PI3-kinase pathways. This will provide additional flexibility for the characterization in multiple assays of drugs in development, offering a broader toolbox for the characterization of the increasing array of GPCR-mediated signaling mechanisms.
Footnotes
Ronald I. W. Osmond is employed by TGR BioSciences. M. Henry Martin-Harris, Michael F. Crouch, Janet Park, Eric Morreale, and Vincent J. Dupriez are employed by PerkinElmer. PerkinElmer manufactures the irradiated cells examined in this study.
The work represented in this article was funded by TGR BioSciences and PerkinElmer.