
Abstract
The discovery of CRISPR-Cas9 systems has fueled a rapid expansion of gene editing adoption and has impacted pharmaceutical and biotechnology research substantially. Here, gene editing is used at an industrial scale to identify and validate new biological targets for precision medicines, with functional genomic screening having an increasingly important role. Functional genomic strategies provide a crucial link between observed biological phenomena and the genes that influence and drive those phenomena. Although such studies are not new, the use of CRISPR-Cas9 systems in this arena is providing more robust datasets for target identification and validation. CRISPR-based screening approaches are also useful later in the drug development pipeline for understanding drug resistance and sensitivity ahead of entering clinical trials. This review examines the developing landscape for CRISPR screening technologies within the pharmaceutical industry and explores the next steps for this constantly evolving screening platform.
Introduction: The Evolution of CRISPR Screening
Genomic screens can provide a rapid and unbiased approach to elucidating gene function, and their impact on drug discovery stretches back to the advent of RNAi-based gene perturbation. Although RNAi has been honed and adapted over the years to support an impressive array of analytical paradigms,1,2 there remain challenges associated with the variability and off-target effects inherent to this technology. 3 CRISPR-Cas9 approaches appear not to suffer from the variability evident with RNAi,2,3 and as such this technology provides both new and compelling opportunities for drug discovery.
CRISPR-based technologies use an RNA-guided endonuclease, such as Cas9, in order to drive genomic perturbation. Functional genomic screening with CRISPR was first demonstrated using a fully active Cas9 endonuclease, which was used to knock out specific gene loci and evaluate the phenotypic effect of the deletion.4–6 In these screens, the gene edits or knockouts driven by Cas9 are enabled by the repair of the double-strand break by the endogenous DNA repair machinery. This most often involves nonhomologous end joining, an error-prone repair mechanism that introduces insertions or deletions (InDels) at the repaired locus, causing frameshifts or premature stop codons that more often than not ablate gene function. 7 This approach provides the substantial benefit of driving gene deletion to homozygosity at a high frequency, 8 maximizing the phenotypic impact of the perturbation. This is distinct from the previous state-of-the-art approaches using RNAi, which can suppress gene expression but rarely eliminate it entirely. In addition, loci where gene editing is successful—an outcome contingent on study design, Cas9 expression, and guide library quality—the impact on phenotype on a per cell and per guide basis is impressively consistent. 9 This again is distinct from RNAi, where individual hairpins can introduce variable gene suppression, causing the response to vary accordingly and generating a more diverse/noisy response signature. These variabilities can be controlled by ultracomplex RNAi libraries, which target each gene with more than 20 hairpins in each case, 10 and they also provide the opportunity for evaluation of gene dosage effects, which CRISPR knockout (CRISPRko) screens rarely provide. But for most initial screens, a common goal is high reproducibility to drive robust hit identification, and this can often be better accomplished by a CRISPR approach. 3
There are, however, some nuances of the knockout approach that provide room for alternative systems. For instance, elimination of gene transcription during a CRISPRko screen can identify genes that mediate a specific phenotype, but recapitulating this effect pharmacologically could prove challenging, especially where complete loss of protein expression is needed rather than a reduction in protein function, creating a potential pool of unactionable drug targets. An alternative set of CRISPR-based tools for screening—CRISPR interference (CRISPRi)11–15 and CRISPR activation (CRISPRa)15–21—have been developed. These approaches enable the repression or overexpression, respectively, of target genes (
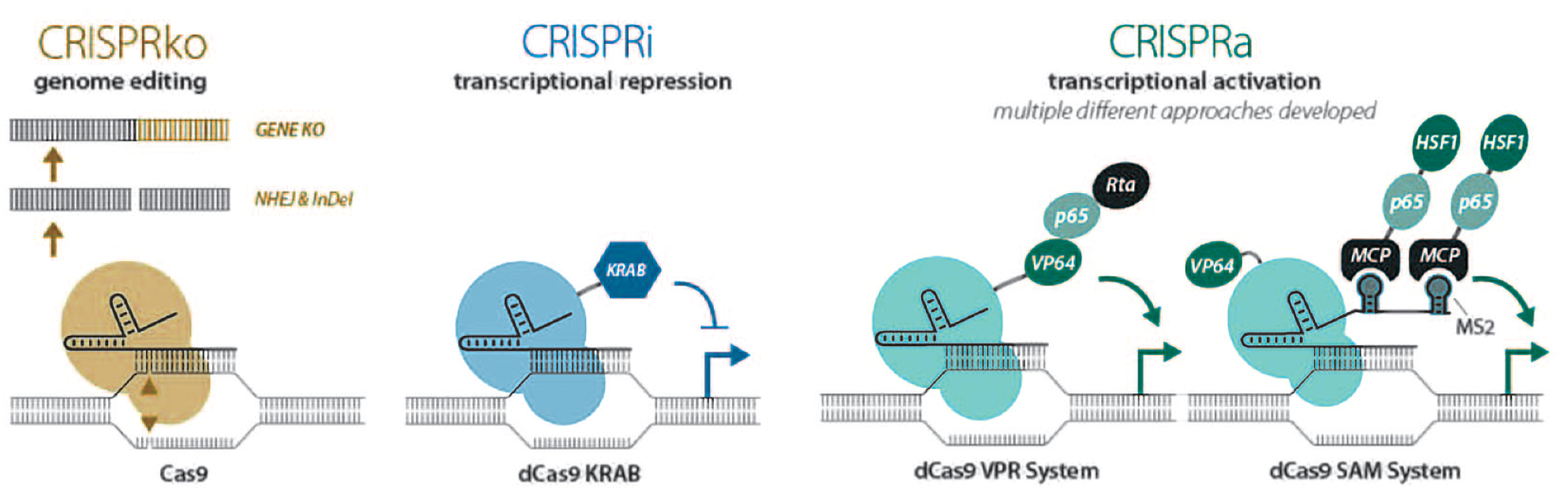
CRISPR screening’s main modalities are for either complete gene ablation via a fully active Cas9 (CRISPRko), a transcriptional suppression approach (CRISPRi) that uses catalytically inactive Cas9 fused to a KRAB domain, or CRISPRa that can drive site-specific transcriptional activation, and has multiple tools published.
The Pooled Approach
Pooled CRISPR screens offer the opportunity to efficiently interrogate cell systems at the whole-genome level with high data resolution. This approach is used to address multiple biological questions, including mechanistic insights into the action of drugs, toxins, and pathogens, or the definition of gene essentiality. However, the experimental design of these screens is key to extracting the most robust and quantitative phenotypic data.
The simplest application of pooled screening is the use of next-generation sequencing (NGS) data to effectively count genotype abundance in each sample or condition (
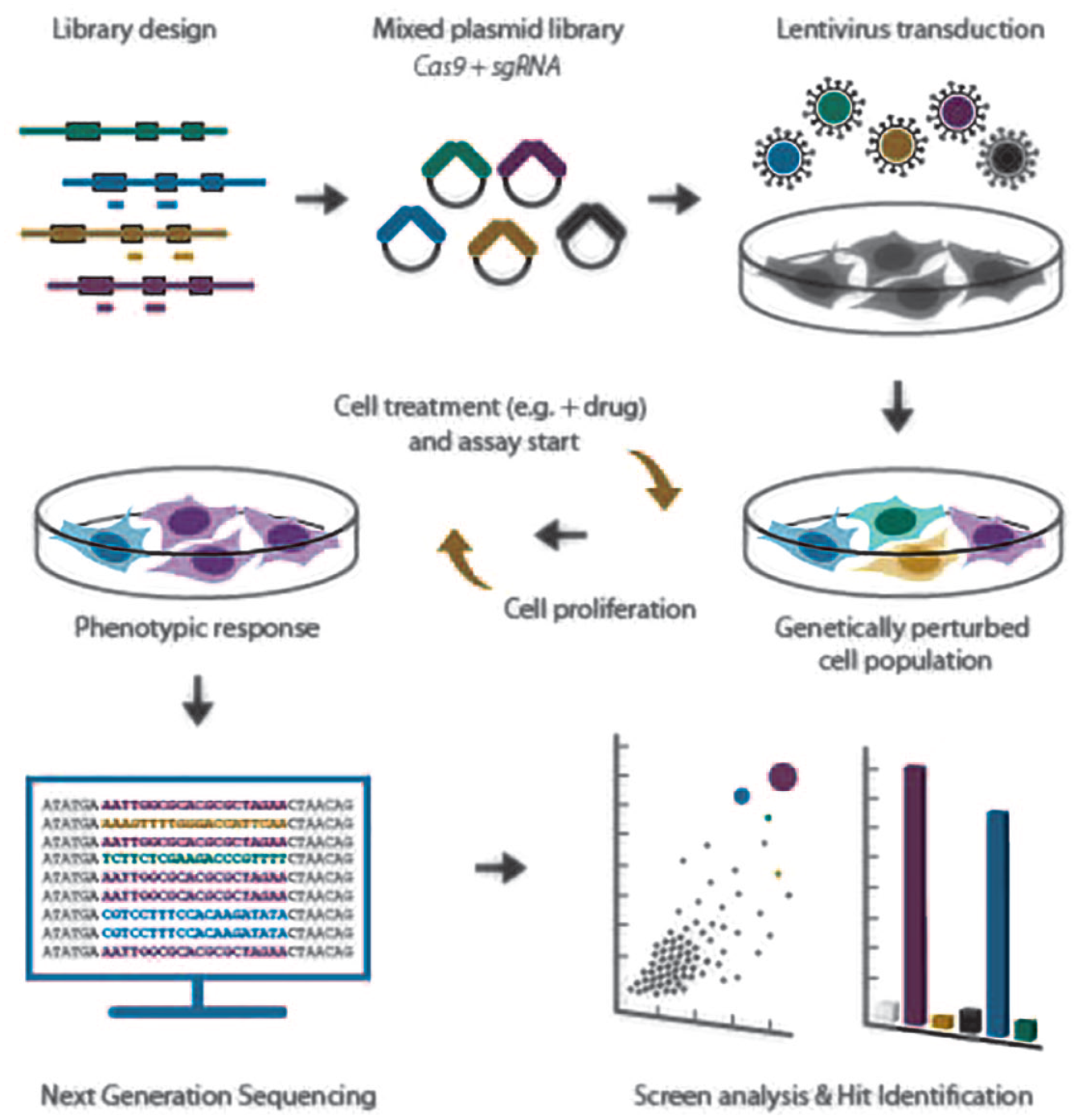
Pooled CRISPR screening workflows begin with the selection of targets and the design of guides associated with those genes. Once synthesized, these guides are then cloned into a suitable library vector and introduced into cells by a lentiviral cassette. Screening assays often involve the addition of a drug (drug–gene interaction screening) or the comparative analysis of variable genotype cells (e.g., genetic interaction screening), but in all cases samples are analyzed by deep sequencing the sgRNA span to quantitatively determine barcode/genotype abundance.
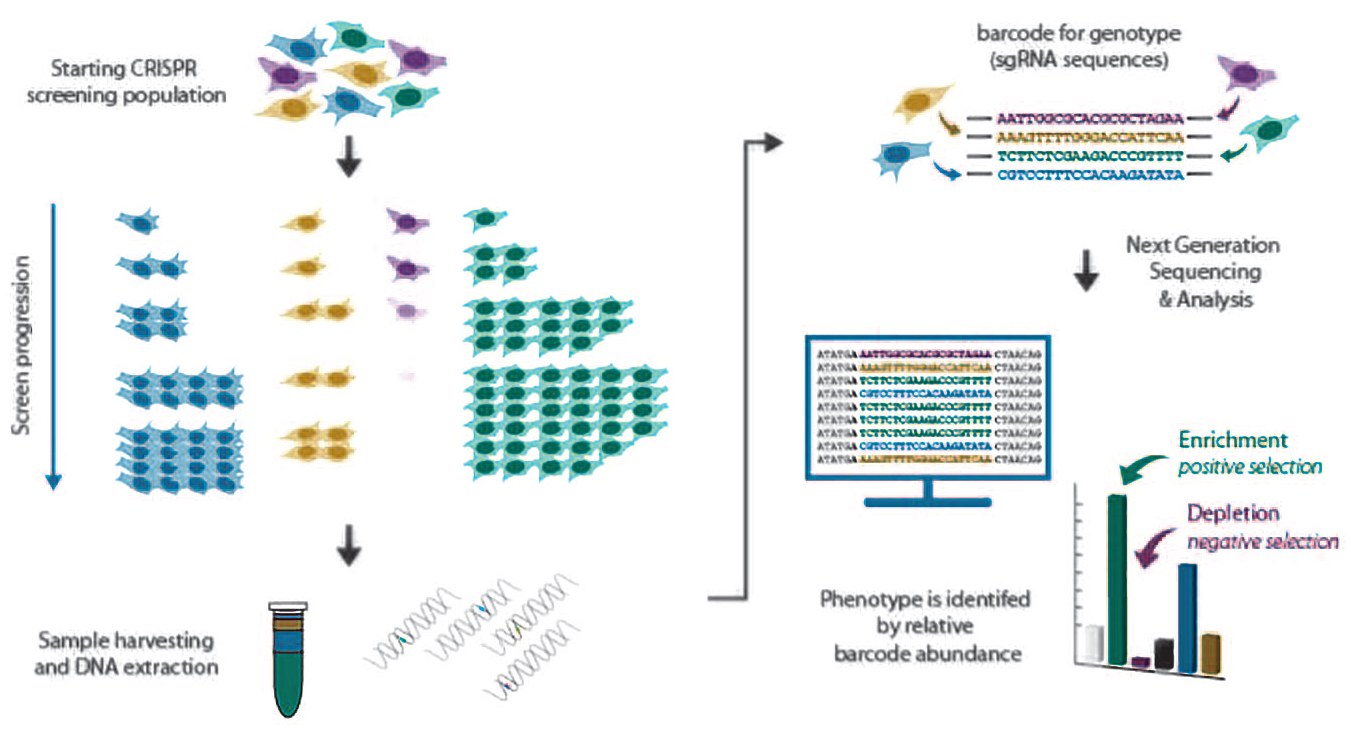
Fundamental properties of pooled CRISPR screening. Depicted are sgRNAs (barcodes) with differential impact on cellular physiology. The blue sgRNAs target gene ablation, which has no positive or negative influence on cell proliferation. The yellow and purple sgRNAs target genes important for cellular fitness and viability, respectively, the loss of which will negatively impact cell survival within the cell population. The green sgRNAs eliminate genes that are normally inhibitory to cell proliferation. The overall impact of sgRNAs on cell physiology is translated into sgRNA abundance, which can be directly measured by NGS.
In the case of positive selection, the most common application is in the discovery of genes that provide resistance to a given cytotoxic agent.19,23,24 Often the cytotoxic agent of interest is a late-stage clinical oncology asset, and pooled screens can be used to start to define the response landscape of the drug. For instance, if you can predict in advance the genotypes that are going to be refractory to the therapy, then the benefits to the clinical study design are obvious. This yields both efficiency and economy, and running a preclinical CRISPR resistance screen for patient stratification is becoming a frequent act of due diligence in drug development.
A good example of a negative selection screen is in the measurement of gene essentiality and the increasingly robust definition of the contribution of each gene in the human genome to cell fitness, which when applied to cancer cells can also be used to map a landscape of gene dependency in cancer.25–28 Gene essentiality studies are arguably the low-hanging fruit for negative selection screening, since loss of cells from a population is the simplest response to monitor and discovery is substantially aided by the a priori knowledge of dozens of highly robust control genes (e.g., genes encoding essential ribosomal components). What has proved more challenging is the adaptation of negative selection to discover drug–gene interactions. Such an approach can be used to identify mechanisms of action or more pertinently to find genes whose loss improves the response to a specific drug (an enhancer screen). The challenge with these approaches is that the datasets are not usually supported by obvious or robust expected outcomes, and the technical aspects of accurately measuring a rare or decreasingly abundant event with NGS render the approach a little noisy.
Overcoming this hurdle has been a major focus of drug discovery CRISPR screeners everywhere, and thankfully there are a number of possible adaptations that can help. The first and simplest is scope: increase the size of the experiment; whether in the number of guides, the cellular coverage, the number of replicates, or the number of cell lines in the study, all of these will improve the chances of robust hit ID by enhancing data reproducibility. 29 Library quality also provides some clear advantages, since the more active elements there are contributing to hit scoring, the greater the sensitivity of the screen can be.30,31 Additionally, some adaptations to the first screening tools used have made a major impact on the screening performance quality and knockout rates.26,32,33 Finally, it is increasingly popular to use multiple independent screening systems to augment the datasets and provide better pathway validation (see below).34,35
The necessity to investigate diseases such as cancer in an environment that recapitulates human disease has led to the development of pooled CRISPR screens in vivo using either cell line-based approaches or patient-derived xenografts (PDXs). 36 In these experiments, cancer cells that have been transduced with a pooled lentiviral library ex vivo can be transplanted into immunodeficient animals. The resulting primary tumor and/or metastatic sites can be harvested and analyzed by NGS to identify genes that selectively alter the growth of the tumor, perhaps in the presence of a late-stage preclinical asset. Better modeling of organism physiology through this approach is a welcome additional tool to pooled CRISPR screening, but results have so far been limited by the minimal implantation rate for most cell systems. This necessarily results in either a reduced complexity library, introducing more noise into the experiment, or a smaller library that targets only a subset of genes. These xenograft approaches contrast with direct in vivo screens, which are more complex and require direct injection of pooled lentiviral or pooled adeno-associated viral particles at the anatomical site of interest to evaluate the impact of the gene perturbation. 37 Both offer alternative approaches to examine functional genomics in more complex physiological systems.
As interest grows in the adaptation of the immune response for healthcare and biotechnology, the value of precise and robust functional genomic approaches in primary immune cells also escalates. Adapting CRISPR screening tools to primary immune cells is challenging and has not been helped by the observation in many labs that the introduction of a nuclease-proficient Cas9 is only feasible using certain delivery routes. In particular, the adaptation of low-throughput electroporation methods for immune cell transfection to high-throughput and high-scale experimentation has facilitated CRISPR screens in primary T cells.38,39 With these tools, a vast array of new analyses in T cells are now possible, and researchers have started to investigate the feasibility of efficient gene editing in other primary tissues and immune cells as a potential prelude to CRISPR screening. 40
Pooled Phenotypic Screening for Complex Disease Analysis
Pooled phenotypic CRISPR screening is an adaptable method to interrogate the phenotypic consequences of gene loss with a high-throughput genomic-based strategy. The more straightforward analysis of screen data by direct NGS quantitation of the preponderance of key sgRNA barcodes can be limiting when it comes to more complex diseases, and a screening mechanism is needed for phenotypes where cellular pathophysiology is uncoupled from cell health. The simplest example of this approach is the use of high-throughput fluorescence-activated cell sorting (FACS) to determine cell phenotypes based on a biomarker signal (
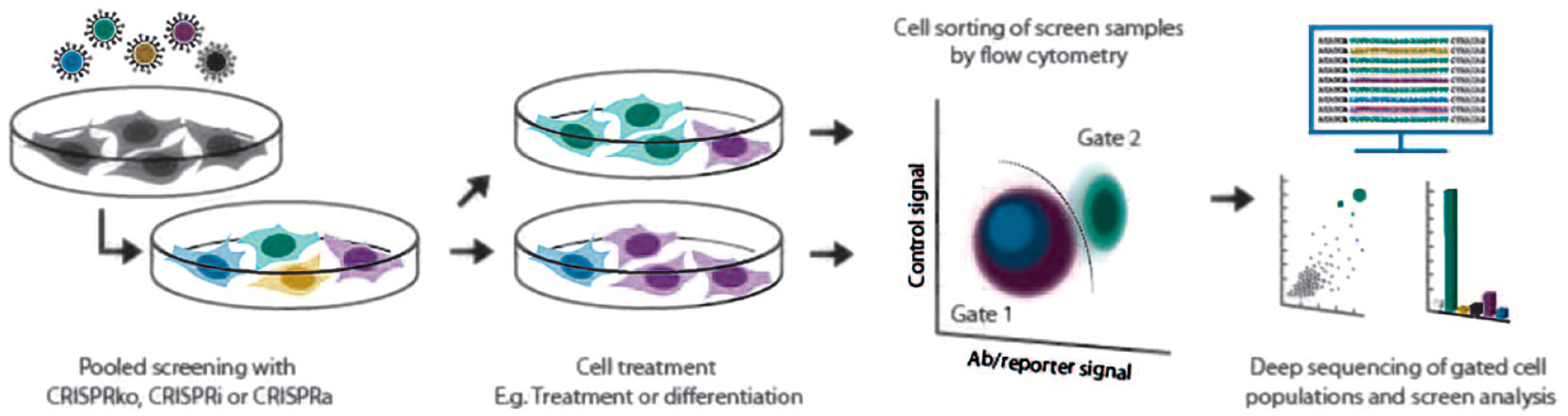
Schematic of a pooled phenotypic CRISPR screen. Detection of changes in the expression of a biomarker, that is, through antibody-based detection of a target molecule, or an endogenous reporter system can be visualized and specifically selected through FACS methods. The sorted populations are then analyzed for enrichment or loss of CRISPR genotypes.
FACS-linked screening has also been used to more precisely monitor genes impacting cell death or cell death pathways. In these cases, a bead-based approach was used to capture the dead cells before they were lost from the population.
42
The value of this approach was not just in the pathway specificity of the readout, but also in the kinetics: proliferation-based readouts often require the cells to undergo several rounds of division to amplify the impact of cell death (
Recently developed platforms that massively increase the phenotypic throughput of CRISPR screening combine CRISPR-based pooled genetic screening with single-cell RNA sequencing (scRNA-seq).43–46 Perturb-seq, CRISP-seq, and CROP-seq are reverse genetic methods that directly link CRISPR-driven genetic perturbations to gene expression phenotypes at the single-cell level (
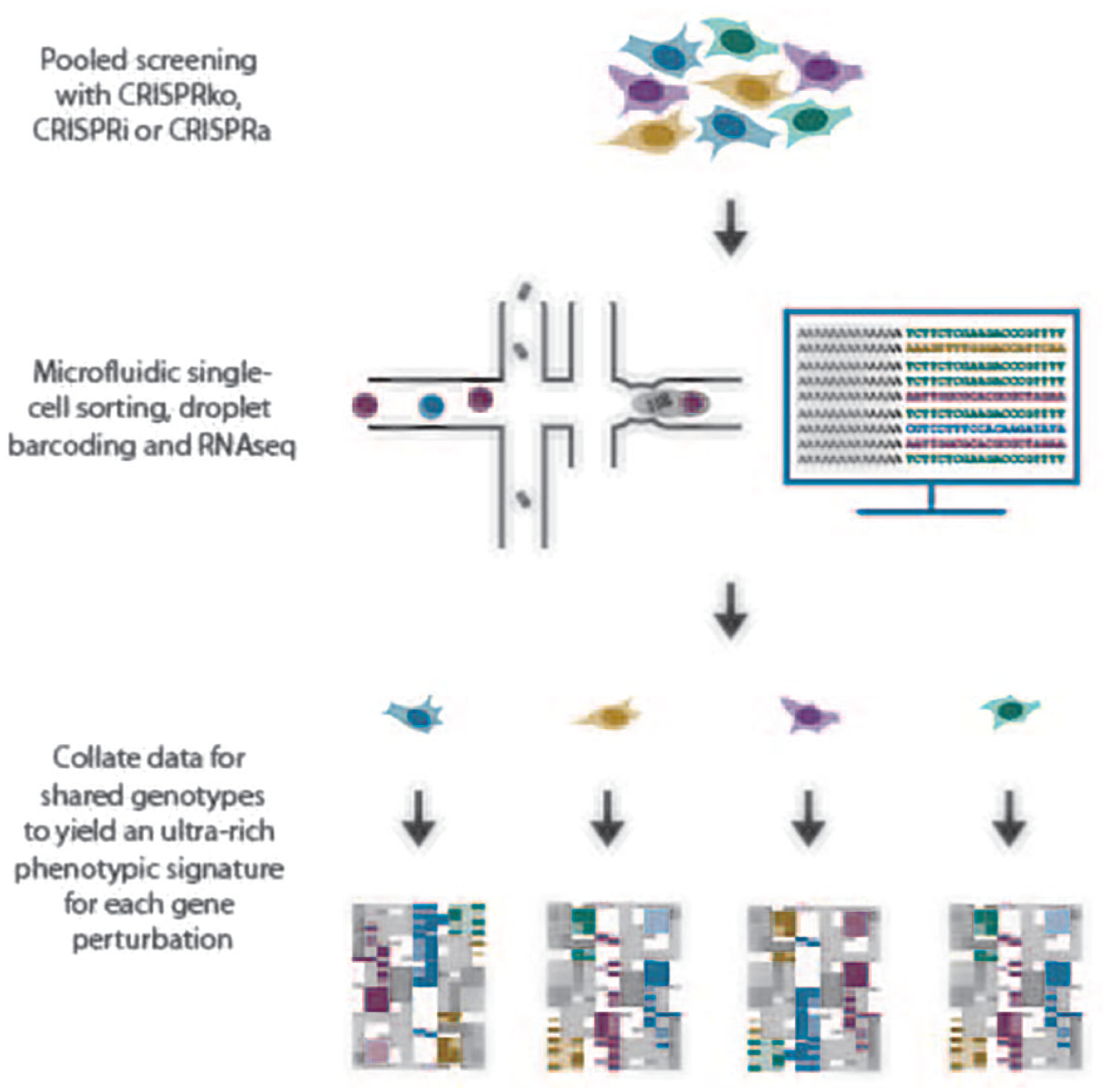
Overview of droplet-based single-cell sorting and subsequent RNA-seq of individual cells originating from a pooled CRISPR screen population. For each genotype introduced into the screen by CRISPR perturbation, a full transcriptomic profile is developed to determine cellular signatures in response to perturbation and treatment.
Arrayed Phenotypic CRISPR Screening: Plugging the Gap
Functional genomic screening in arrayed format offers the possibility to more directly explore complex functional assays, or phenotypic readouts that are less compatible with pooled screening approaches. Arrayed loss- or gain-of-function screens using CRISPRko and CRISPRa reagents offer alternative means to perturbing gene function with either siRNA or cDNA overexpression reagents. An important distinction of CRISPR-based arrayed screens from RNAi, however, is that cells also require Cas9 (or dCas9) present in the cells, adding an extra element to the procedure. This can be achieved by generating a stable cell line expressing Cas9 using a lentivirus prior to screening or by transient introduction of either Cas9 protein or mRNA into the cells (
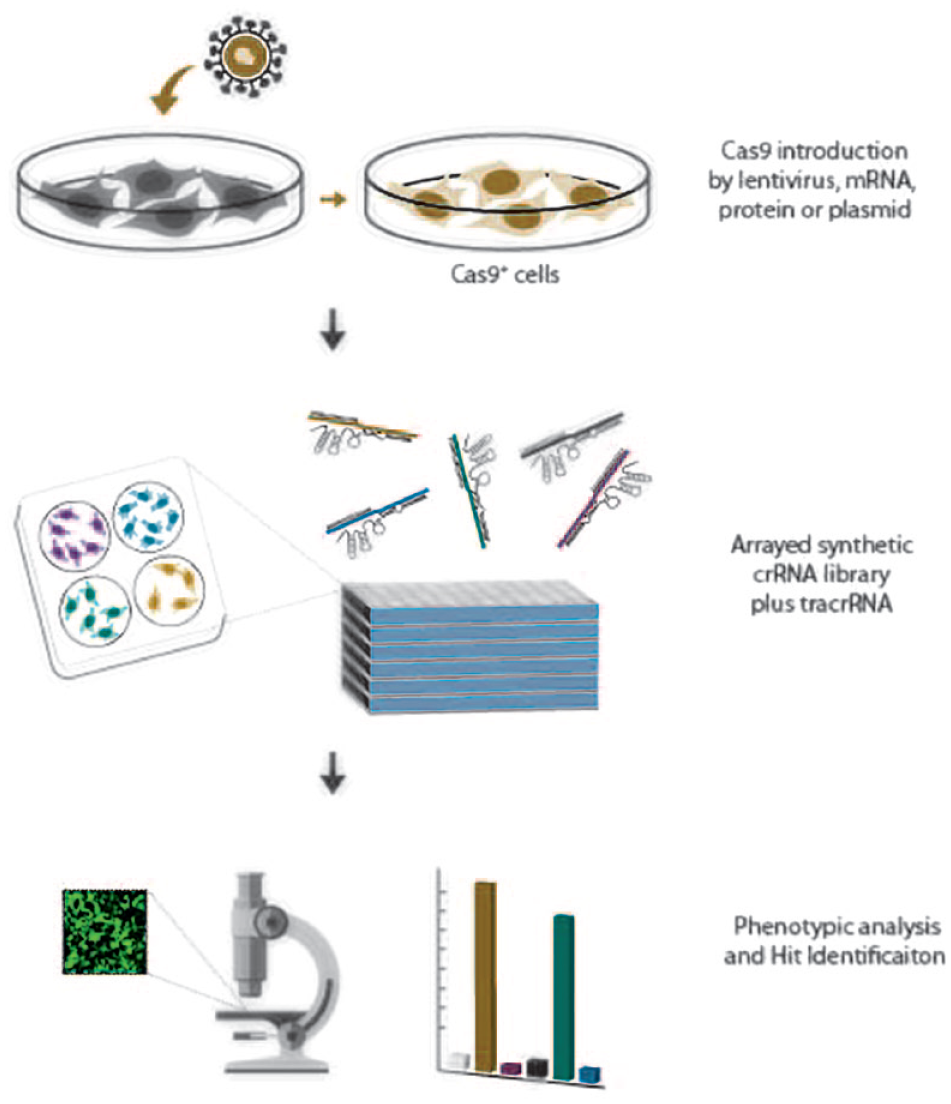
Arrayed CRISPR screening workflow. CRISPR-ready cells can be generated by a two-step process where cells are engineered to express Cas9, or by a one-shot approach where the Cas9 is introduced concomitantly with the sgRNA or crRNA:tracrRNA components. Either way, CRISPR-Cas9 reagents are added to cells in individual wells to allow perturbations to be constrained within individual wells. This has the disadvantage of lower throughput than pooled screening, but the distinct advantage of allowing multiplexed readouts or the monitoring of non-cell-autonomous effects on a well-by-well basis, directly linking phenotype(s) to genotype.
Overall, arrayed CRISPR screening tools allow researchers to conduct a diverse range of complex assays and readouts, including measuring changes in biomarker expression/localization or cell/organelle morphology. The possibility of highly multiplexed assay readouts also significantly increases the value of arrayed approaches.50–52
Doubling Up: Two Heads Are Better than One
One final additional tool in the armory of the CRISPR screener is the recently developed dual screening approach. Simultaneously developed by scientists at Horizon Discovery and the University of California, San Francisco, this technique uses parallel loss- and gain-of-function analyses (e.g., CRISPRi and CRISPRa) and provides the ability to identify genetic interactions through paired gene perturbation analysis. This approach offers researchers a novel, powerful, and systematic method to explore drug mechanisms of action; enables the identification of novel biomarkers; and can provide compelling targets for the development of combination therapies. It can also be used to increase the value of the data from phenotypic screening or even in an in vivo analysis.
Combining screening platforms substantially augments the quality and value of data derived from the screening campaigns, as well as providing novel insights not accessible when using one technology in isolation. Thus, with two opposing orientation datasets in hand, one can cross-validate hits. This approach is of particular value in enrichment-based screening (e.g., resistance screening). One can look for genes that lead to resistance when lost in a CRISPRko screen, but which have no effect or lead to sensitization in a CRISPRa screen. Depleting a target gene and examining the effect of its loss on cell viability might be harder to study with simple loss-of-function screening; in a dual screen the response of cells to hyperactivating a gene on the opposing phenotypic response can be exploited for more robust target ID.
Having both directions of perturbation for each gene also allows for novel hit ID. Gene interactions are complex, and adding a drug response into the mix further complicates the analysis. In a single-direction screen, one depends on the ID of crucial hits based on their variable response to perturbation in one sample versus another. But in cases where deletion is functionally silent due to phenotypic masking, epistasis, or gene dosage effects, the ability to also query gain-of-function can expose an otherwise opaque but critical gene interaction. Dual-direction screening is therefore a potent new tool in the discovery toolbox and is quickly becoming an industry standard for CRISPR-based functional genomic screening.
Conclusions and Outlook
CRISPR has undoubtedly provided researchers with augmented tools for functional genomics, and with new adaptations and innovations arriving all the time, it is an exciting time to be a CRISPR screener. Combining CRISPR screening platforms improves the robustness of screening datasets, and the impact of these tools for drug discovery is really only just starting to be understood. This also extends to the additional value of other genomic tools, such as RNAi, where notwithstanding the buzz and excitement around CRISPR it is important to understand the value and carefully consider the application of all the tools available. In an age of personalized medicine, functional genomics is key and creativity, expertise, and pragmatism in screening are crucial if these tools are to yield benefits for patients.
Footnotes
Acknowledgements
The authors thank Nicola McCarthy for insightful comments and suggestions while preparing this manuscript.
Declaration of Conflicting Interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: All authors are employees of Horizon Discovery Ltd.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.