
Abstract
Posttranslational modifications of histones play an important role in the regulation of gene expression and chromatin structure in eukaryotes. The balance between chromatin factors depositing (writers) and removing (erasers) histone marks regulates the steady-state levels of chromatin modifications. Here we describe a novel microscopy-based screening method to identify proteins that regulate histone modification levels in a high-throughput fashion. We named our method CROSS, for Chromatin Regulation Ontology SiRNA Screening. CROSS is based on an siRNA library targeting the expression of 529 proteins involved in chromatin regulation. As a proof of principle, we used CROSS to identify chromatin factors involved in histone H3 methylation on either lysine-4 or lysine-27. Furthermore, we show that CROSS can be used to identify chromatin factors that affect growth in cancer cell lines. Taken together, CROSS is a powerful method to identify the writers and erasers of novel and known chromatin marks and facilitates the identification of drugs targeting epigenetic modifications.
Introduction
The accessibility of the eukaryotic genome is compromised by the dense compaction of DNA in the form of chromatin. The nucleosome represents the repeating unit of chromatin and consists of two copies of each of the canonical histones (H3, H4, H2A, and H2B) around which ~146 bp of genomic DNA is wrapped. The molecular structure of the nucleosome revealed the protrusion of the N- and C-terminal tails of histones from the globular particle. 1 A large body of work during the past decade has revealed that these accessible parts of the core histones can be modified by specific enzymes. These modifications include the acetylation and methylation of lysine residues, which are mediated by lysine acetyltransferases (KATs) and lysine methyltransferases (KMTs), respectively. Enzymes that deposit histone modifications are commonly referred to as chromatin “writers.” 2 Although often referred to as epigenetic marks, all types of histone modifications are biochemically reversible. 3 Histone acetylation is removed by histone deacetylases (HDACs and sirtuins), whereas histone methylation is erased by lysine demethylases (KDMs). These enzymes are classified as chromatin “erasers.” It has become increasingly clear that mutations in chromatin-modifying enzymes are important drivers of tumorigenesis.4,5 Together, these findings also indicate that the modification state of chromatin is highly dynamic and results from the interplay between enzymes depositing and removing chemical groups. Further-more, disturbance of the epigenetic landscape can severely affect cell fate and homeostasis.
A third class of chromatin proteins is the so-called readers. A chromatin reader is defined as a protein bearing a domain that can specifically recognize a particular histone modification. Examples of such reader domains include bromodomains, which bind to acetylated lysines, and PHD fingers, which “read” methylated lysines. Other described readers include Chromo, Tudor, MBT, and PWWP domains, each of which displays affinity for a particular histone modification.3,6–8 Chromatin-binding protein complexes have often evolved to contain multiple reader domains for different modifications. Histone modifications co-occurring on active or repressive chromatin can be recognized by readers in a combinatorial manner, thus giving rise to a high-affinity interaction driven by multiple relatively low-affinity single interactions. This also allows for a sophisticated fine-tuning of interactions rather than simple binary kinetics. Together, this illustrates the combinatorial nature or “crosstalk” of histone modifications. 9 An example is the binding of TFIID to chromatin. The TFIID subunit TAF3 reads trimethylated histone H3 lysine-4 (H3K4me3) via a PHD finger in its C-terminus. 10 Binding of TFIID to chromatin is further enhanced by acetylation of histone H3. These marks are recognized by the double bromodomain of TAF1. 10 Deregulation of chromatin readers can also result in cancer as exemplified by the oncogenic fusion of the nuclear pore complex subunit NUP98 to the C-terminal PHD domain of the KDM5A/JARID1A demethylase for H3K4 or the PHD domain of PHF23 protein. This fusion of NUP98 to a PHD domain is one of the most prevalent gene rearrangements found in various forms of hematopoietic malignancies. 4 The oncogenic potential of the fusion protein relies on its ability to bind to H3K4me2/3-modified nucleosomes, preventing demethylation and thereby inactivation of developmentally crucial genes. 4
Efforts to determine which histone modifications co-occur on chromatin and therefore function in chromatin signaling pathways have revealed crosstalk between distinct histone modifications. 3 Relevant in this respect are seminal contributions made by GPS (global proteomic analysis in Saccharomyces cerevisiae) developed by Schneider and colleagues. 11 GPS relies on screening protein extracts of the yeast deletome library (~4800 viable single-gene knockout strains) by immunoblotting with histone modification-specific antibodies. This unbiased approach revealed that the PAF1 elongation complex and H2B mono-ubiquitylation by Rad6/Bre1 are upstream of H3K4 methylation by the Set1/COMPASS complex. 12 The GPS screening method involved a significant amount of experimental work (~4800 lysates on ~300 separate blots per antibody tested). Coverage of mammalian genomes would increase the effort by a factor of four.
Here we describe a novel approach based on high-throughput microscopy to investigate histone modification pathways in human cells. We generated a small interfering RNA (siRNA) library targeting the expression of 529 genes, including annotated writers, erasers, readers, and cofactors of chromatin-modifying complexes. Following siRNA-mediated knockdown in a 96-well format, quantitative immunofluorescence microscopy with histone modification-specific antibodies is used to determine changes in global histone modification levels. We name this method CROSS for Chromatin Regulation Ontology SiRNA Screening. As a proof of principle, we used CROSS to screen H3K4 and H3K27 methylation in U2OS and MCF7 cells. In both cell lines, we identified the writers and erasers for these marks. We also used CROSS to identify chromatin factors that affect cell growth, which in the future could provide novel candidates for the development of drugs targeting epigenetic modifications in relation to cancer.
Materials and Methods
Antibodies
Antibodies used are listed here: H3 (ab1791; Abcam, Cambridge, UK), H3K4me1 (pAb037-050; Diagenode, Liège, Belgium), H3K4me2 (pAb035-050; Diagenode), H3K4me3 (pAb003-050 [Diagenode] or 04-745 [Millipore, Billerica, MA]), H3K27me1 (pAb045-050; Diagenode), H3K27me3 (pAb069-050; Diagenode), H3K27ac (pAb174-050; Diagenode), H3K9me1 (ab8896; Abcam), H3K9me3 (ab8898; Abcam), ASH2L (kind gift from Winship Herr), RBBP5 (BL766; Bethyl Laboratories, Montgomery, TX), GAPDH (clone 6C5, mAb374; Millipore), and EZH2 (clone AC22, #3147; Cell Signaling Technology, Danvers, MA).
Cell Lines
Osteosarcoma cells (U2OS) and breast carcinoma cells (MCF7) were obtained from the ATCC (American Type Culture Collection, Manassas, VA) and cultured in Dulbecco’s modified Eagle’s medium (DMEM; Lonza, Verviers, Belgium) supplemented with 10% fetal bovine serum (Lonza) and antibiotics (100 U/mL penicillin and 100 µg/mL streptomycin, Lonza) at 37 °C, 5% CO2 in a humidified atmosphere.
siRNA Library
Based on the chromatin library described in Mulder et al., 13 we included complex members of chromatin modifiers and binders. The chromatin library, targeting expression of 529 genes, was purchased from Thermo Fisher Scientific Dharmacon (Lafayette, CO, custom-made library, order numbers 245120-245146, 245170, and 246203). A full gene list is given in the supplemental data.
siRNA Transfection
Cells were transfected in a reverse transfection protocol using Hiperfect (Qiagen, Valencia, CA) according to the manufacturer’s instructions. In short: 5 µL of a SMARTpool of four siRNAs (25 nM each) from the chromatin library was dispensed in 96-wells plates (Costar, 3599; Corning, New York, NY) (triplicates for each screen). Transfection mix (0.4 µL Hiperfect in total volume of 15 µL Opti-MEM [Invitrogen, Carlsbad, CA] per well) was added using the multidrop dispenser (Multidrop combi Type 836; Thermo Scientific, Waltham, MA), followed by a 30-min incubation at room temperature (RT). Meanwhile, cells were counted and 3000 cells/well were added (in 80 µL). Cells were fixed 72 h after transfection.
Fluorescent Microscopy
Depending on the antibody, cells were fixed using formaldehyde or cold methanol. In case of formaldehyde, cells were fixed for 20 min at RT with 4% formaldehyde in phosphate-buffered saline (PBS), followed by a 30-min permeabilization with 0.5% Triton X-100 in PBS and a 10-min incubation with 50 mM glycine in PBS. In case of methanol, cells were fixed and permeabilized by a 5-min incubation with cold methanol, after removal cells were rehydrated by washing twice with PBS, followed by washing once with PBS/0.1% Triton X-100. Cells were blocked using 5% NGS in PBS/0.1% Triton X-100, followed by incubation with appropriate antibodies diluted in blocking solution. After antibody staining, cells were incubated with DAPI solution (2 mg/L 4′,6-diamidino-2-phenylindole [Sigma-Aldrich, St. Louis, MO] in 50 mM Tris-HCl [pH 7.4], 100 mM NaCl).
Antibody Quality Control
Antigen specificity was tested using in-house synthesized biotin-labeled peptides, both in immunofluorescence microscopy competition assays as well as on immunoblots with the appropriate peptides. For competition assays, peptides were added in a 2-µM concentration to the primary antibody solution. To prepare immunoblots, peptides were bound to streptavidin, run on a 10% SDS-PAGE, and transferred onto PVDF membranes. The membrane was developed with the appropriate antibodies and ECL (Pierce, Thermo Fisher Scientific Inc., Rockford, IL).
CROSS Data Analysis
Average nucleus fluorescence intensity values and cell count per field were extracted from the microscopic images using Cellomics vHCS: View 1.6.1.613 (Thermo Fisher Scientific Inc., Rockford, IL). Robust z scores were calculated using R version 2.15.1 (2012-06-22) for all variables per plate for all chromatin library siRNAs using equation (1):
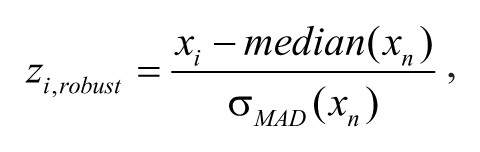
whereby
CROSS robust z score data sets of the same residues were merged. Genes that were significantly up- or downregulating global histone modification levels (z < –1.96 or z > 1.96 unless otherwise noted) in two of three replicates in at least one screen were selected. To exclude effects caused by cell death or growth effects, we excluded genes showing a significant reduction in objects per field. When necessary, known associated genes were added to the list (WDR5, RBBP5, ASH2L, and DPY30 for H3K4 and EZH2, EED, SUZ12, and KDM6A for H3K27).
Clustering of Growth Affecting Genes
CROSS H3K4 robust z score data sets were merged, and genes that were significantly downregulating valid objects per microscopic field (z < –1.96) in two of three replicates in at least one screen were selected. Clustering was performed on Euclidean distance using Ward’s method.
MTT Assay
Wild-type U2OS and MCF7 cells were seeded at 1000 cells per well in 100 µL normal DMEM medium in a 96-well plate. After 24 h, medium was replaced by medium containing either I-BET inhibitor (I-BET151 at concentrations of 0.1, 0.3, 1, 3, or 10 µM), 0.1% DMSO, 2 mg/L puromycin, or medium without addition. Seventy-two hours after adding I-BET, MTT (3-(4,5-dimethylthiazolyl-2)-2, 5-diphenyltetrazoliumbromide; Sigma-Aldrich) was added to a final concentration of 0.5 mg/mL. 14 Cells were incubated for 4 h under normal culturing conditions, followed by careful removal of medium, and 200 µL DMSO was added to dissolve the formazan crystals. Spectramax M5E (Molecular Devices, Sunnyvale, CA) was used to measure absorbance at 570-nm and 630-nm wavelengths. Relative growth was calculated using equation (2):
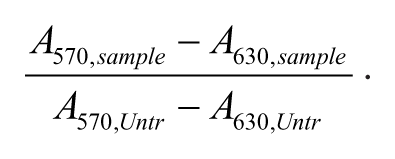
Crystal Violet Staining
For long-term colony formation assays, 2000 cells/well were seeded in 12-well plates. I-BET151 was added at 0.5 and 1 µM; DMSO was used as vehicle control. Cells were incubated at 37 °C for 6 days. After the incubation period, the medium was aspirated and cells were fixed with ice-cold methanol (100%) for 10 min at room temperature, with a subsequent staining with crystal violet (0.25% w/v crystal violet in 25% methanol) for 10 min. Cells were washed with water and air-dried overnight.
Results
CROSS for H3K4 Methylation Identifies Known Major Methyltransferase and Demethylase Complexes
Epigenetic modifications are affected in a variety of tumors, and these aberrant modification patterns can significantly alter gene expression patterns. We therefore decided to profile a subset of histone modifications in two distinct human cancer cell lines, MCF7 (breast carcinoma derived) and U2OS (osteosarcoma derived), using immunoblotting and immunofluorescence ( Fig. 1A , B ). Immunoblotting revealed comparable levels of H3K4 methylation (mono, di, and tri), as well as H3K27me1 and H3K9 methylation (mono and tri) between U2OS and MCF7 ( Fig. 1A ). Interestingly, H3K27me3 levels are greatly reduced in U2OS compared with MCF7, while H3K27ac is higher in U2OS. The difference in H3K27me3 levels was confirmed by immunofluorescence ( Fig. 1B ). EZH2 expression, however, is similar in both cell lines ( Fig. 1A ). This indicates that factors other than EZH2 may be responsible for the reduced H3K27me3 levels in U2OS cells compared with MCF7.
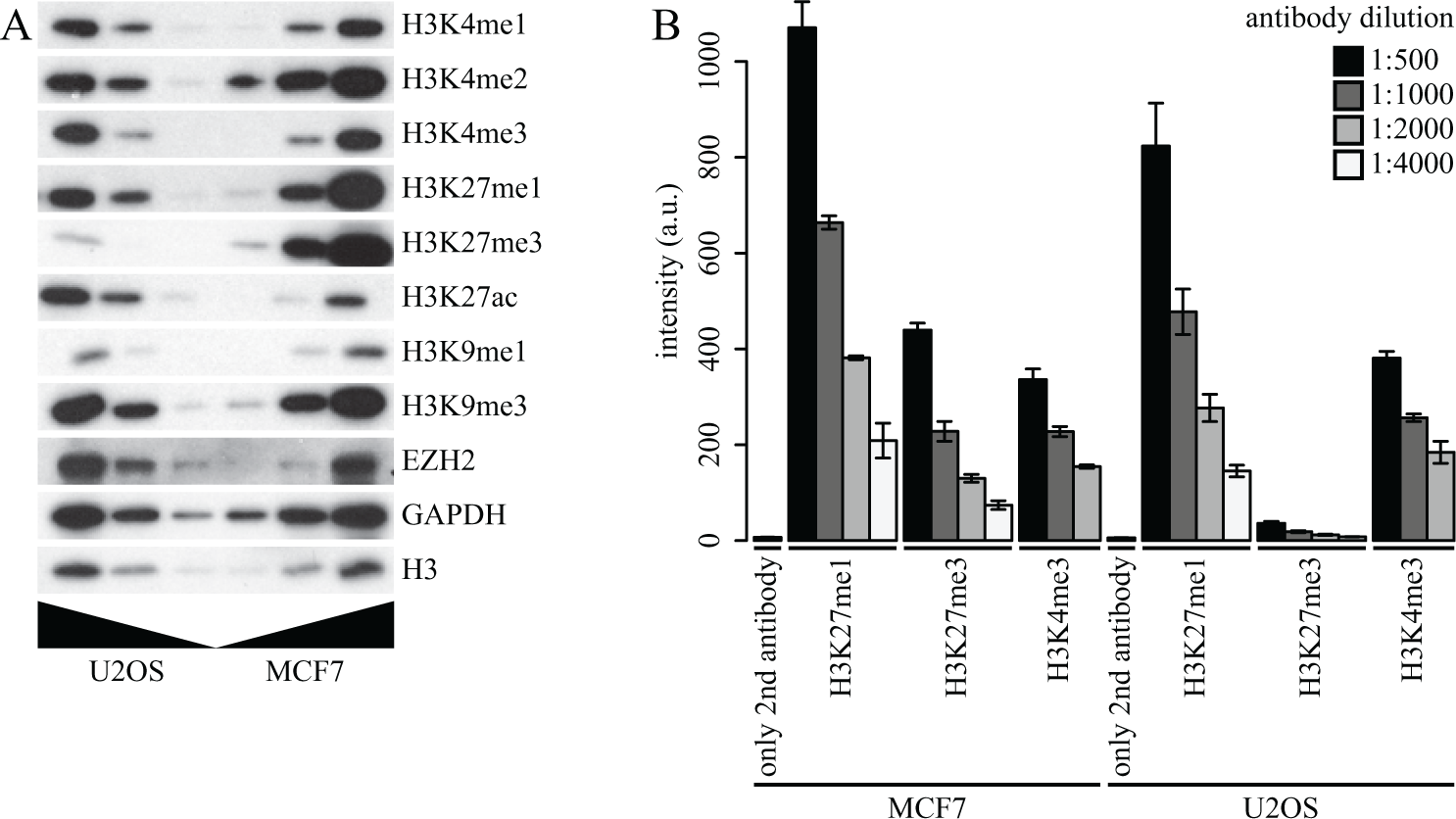
Global histone mark levels in U2OS and MCF7. (
To identify complexes or cofactors that influence the global levels of chromatin modifications, we combined siRNA-mediated knockdown with high-throughput immunocytochemistry and automated microscopy. We named this method CROSS for Chromatin Regulation Ontology SiRNA Screening. To this end, a custom siRNA library was designed targeting human chromatin-associated proteins, including writers, erasers, readers, and cofactors of known chromatin-modifying complexes, resulting in a gene list of 529 genes (full list in
We applied CROSS first to H3K4 methylation, a well-characterized histone modification for which the enzymes, their complexes, and even regulatory pathways are known.
15
In short, U2OS and MCF7 cells were reverse transfected in triplicate using the chromatin library consisting of 529 siRNAs divided over a series of 96-well plates (21 plates per antibody staining). Cells were fixed 72 h after transfection and stained using three antibodies that recognize respectively histone H3K4 mono-, di-, or trimethylation (
Each individual plate also contained nontargeting, ASH2L, and PLK1 siRNA controls. These controls are expected to respectively show no effect on H3K4 methylation signal (negative control, nontargeting siRNA), a reduction in H3K4 methylation signal (positive control, siRNA targeting ASH2L), and cell mitotic defects resulting in reduction of cell count and an increase of H3K4 methylation signal caused by nuclear condensation (siRNA targeting PLK1). Robust z scores for these controls are plotted in
Occasionally, knockdown of a gene causes growth effects such as a cell cycle arrest or even apoptosis. In these cases, chromatin structure and function are severely altered, thereby influencing the readout of CROSS. Therefore, we also calculated robust z scores on the amount of selected cell nuclei per field. This allowed us to filter out genes that induce cell cycle arrest or apoptosis. Remaining genes that influenced global H3K4 methylation levels with a z score of 1.96 (corresponding to the 95% confidence interval) in either positive or negative direction in at least two of three technical replicates in at least one of the three modification screens (H3K4 mono-, di-, or trimethyl) were selected and subjected to hierarchical clustering (
Fig. 2A
and
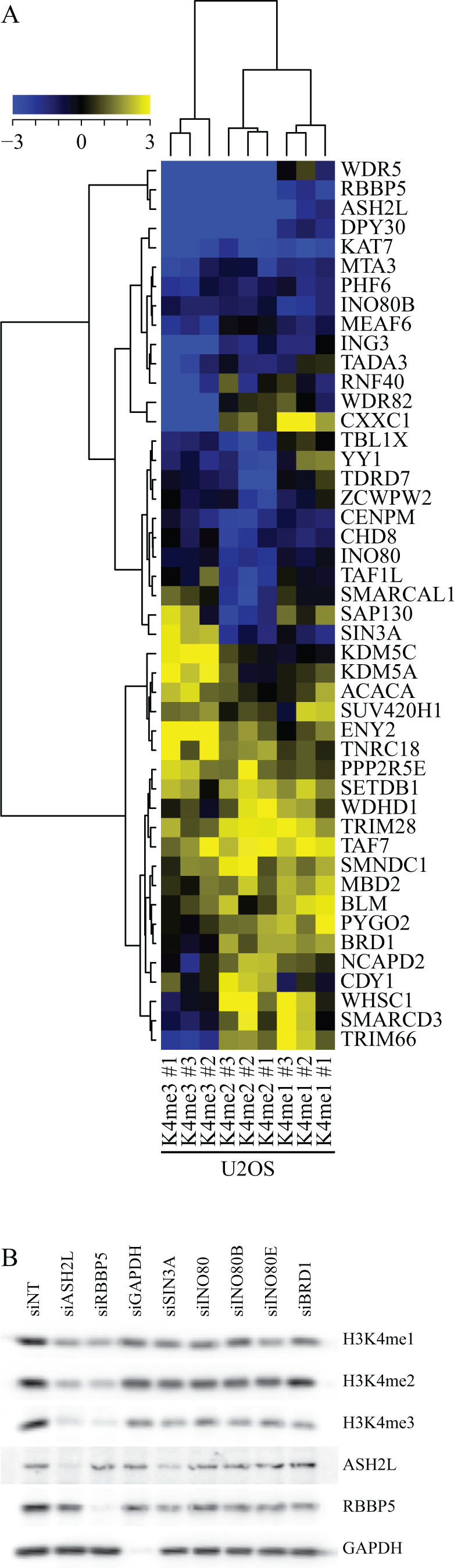
CROSS for H3K4 methylation in U2OS cells. (
In addition to these expected methyltransferase and demethylase complexes, a number of other proteins that affect H3K4 methylation levels either positively or negatively were identified. Examples include subunits of the INO80 chromatin remodeling complex as well as subunits of the SIN3 histone deacetylase complex and the BRD1 bromo-domain protein. Currently, it is not clear how the known function of these proteins could affect H3K4 methylation levels, but further experiments could help to shed light on this.
As a confirmation of our CROSS results, we performed immunoblot with a selection of hits from CROSS. Knockdown of ASH2L and RBBP5 reduced all three methylation states of H3K4 (
Fig. 2B
). However, no effect on methylation levels detected by immunoblotting was obvious after knockdown of INO80 complex, SIN3A, or BRD1 (
Fig. 2B
and
Cell-Type Specific K27 Methylation Patterns and Regulation in U2OS and MCF7 Cells
Using CROSS, genes affecting global levels of histone modifications can be identified. To measure these changes, a histone modification of interest should be of reasonable abundance under normal culturing conditions. As H3K27me3 is of low abundance in U2OS cells ( Fig. 1A , B ), we applied CROSS for H3K27me3 only in MCF7 cells, whereas CROSS was applied for H3K27me1 in both cell lines ( Fig. 3 ). As expected, knockdown of PRC2 complex subunits EZH2, SUZ12, and EED results in a reduction of global H3K27me3 levels in MCF7 cells, confirming the ability of CROSS to reveal the enzyme(s) and associated factors that regulate particular histone modification(s). Interestingly, knockdown of PRC2 subunits has different effects on H3K27me1 in MCF7 and U2OS cells. While depletion of PRC2 results in a downregulation of H3K27me1 in U2OS cells, this effect is not observed in MCF7 cells. This may imply that in MCF7 cells, H3K27me1 may be (partially) catalyzed by a PRC2-unrelated methyltransferase. Knockdown of the known H3K27 demethylase KDM6A (aka UTX) showed an overall increase in H3K27 methylation levels in MCF7 but not U2OS, again showing the potential of CROSS to identify novel epigenetic regulators.

CROSS for H3K27 methylation in U2OS and MCF7. Hierarchical clustering of mono- and trimethylation of H3K27 in U2OS and MCF7. Robust z scores were calculated using an R script (see Materials and Methods section, with negative z < –1.20). Replicates are indicated on the bottom and identified hits are on the right. Coloring (ascending from blue to yellow via black) is based on the robust z score for the indicated antibody.
CROSS Can Reveal Cell Line–Specific Growth Effects on Gene Knockdown
Due to their general importance inside the nucleus, knocking down chromatin-associated proteins can in some cases induce a growth defect. To measure these effects in U2OS and MCF7 cells, we calculated robust z scores for the amount of cells that were present per optical field. Clustering analysis of these data revealed a clear separation of the two cell lines (
Fig. 4A
). Three clusters of genes can be identified: genes that specifically reduce cell numbers in U2OS (majority of the genes), genes leading to a reduction in cell number in MCF7, and genes that affect growth in both cell lines (PLK1, SGOL1, and ESPL1). In the cluster of genes affecting growth in MCF7, the BET proteins BRD2 and BRD4 are found. BRD4 also reduces growth in U2OS, although to a lesser extent than in MCF7. In contrast, BRD2 does not affect cell growth in U2OS (
Fig. 4A
), while knockdown is efficient (
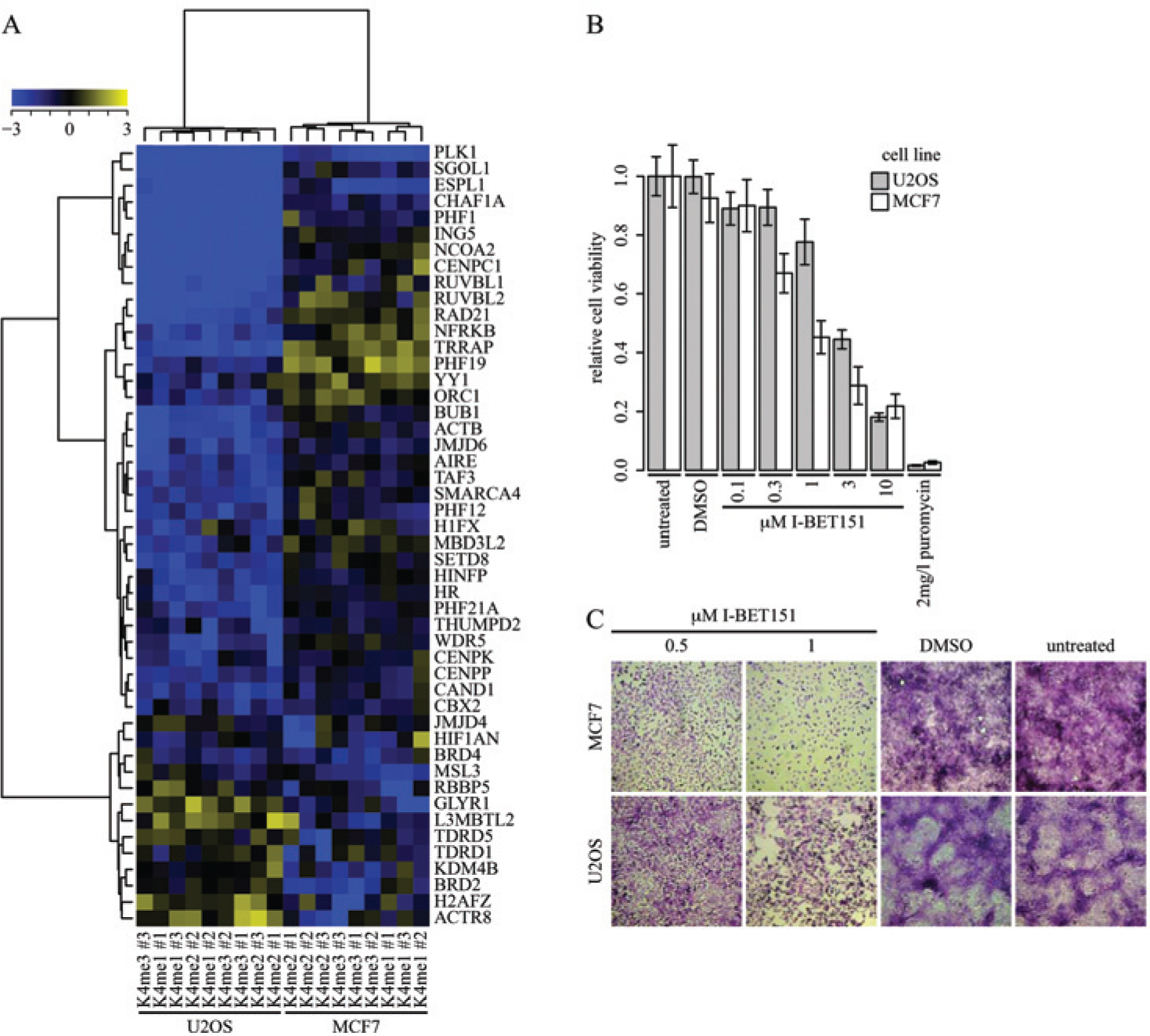
Effect on cell growth on U2OS and MCF7 by siRNA transfection and I-BET151 treatment. (
Discussion
Here we describe the development of a new method (CROSS) to identify writers and erasers of epigenetic marks. As a proof of principle, we identified the SET/MLL complexes to be involved in the methylation of histone H3K4, as well as the lysine demethylases KDM5A and KDM5C in the removal of H3K4me3, but not H3K4me1 or -me2. Similarly, we used CROSS to identify the writers and erasers of H3K27 methylation (PRC2 and KDM6A/UTX, respectively). Finally, we show that CROSS can be used to identify chromatin proteins that are essential for growth in cancer cell lines.
Histone H3K4 Methylation
Using CROSS, we identified the interaction partners of the writers involved in H3K4 methylation. The H3K4 methyltransferases themselves, however, do not show reduced global methylation levels when depleted in cells. A single methyltransferase complex (COMPASS/Set1) catalyzes H3K4 methylation in yeast, while in mammals, at least six different COMPASS-related complexes are known to methylate H3K4. Possibly each of these methyltransferase complexes catalyzes only a minority of the total amount of H3K4 methylation in the cell lines that we used, and CROSS is not sensitive enough to detect such moderate reductions in global modification levels. In support of this, the knockdown of subunits that are shared by multiple methyltransferase complexes such as WDR82 and CXXC1 (SET1A/SET1B complexes) resulted in a detectable reduction of global H3K4me3 levels. These observations are also in agreement with observations from the Shilatifard laboratory. 23 Interestingly, we found that knockdown of RNF40 results in reduced H3K4me3 levels in U2OS ( Fig. 2A ). This is in agreement with the observation that ubiquitination of H2B is required for H3K4 trimethylation. 12 Thus, CROSS can reveal crosstalk between different histone modifications. Recently, it was found that subunits of the Drosophila methyltransferase Trr complex (the MLL3/4 homolog) are involved in the monomethylation of H3K4 in chromatin areas with an enhancer-like signature. 24 However, we did not find any effect of the MLL3/4-specific subunits (KDM6A/UTX, PA1, or PTIP) on the global levels of monomethylation. Still, removal of these subunits using knockdown by siRNA might have effects at specific genomic locations but not on the global levels of H3K4me1.
Histone H3K27 Methylation
Trimethylation of histone H3K27 is mediated by the PRC2 complex. How monomethylation is established is less clear. 25 We used CROSS to identify the writers and erasers of H3K27me1. In U2OS, knocking down PRC2 components EZH2, SUZ12, and EED results in reduced H3K27me1 levels ( Fig. 3 ). In contrast, knockdown of these proteins in MCF7 has little effect on the levels of H3K27me1. Interestingly, knocking down two H3K9 methyltransferases, EHMT1/EHMT2, reduced H3K27me1 in MCF7 cells ( Fig. 3 ). In the deconvolution of these data (transfecting the single oligonucleotides instead of the gene pool of four siRNAs), these observations could not be confirmed (data not shown). Further experiments are therefore required to investigate these observations.
Interestingly, our experiments revealed that U2OS and MCF7 have markedly different levels of H3K27me3, even though EZH2 protein levels in both cell lines are similar ( Fig. 1A , B ). Although at this point, the molecular mechanism underlying this difference is unclear, aberrant H3K27me3 levels can have a profound impact on cell proliferation. This is evident from recent publications that link altered PRC2 activity to cancer. 26 In hormone-independent growing prostate cancer cells, EZH2 appears to have an activating, PRC2-independent role. 27 Also, H3K27 mutations were linked to gliomas. 28 Given these findings and the recent development of highly specific EZH2 inhibitors,29,30 PRC2 in general and EZH2 in particular will remain a major focus, both in fundamental and clinical research.
CROSS to Identify Targets for Epidrugs
During normal development, the cell fate switch from proliferation to differentiation is driven by epigenetic alterations. It is therefore not surprising that epigenetic regulators, when mutated, are important drivers of cancer. Unlike genetic mutations, altered epigenetic profiles are in principle all reversible, which makes them attractive targets for the development of so-called epidrugs.4,5 Our CROSS setup to screen for growth effects in U2OS and MCF7 ( Fig. 4A ) could be expanded to a larger panel of cell lines corresponding to different tumor stages and also different tumor origins. Such experiments could lead to the identification of novel targets for drugs.
CROSS in Perspective: Limitations and Strength
CROSS analysis on H3K4 and H3K27 methylation identified the known major complexes targeting these marks (
Figs. 2A
and
3
). Inspection of the data reveals that the dispersion in the MCF7 CROSS data is larger than in U2OS (
On the basis of our experiments, we estimate that a knockdown efficiency of >50% is required to have an effect on global histone modification (
Currently, the limitation of our experiments is the use of a dedicated library of 529 genes based on known domains and complexes involved in writing, reading, and erasing histone and DNA modifications, as used for proof of principle of CROSS. Clearly, this library is biased toward “known” histone and chromatin biology, and opportunities lie in the use of a genome-wide siRNA library. Such experiments could reveal the interplay between chromatin and metabolism, for example. This is particularly important given the recently emerging strong link between chromatin, metabolism, and cancer. For example, it was recently shown that threonine levels in embryonic stem cells directly influence H3K4me3 levels. 33 Furthermore, metabolites that are part of the TCA cycle such as succinate directly affect the enzymatic activity of lysine demethylases.34,35 Clearly, additional crosstalk between epigenetics and seemingly unrelated cellular pathways and processes are yet to be found, and our CROSS method will be a powerful tool to facilitate such discoveries.
Footnotes
Acknowledgements
We thank Wienand Omta and Arne Smits for help and useful discussions on mathematics and R scripts, Simona Antonova for technical assistance, and the whole Timmers/Vermeulen group for useful discussions. We thank Jan Hendrickx and Didier Allaer from Diagenode (Belgium) for providing antibodies and sharing with us the H3K27ac antibody before it was commercially available. I-BET151 was a kind gift of Dr. Michael D. Woodrow and Dr. Rab Prinjha from GlaxoSmithKline (United Kingdom). Anti-ASH2L was a kind gift of Professor Winship Herr (UNIL, Switzerland).
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by an NWO-Chemical Sciences TOP grant (700.57.302) and an NWO-Life Sciences grant (821.02.012), both to H.Th.M.T.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.