
Abstract
Gene silencing by RNA interference has become a powerful tool to help identify genes that regulate biological processes. However, the complexity of the biology probed and the incomplete validation of the reagents used make it difficult to interpret the results of genome-wide siRNA screens. To address this challenge and maximize the return on the efforts required for validating genomic screen hits, the screening strategy must be designed to increase the robustness of the primary screening hits and include assays that inform on the mechanism of action of the knocked-down transcripts. Here, we describe the implementation of a small interfering RNA (siRNA) screen to identify genes that sensitize the effect of poly–(ADP ribose)–polymerase (PARP) inhibitor on cell survival. In the strategy we designed for the primary screen, two biological activities, apoptosis and cell viability, were measured simultaneously at different time points in the presence and absence of a PARP inhibitor (PARPi). The multiplexed assay allowed us to identify PARPi sensitizers induced by both caspase-dependent and independent mechanisms. The multiplexed screening strategy yielded robust primary hits with significant enrichment for DNA repair genes, which were further validated using relevant high-content imaging assays and confirmation of transcript knockdown by real-time PCR (rtPCR).
Keywords
Introduction
Gene silencing by RNA interference has become a powerful tool to identify genes that regulate biological processes both in vitro and in vivo. The ability to use automated robotic systems to perform lipid-based small interfering RNA (siRNA) transfections has enabled the large-scale implementation of high-throughput genome-wide siRNA screens. 1 As more experience is gained with siRNA high-throughput screening (HTS), data analysis and interpretation have proven to be significantly complex. The reasons for this include the inherent off-target effects of current siRNA designs, variance in the data measurements inherent in cell-based assays, and biological variance seen from knocking down intended and unintended genes. 2 As a consequence, physiologically relevant genes often have weak activity in phenotypic assays and are missed during primary HTS. It is therefore critical to develop siRNA screening strategies that can counteract and overcome these limitations.
Several strategies have been published to address the issues described above and improve the quality of hits from siRNA HTS. The current practical solution to address siRNA off-target effects due to the knockdown of messenger RNA (mRNA) for unintended genes is to test several different siRNA duplexes targeting different regions of the gene for their ability to induce the same phenotype. 3 A successful probability-based approach has been demonstrated in which six siRNA duplexes targeting each gene were tested and a redundant gene analysis was used for hit selection. 4 Although this method addresses the issue of off-target effects at the primary level, it is resource intensive and does not address the issue of biological variance and the ability to find weak hits in a primary HTS.
Here we describe the design of a strategy involving the use of a multiplex readout and its application in an siRNA HTS campaign. This campaign was aimed at identifying genes synthetically lethal with poly–(ADP-ribose)– polymerase 1 (PARP1) enzyme inhibitors as potential drug targets for tumor suppression. PARP has a well-established role in DNA repair processes, and small-molecule inhibitors of PARP have been developed as chemotherapy sensitizers for the treatment of cancer. 5 It has been demonstrated that BRCA1 and BRCA2 dysfunction sensitized cells to the inhibition of PARP enzymatic activity, resulting in genomic instability, cell cycle arrest, and apoptosis.6,7 These results also indicated that PARP inhibitors target the homologous recombination (HR) deficiency in BRCA-deficient cells. PARP1 is essential for the repair of single-strand DNA breaks (SSBs), and PARP inhibitors cause an increase in persistent SSBs. 8 In addition, SSBs, when encountered by replication forks, cause fork collapse and the formation of double-stranded breaks (DSBs). 5 A reasonable explanation is that in the presence of excessive SSBs and resultant collapsed replication forks, HR-deficient cells are unable to maintain the integrity of the genome and become nonviable. The demonstration that PARP inhibition is selective for BRCA1 or BRCA2 deficiency suggests that PARP inhibitors may be particularly useful for the treatment of cancer with BRCA mutations. In addition, the identification of novel mediators of cellular response to PARP inhibitors may highlight additional patient populations that might benefit from this therapeutic approach. 9
To identify genes synthetically lethal with a PARP inhibitor, we implemented a multiplexed readout, multitemporal, and multiconditional screening strategy to test siRNA pools (three siRNA/pool) targeting the 23 866 gene transcripts encoded by the whole human genome. The data were analyzed to monitor whether selection of siRNA hits using multiple parameters produced genes that were biologically enriched for relevant processes. As part of the hit validation strategy, single siRNA duplexes from the initial siRNA pool hits as well as different siRNA designs were tested using cell viability and caspase activation (apoptosis) multiplexed readouts and additional imaging-based phenotypic assays relevant to DNA damage repair and cell cycle pathways. Genes that were identified as being biologically relevant were also confirmed by real-time PCR (rtPCR) to validate knockdown of the intended targets. Our data indicate that using a ratiometric ± drug cell viability and caspase readout strategy for hit selection in the primary screen returned the most biologically relevant hits. The overlap of hits between the two assays was significantly enriched in genes involved in DNA repair. Hits exclusive to viability showed enrichment in caspase-independent pathways. On-target hit rates were similar as determined by using single siRNA duplexes from initial pools tested and pools containing siRNA duplexes with alternative design. This work illustrates that implementing methods to enrich the primary screen for biologically relevant genes allows for the identification of genes to define nodes of cellular pathways.
Materials and Methods
Cell line
Hela cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM; cat. 11965-092; Gibco, Carlsbad, CA) with 10% fetal bovine serum (cat. 10082-147; Gibco) and 1% penicillin-streptomycin (cat. 15140-148; Gibco). Cells were dissociated using TripLEexpress (cat. 12605-010; Gibco) and resuspended at a final concentration of 15 000 cells/mL. A MultiDrop instrument (Thermo Electron Corp., Waltham, MA) was used to dispense 40 µL of cell suspension to each well of a black, clear-bottom tissue culture–treated 384-well assay plate (cat. 353988; BD, Franklin Lakes, NJ) to give a final cell density of 600 cells/well.
siRNA transfection
The siRNA transfection was carried out 24 h after cell plating. A 384-well tissue culture–treated assay plate (cat. 3712; Corning, Lowell, MA) served as an intermediate plate for preparation of the siRNA/lipid complex. siLentFect (cat. 170-3362; Bio-Rad, Hercules, CA) was diluted at a 1:80 concentration into OptiMEM media (cat. 31985-070; Invitrogen, Carlsbad, CA), and 30 µL was added to each well of the intermediate plate with a CyBi Well instrument (CyBio, Jena, Germany). Next, 3 µL of 5 µM siRNA was added to each well of the intermediate plate using the CyBi Well Replicator (CyBio). Following a 40-min incubation for siRNA/lipid complex formation, 5 µL of this mixture was added to a 384-well assay plate and thoroughly mixed using a CyBi Well instrument (CyBio). Four replicate assay plates were prepared from each of the intermediate plates. In addition to siRNA library test sequences, four control sequences were added to each assay plate. These control sequences targeted PLK, luciferase, BRCA1, and RAD51 mRNA and were included for their biological relevance.
Drug addition
Four hours following transfection, two plates from each set of replicate assay plates received PARP inhibitor (“plus-drug” condition), whereas the other two received DMSO control (“no-drug” condition). PARP inhibitor and DMSO were diluted to a final concentration of 10 µM in complete media and added to the cells using a Multidrop (Thermo Electron Corp.). One set of each no-drug/plus-drug plate pair was screened after a 48-h incubation at 37 °C, whereas the other plate pair was screened after 96 h.
Cell viability and apoptosis assays
Following an incubation of either 48 or 96 h after drug addition, an Alamar Blue–based viability assay was performed and multiplexed with a caspase-3/7–based apoptosis assay. Cell growth media were removed from the assay plate using a CyBi Well (CyBio). Then, 40 µL of 10% of Alamar Blue cell viability reagent (cat. DAL1100; BioSource, Chevy Chase, MD) diluted with OptiMEM (cat. 31985-070; Invitrogen) and 2% fetal bovine serum (FBS; cat. 10082-147; Invitrogen) was dispensed to assay plates with a MultiDrop (Thermo Electron Corp.) Plates were incubated for 2 h at 37 °C. Plates were then read on the AcQuest plate reader (Molecular Devices, Sunnyvale, CA) using fluorescence intensity with an excitation wavelength of 545 nm and emission wavelength of 590 nm. Following this read, Alamar Blue solution was removed using a CyBi Well (CyBio). Then, 60 µL of Caspase-Glo (cat. G8092; Promega, Madison, WI) diluted with 1× phosphate-buffered saline (PBS; cat. 14040-133; Gibco) in a 1:1 ratio was then dispensed to the assay plates with a MultiDrop (Thermo Electron Corp.) Plates were incubated for 2 h at room temperature and luminescence signal was measured using an EnVision plate reader (PerkinElmer, Waltham, MA), with a 1-s exposure time.
siRNA HTS
In the primary screen, 23 886 pools of three different siRNA oligonucleotides targeting the same gene transcript were tested under four different conditions: 48-h incubation ± PARP inhibitor and 96-h incubation ± PARP inhibitor. %Viability was calculated from the Alamar Blue cell viability data, whereas a ratio of apoptosis activity over cell viability was calculated from the Caspase-Glo and Alamar Blue assays. Each assay plate also contained positive controls for transfection efficiency and drug sensitivity. Each plate contained eight wells each of BRCA1 and RAD51 siRNA pools to serve as biological controls for drug sensitivity. Each plate contained 16 wells of PLK1 to serve as positive controls for transfection efficiency. Each plate also contained 16 wells of luciferase and mock control, which served as indicators of toxicity of the siRNA and transfection lipid.
Cell imaging assays
Transfections and drug additions were carried out using the same methods as for the primary screen. Following a 72-h incubation period, 25 µL of 12% paraformaldehyde (PFA) solution was added to each assay plate using a WellMate (Thermo Fisher, Waltham, MA). Assay plates were incubated for 20 min at room temperature and then washed three times with Tris-buffered saline (TBS; cat. T664; Sigma-Aldrich, St. Louis, MO) using an Embla 384 plate washer (Molecular Devices). Then, 20 µL of 2% bovine serum albumin (BSA; cat. A9576; Sigma-Aldrich) with 0.5% Triton X-100 (cat. 234729; Sigma-Aldrich) was added with a WellMate (Thermo Fisher) and plates were incubated for 20 min at room temperature. Next, 20 µL of primary antibody was added with a WellMate (Thermo Fisher) and plates incubated for 1 h at 37 °C. Assay plates were washed three times with TBS using an Embla 384 (Molecular Devices). Then, 20 µL of secondary antibody was added with the WellMate (Thermo Fisher). Assay plates were incubated for 1 h at 37 °C. Assay plates were washed three times with TBS using an Embla 384 (Molecular Devices). Then, 20 µL DAPI (cat. D1306; Invitrogen) was added to the assay plates for a final concentration of 250 ng/mL with the WellMate. Following a 20-min incubation, assay plates were washed three times with TBS on the Embla 384 (Molecular Devices) and images were acquired. Primary/secondary antibody combinations used were as follows: anti–cleaved caspase-3 (D175) (CST; rabbit, 1:200)/Alexa 488 antirabbit IgG (goat, 1:500; Molecular Probes, Eugene, OR), anti–phosphohistone H3 (S10) (PH3; CST, mouse, 1:500)/Alexa 647 antimouse IgG (goat, 1:500; Molecular Probes), anti-Rad51 (H-92) (SCBT; rabbit, 1:200)/Alexa 488 antirabbit IgG (goat, 1:500; Molecular Probes), and anti–phosphohistone H2A.X (S139) (PH2A.X; mouse, 1:500; Upstate Scientific, Lake Placid, NY)/Alexa 647 antimouse IgG (goat, 1:500; Molecular Probes). Detection of cleaved caspase-3 was multiplexed with PH3, as well as the detection of Rad51 with PH2A.X.
Image acquisition and analysis
Images were acquired on the InCell 3000 automated confocal microscope and then followed by iCyte (Berkeley, CA) in situ cytometric measurement for cell cycle profiling. Individual nuclei were identified based on size and intensity above background, and the boundary was dilated to include both nuclear and cytoplasmic areas. The cells with signals above a set threshold for each of the stains were classified as “positive” or “negative.” The percentage of positive cells (%pos) was calculated for each well, for each of the four markers. The total number of cells was reported as Ntot. An additional algorithm for the identification of foci associated with RAD51 or PH2A.X was developed and reported as Fgran. Manual fitting of DNA content histograms was generated on iCyte by setting the regions for G1, S, and G2/M cell cycle stages. Data were generated from image analysis, and statistical analyses were performed on triplicate results. Results from the imaging assays were rescaled between 0 and 1. A hierarchical clustering analysis was performed using unweighted average method and Pearson similarity measure.
Validation of knockdown by rtPCR
Transfection conditions were carried out using the same methods as in the primary screen. One difference is that cells were plated at a density of 900 cells/well to detect sufficient gene expression. Forty-eight hours later, siRNA transfected cells were lysed using cells-to-ct reagents (4399002; Life Technologies, Carlsbad, CA) in triplicate and followed by reverse transcription and PCR reactions on the 7900HT thermal cycler (Applied Biosystems, Foster City, CA). %mRNA remaining was calculated from the delta Ct values for transfected and untransfected cells.
Results
siRNA HTS to identify genes that enhance the cellular antiproliferative activity of PARP inhibitors
A multiplex cell-based assay that measures cell viability and apoptosis in the same plate was designed for a genome-scale siRNA screen. Using this assay in 384-well assay plate format, siRNA pools to 23 886 unique genes were screened in Hela cells to identify genes that, when silenced, enhance cell killing in the presence of a PARP inhibitor. Cell viability was measured with the Alamar Blue assay, whereas apoptosis was measured by detecting caspase activity via the Caspase-Glo 3/7 assay. This assay detects the activities of caspases-3 and -7, both of which are effector proteases involved in apoptosis. To increase the probability of the screen to identify genes with a weak phenotype and/or requiring the cells to go through few cell cycles to manifest their genotoxic effect, we ran the multiplex viability/apoptosis assay at multiple time points, 48 and 96 h posttransfection. These time points were determined to be optimal during assay development for finding both fast-acting and slow-acting genes that sensitize in the presence of PARP inhibitor (PARPi). This screening strategy is summarized in
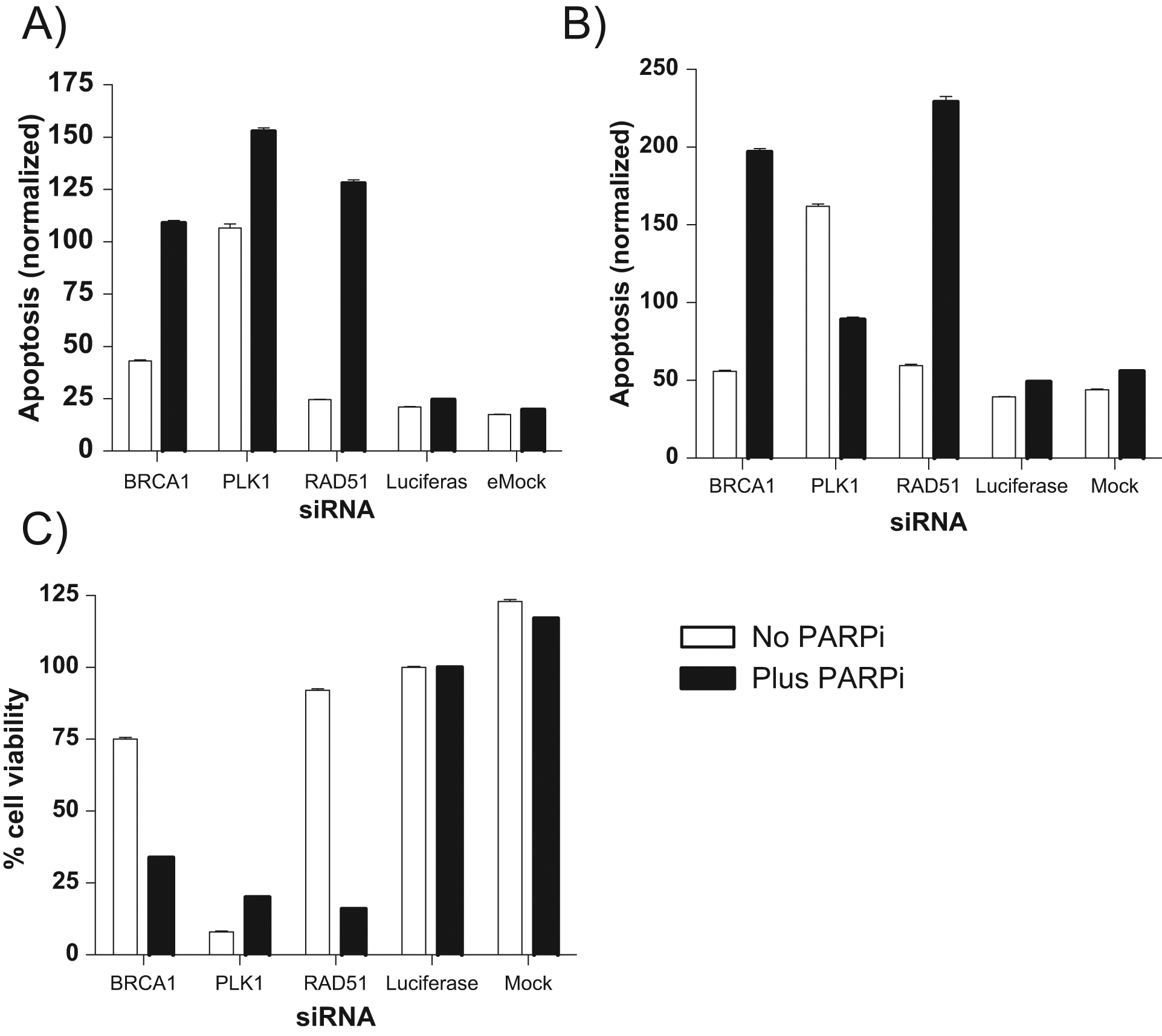
Activity profiles of known control genes. Activity profiles of BRCA1 and RAD51, both known to be sensitized in the presence of poly–(ADP ribose)–polymerase inhibitor (PARPi). Polo-like kinase 1 (PLK1) was used as a positive control for transfection efficiency. Luciferase was used as a control for small interfering RNA (siRNA) toxicity. Mock was used as a control for transfection toxicity. (
The results from the screen are shown in Figure 2A . Here, each pair of scatterplots represents the correlation between cell viability (x-axis) and caspase activity (y-axis) at a 48-h (top plots) or 96-h (bottom plots) time point, plus or minus the PARPi. It is clear from these plots that the positive siRNA controls BRCA1 (data points in green), and RAD51 (data points in red) induces cell sensitization to PARPi. The separation of the positive controls (BRCA1 and RAD51) from the negative control luciferase (blue data points) is good at both time points, with a marked left and upward shift of the positive control in the assay readouts.
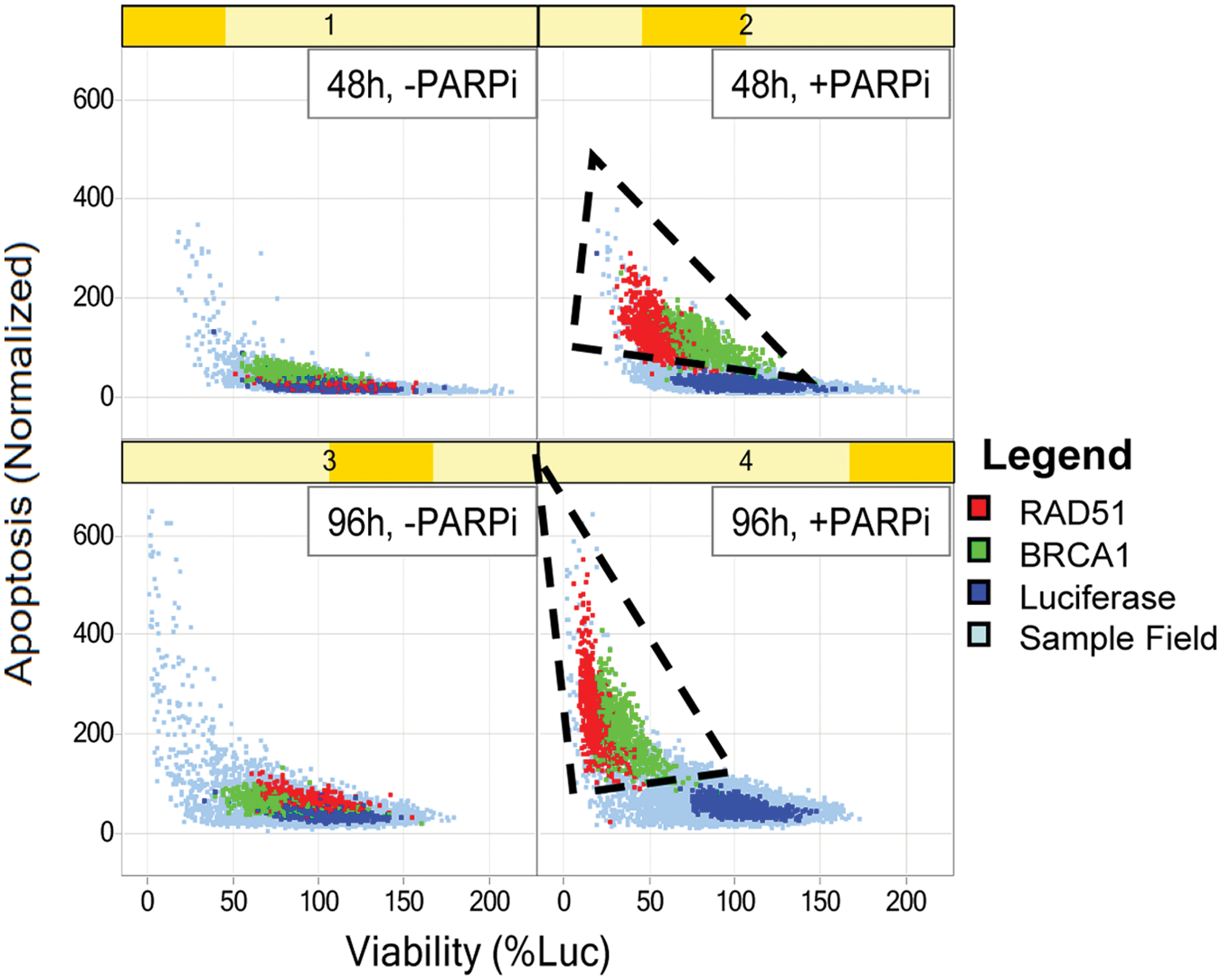
%Viability and apoptosis scatterplots of primary screen. In these scatterplots, %Viability is plotted on the x-axis, whereas apoptosis is plotted on the y-axis. They show the behavior of the control small interfering RNA (siRNA) used in this assay, at each time point, for both plus and minus poly–(ADP ribose)–polymerase inhibitor (PARPi).
Hit selection strategy
To score hits, we recalculated viability and apoptosis results as a log ratio (base 2) of +PARPi over −PARPi. Following the generation of ratios, z* scores, a statistical scope for the distance between individual well data and sample median expressed in multiples of MAD (median absolute deviation), were calculated at both “local” (per plate) and “global” (entire screen) levels using the following equation10,11:

This metric was used for hit selection as it is a more favorable method to use in a primary screen without replicates. 12 The local scores compensate for any plate-to-plate variations that might have been encountered during the screen, whereas the global scores ensure the selection of true positives in siRNA library plates that are enriched with specific pathways. 11 Scatterplots in Figure 3A , B , 96-h viability data, show an overlay of z* scores over the ratio of +drug/−drug calculation to illustrate these two hit selection methods. Figure 3A shows how hits can be selected in less noisy plates, by doing so on a plate-by-plate basis. Figure 3B shows how hits can be selected on a global basis to account for noisier plates in which individual siRNA library plates might have been enriched in specific pathways. A similar procedure was carried out for the caspase data at both the 48-h and 96-h time points. z* scores <−3 (viability) and >3 (apoptosis) were the cutoffs used to select genes that sensitized in the presence of PARPi. This hit selection strategy likely increased the false-positive rate in the confirmation screen but at the same time decreased false negatives, which is of higher importance in any nonreplicated, primary screen. 13 In total, 808 siRNA pools were selected as sensitizers for confirmation. The Venn diagram in Figure 3C indicates the assay that was the source of the selected hits. We evaluated our multiplexed screening strategy by analyzing the hits exclusive to viability and caspase and those that were in common between the two assays. Through analysis of the hits exclusive to viability ( Fig. 4A ), we found enrichment in DNA repair, response to DNA damage, and various elements of the cell cycle. Through analysis of the hits exclusive to caspase activity (apoptosis) ( Fig. 4B ), we found enrichment in DNA repair and sister chromatid cohesion. Through analysis of the hits in common between the two assays, we found enrichment in genes that were hits in both the viability and apoptosis assays that were specifically enriched for the double-stranded break repair process, which is the specific process in which PARP participates ( Fig. 4C ). Individually, both caspase and viability hits showed enrichment in common pathways expected to be found in the PARP pathway. Taking the two assays together, we found additional significance based on higher expectation values in the hits that overlapped in both assays, rather than a single assay.
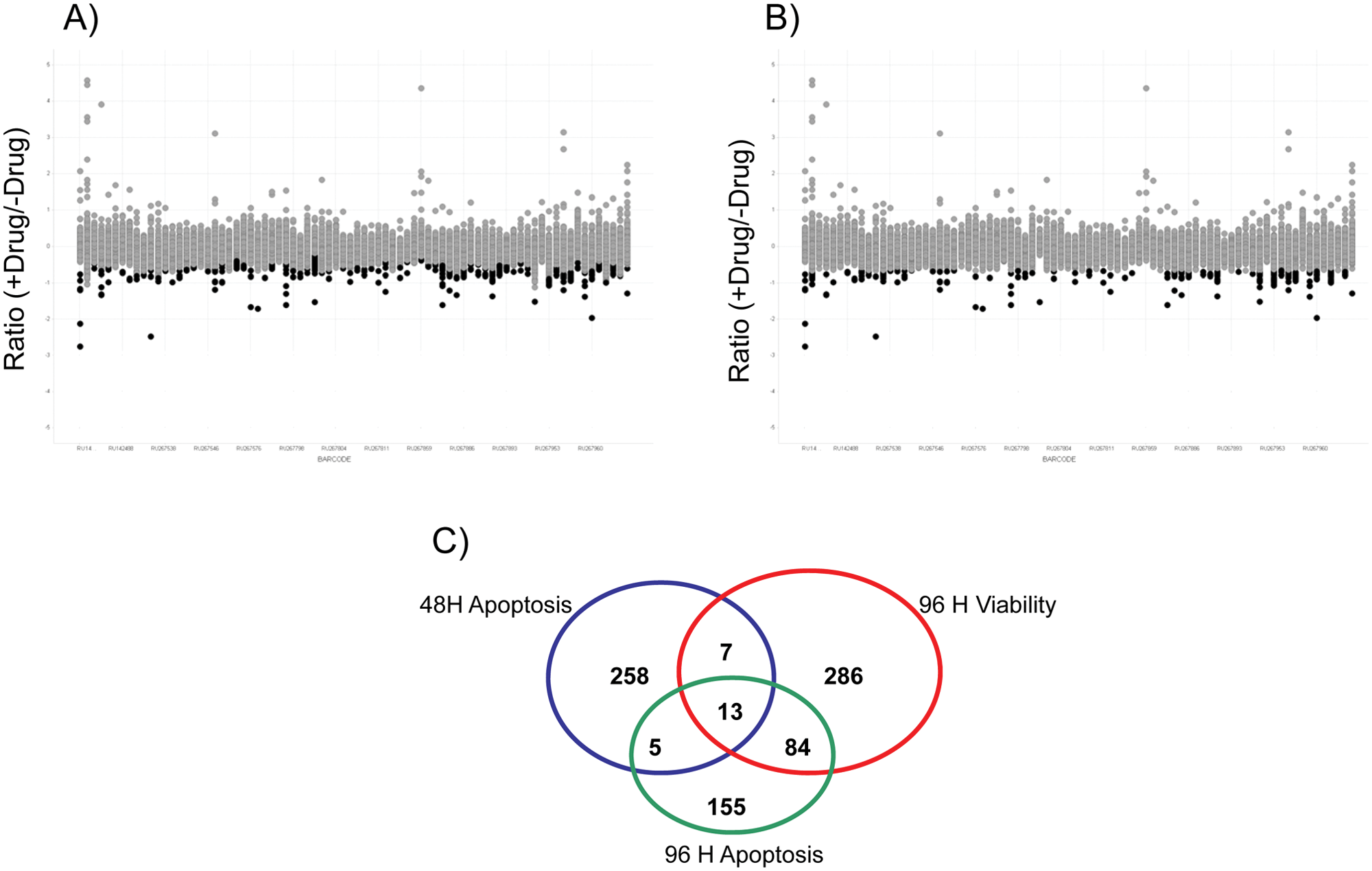
Hit selection strategies. (
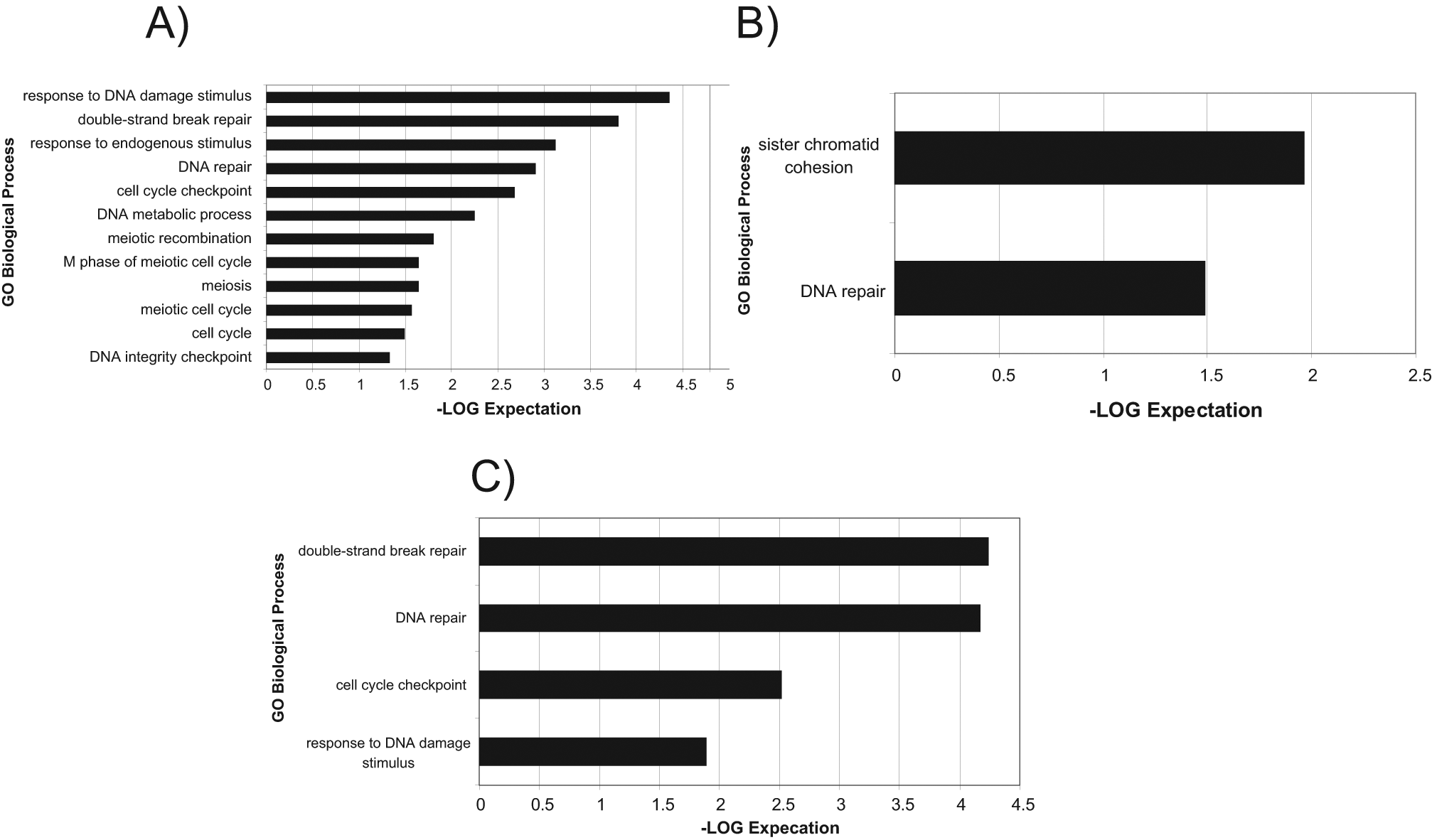
Enrichment of the selected hits from the primary screen. Hit lists generated from the hit selection process were analyzed for pathway enrichment using the GO Biological Process database. Pathways with expectation values smaller than 0.01 and with a gene set overlap count 3 or greater are shown. Note the list of genes generated from the 48-h apoptosis assay was not enriched in any pathways (data not shown). (
Confirmation of primary HTS hits
The 808 siRNA pools selected from the primary screen were confirmed in triplicate in the same assay format as the primary HTS assay. For purposes of selecting hits, the z* score was calculated for the sample field, and controls on a global level based on the variability in the luciferase siRNA control z* score >3 (apoptosis) and <−3 (viability) were used as the hit cutoff. To increase the stringency of the hit selection, we calculated the p-value (t-test against the pooled negative controls, luciferase siRNA) of the replicate data. Using the z* cutoffs and a p-value of <0.05, 83 genes were chosen in the apoptosis assay and 72 genes were chosen in the viability assay. We also selected an additional 18 genes that were classified as hits in two or more assays, regardless of their p-value. We reasoned that any gene positive in more than one assay was relevant, even if the p-value was too high to be statistically significant in the individual assays. From this hit selection strategy, a final list of 173 confirmed genes was compiled for follow-up. Analysis by the GO Biological Process conducted on these genes indicated a measurable enrichment in genes involved in DNA repair, further strengthening the screen resolving power. Specifically, genes that confirmed in both viability and apoptosis assays were enriched in processes involving double-stranded break repair via homologous recombination ( Fig. 5A ). On the other hand, genes that hit only in the viability assay were specifically enriched in processes involving the regulation of ubiquitin-protein ligase activity ( Fig. 5B ).
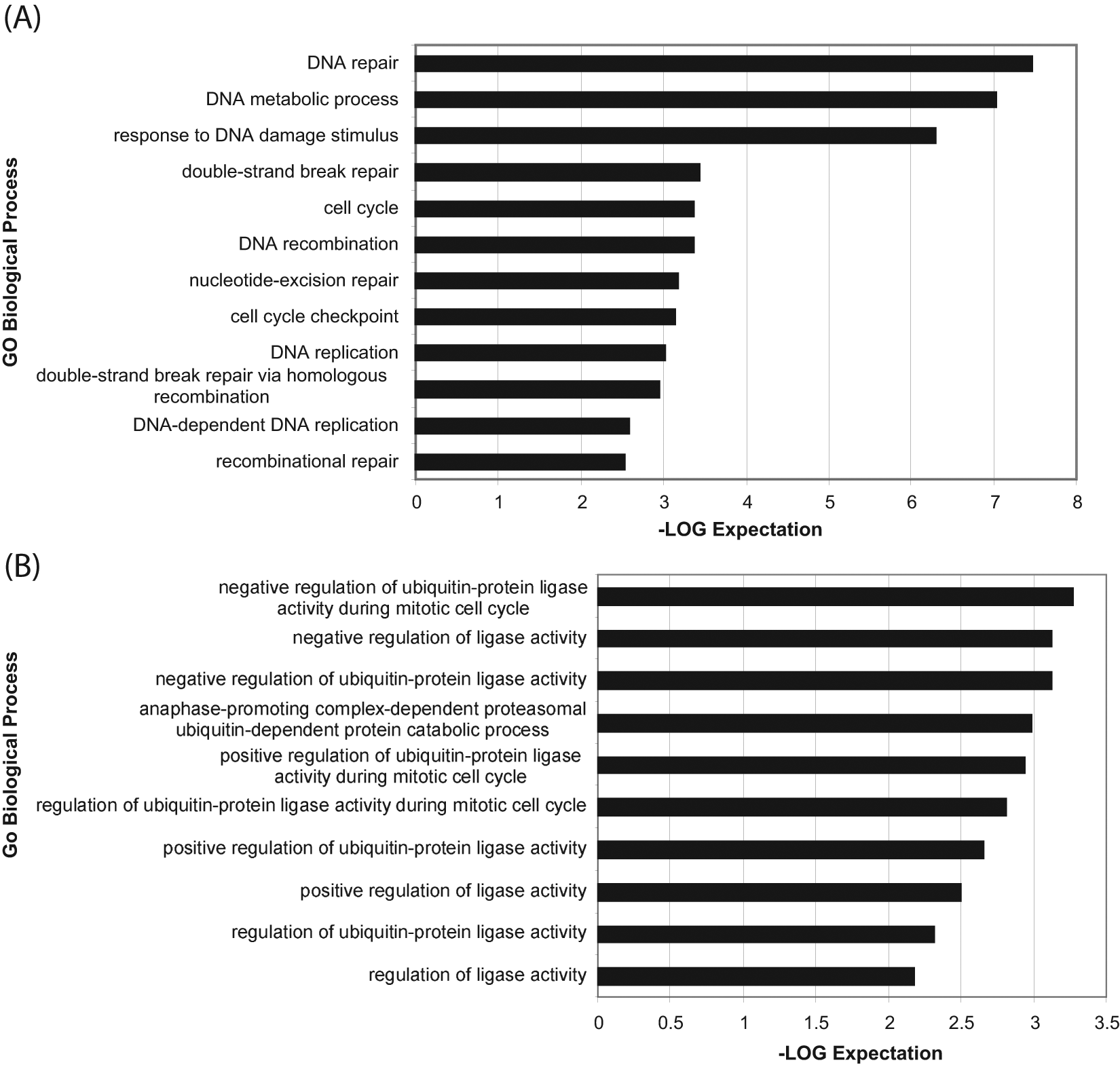
Enrichment of the selected hits from the confirmation screen. (
Validation of siRNA hits
In an effort to further validate the 173 confirmed genes, two strategies were employed: (1) screen each of the three siRNA oligos that comprised the original pools used in HTS individually (“single” siRNA screen) and (2) screen additional pools of siRNAs from siRNA libraries using alternative oligo designs. In this case, we chose to use pools designed by Ambion (Austin, TX) and Qiagen (Valencia, CA) (“alternative design” screen). If there were two out of three or three out of three single oligos for an intended target, or if the related pools from alternative designs produced the same phenotype, then the RNA was considered to have a high probability of being “on target” and the cell phenotype to be a result of the silenced gene transcript.
From our original pooled siRNA library screen, the single oligonucleotides comprising each pool were tested individually for activity. Seventy-six genes had two out of three single oligos confirm, and 41 genes had three out of three single oligos confirm. Therefore, we could confirm 71% of the siRNA hits as having a high probability of being “on target” by our criteria of activity in more than one single oligo tested.
Of the 164 siRNA hits with unique gene IDs tested, 89 genes had two of three alternative designs confirm in at least one of the assays measured. An additional 38 genes had all three alternative designs confirm in at least one assay. Therefore, we could confirm that 73% of the siRNA hits have a high probably of being “on target” by our criteria of activity in more than one library design tested.
Relevance of siRNA hits to DNA repair and cell division pathways
The 173 confirmed pools were also screened using high-content cell imaging. Several multiplexed immunofluorescence assays were developed to detect intracellular levels of cleaved caspase-3 (a marker of apoptosis), Rad51 (a marker of DNA damage response), phosphohistone H3 (a marker of mitosis), and phosphohistone H2A.X (a marker of DNA double-strand break damage response). Following image acquisition, high-content image analysis was performed to localize and quantitate the fluorescent signal of the individual markers. Signal thresholds were selected using the luciferase siRNA as a negative control and used to gate and classify cells in sample siRNA wells as “negative” or “positive” depending on whether they were at the same or a different level with respect to this threshold. In addition, morphological parameters such as nuclear foci number and size were also acquired. Cell number and cell cycle analysis were also performed using a DNA-binding nuclear dye. The total number of parameters generated from this high-content screen was 48 for each siRNA pool screened. Results from different assays and parameters measured were normalized to have a value between 0 and 1, and each was equally weighted. A hierarchical clustering analysis was performed using an unweighted average method and a Pearson correlation similarity measure. A cluster of genes was identified that, when silenced by siRNA, showed higher cell viability in the absence of PARPi and lower viability with PARPi treatment at the 96-h time point. Further examination of the parameters identifying this cluster revealed that although there was a strong correlation between viability ( Fig. 6A ), PH2A.X ( Fig. 6B ), and cell cycle assays ( Fig. 6D ), significant differences were also observed among these siRNA in modulating the levels of Rad51 protein ( Fig. 6C ). When the parameters from all the assays run on this set of siRNA were included in the clustering analysis, three distinct clusters could be identified. Representative examples of the phenotypes of the three clusters are shown in Figure 6A , whereas cumulative performance of the siRNA comprising each cluster across PH2A.X, RAD51, G2/M, and viability is shown in Figure 6B . Cluster 1 siRNA pools behaved similarly to Rad51 siRNA and generated constitutively low levels of Rad51 regardless of PARPi treatment. The siRNAs in this cluster were strong PARPi sensitizers, increasing both the level of PH2A.X and the number of H2A.X foci upon PARPi treatment ( Fig. 7 ). Cluster 2 siRNA pools were different from those in cluster 1 because they generated high basal levels of Rad51 regardless of PARPi treatment. Cluster 2 contained 36 siRNAs, including many such as BRCA1/2, BARD1, SHFM1, PALB2, and RBBP8, which were already known to participate in PARP-mediated DNA repair processes and were expected to be hits prior to running the genome-scale screen. Cluster 2 also included 20 new siRNAs such as ESPNL, TTC30B, and WDR77 that had not been previously shown to be linked to DNA repair processes. Cluster 3 siRNA pools were classified as moderate PARPi sensitizers, showing low basal Rad51 levels that were induced by PARPi. This third cluster contained nine siRNAs that were not previously identified as participating in PARP-involving processes.
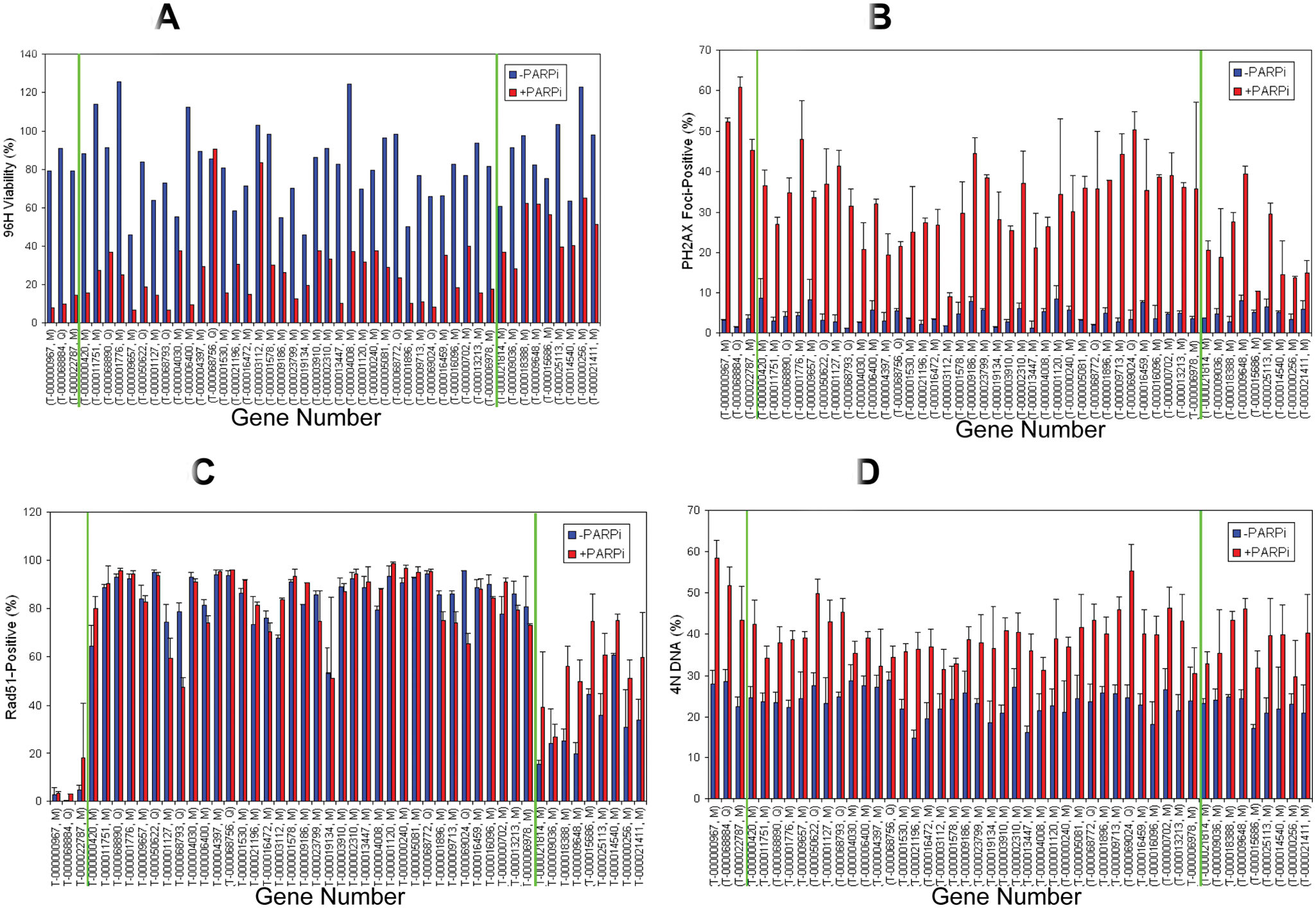
Hierarchical clustering analysis of high-content imaging assay results. (
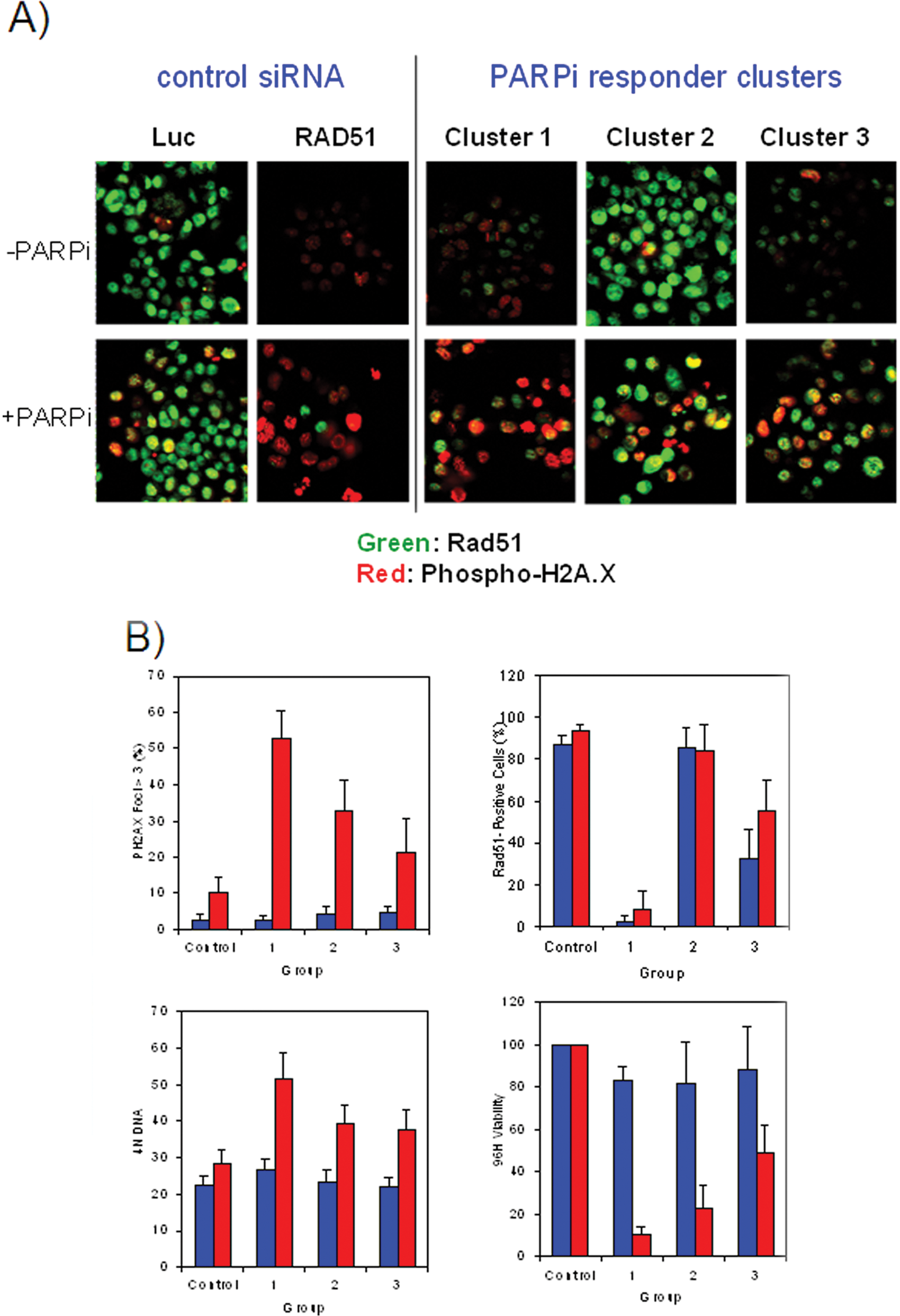
(
Confirmation of siRNA knockdown by rtPCR
To further validate our screening strategy and hypothesis, we selected several genes from each of the imaging clusters for validation of knockdown by rtPCR. Both pools confirmed from the primary screen and the singles that comprised these pools were selected. To measure sufficient gene expression, the cell density needed to be doubled. Table 1 shows the efficiency of gene knockdown as a percentage of mRNA remaining. The control siRNA used in the screen showed greater than 50% knockdown for the pools. Some of the singles had greater transfection efficiency than others. Four of the six genes tested from cluster 2 showed greater than 50% knockdown in the pools and singles. Several genes, including C5orf5 and PTCH2, did not show efficient knockdown and may have been selected as hits as a result of off-target activity. One gene, SUZ12P, did not have sufficient expression in the cell line screened. Two of the four genes tested from cluster 3 showed greater than 50% knockdown and one other around 40%. Overall, we were able to confirm that the phenotypes expressed by the three imaging clusters identified were due to sufficient biological knockdown of the targets of interest.
Taqman Data from Selected Genes of Each Cluster Identified from the High-Content Follow-up Screen
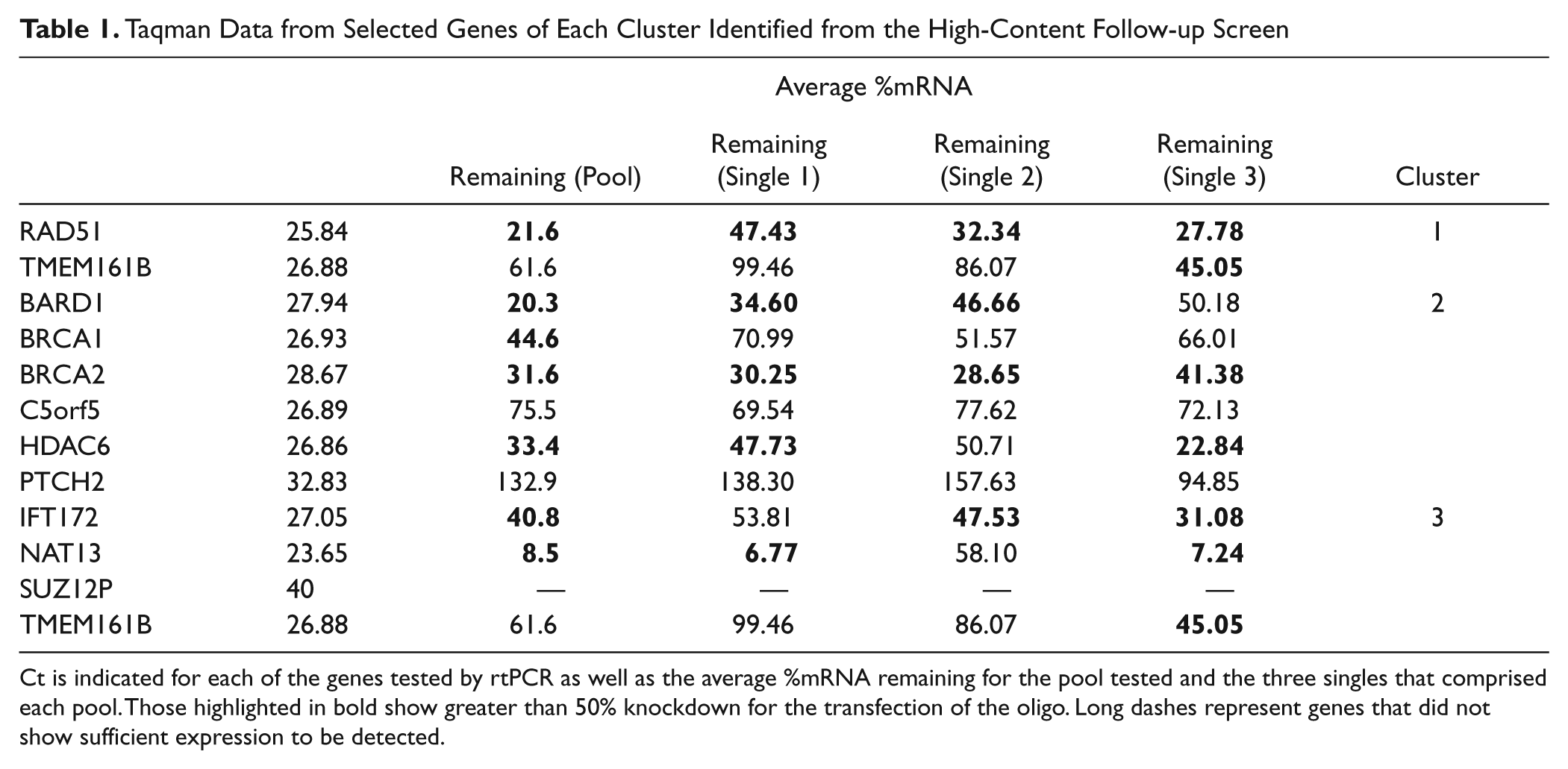
Ct is indicated for each of the genes tested by rtPCR as well as the average %mRNA remaining for the pool tested and the three singles that comprised each pool. Those highlighted in bold show greater than 50% knockdown for the transfection of the oligo. Long dashes represent genes that did not show sufficient expression to be detected.
Discussion
We have performed a multiparametric, genome-scale RNAi screen to identify chemosensitizing drug targets for a PARP inhibitor. The screening strategy we designed exploited multiple readouts at multiple time points to find the most biologically relevant targets in the PARP pathway. The primary screen was carried out by measuring cell viability and apoptosis at both 48 and 96 h post-siRNA transfection and PARPi treatment. This was accomplished by developing a protocol that multiplexed the Caspase-Glo luminescence assay for apoptosis with the Alamar Blue assay for cell viability. The resulting dual-readout assay was easily implemented for the screening of the genome-wide siRNA library. The initial validation of the dual assay with BRCA1 and RAD51 controls indicated that using apoptosis and viability readouts provided better sensitivity. PARPi sensitizers with a weak phenotype such as BRCA1 were clearly resolved from the sample field by the apoptosis readout at time points when viability would have identified only robust sensitizers such as RAD51. Thus, these two assays effectively corroborated one another and provided substantially different methods of identifying hits from different pathways.
The GO Biological Process analysis was performed during hit selection at each stage of the screening process to validate the screening method that was chosen and to determine the effectiveness of our hit selection strategy. Initial GO analysis of the primary hits indicated that viability and apoptosis assays found genes that were most relevant to the PARPi pathway. By looking for what we expected to find, we established that this screen did not just return a random set of genes but rather genes that were enriched in pathways of interest. We saw enrichment in genes associated with DNA repair and, more specifically, double-stranded repair, the main process in the PARP pathway. We also saw additional significance in genes that were identified as hits in both assays performed. This could be of particular importance if one is limited in the numbers of genes to carry forward into confirmation assays. A multiplex strategy could identify those that have a higher probability of being involved in the pathway of interest, providing greater confidence in the genes that were identified by both assays.
The primary hits from the screen that confirmed in triplicate could be further divided into two groups: (1) those that confirmed in both the viability and caspase assays and (2) those that only scored as hits in the viability assay. This partition is noteworthy, as it suggests that genes in the first group have induced a canonical caspase-dependent cell death ( Fig. 4A ), whereas those in the second group could represent PARP sensitizer genes that have induced cell death in a caspase-3/7–independent mechanism. The GeneGO analysis of the second group of hits confirming only in viability finds an unusual enrichment in genes involved in the ubiquitin pathway, specifically in genes associated with ubiquitin ligase activity ( Fig. 4B ). Caspase-independent apoptosis is known to arise from diminished protein degradation such as that resulting from the inhibition of the ubiquitin/proteosome protein degradation pathway. 14 This cluster of genes could sensitize cells to the PARP inhibitor through their negative effects on protein degradation. Alternatively, these genes could highlight a new and yet more direct role for ubiquitinating/deubiquitinating enzymes in regulating DNA repair by HR. This is the case of the ubiquitin-specific peptidase USP-11, which has recently been shown to participate in HR repair of DNA double-strand breaks and whose silencing by siRNA sensitizes cells to genotoxic stress. 15 The identification and segregation of these hits demonstrate how the multiplex viability/caspase activation readout paradigm not only allowed the identification of new PARPi sensitizers but also provided information on the different apoptotic pathways.
Independent of assay readout, we attempted to determine if the siRNAs confirming had a high probability of being on target by employing a twofold strategy of screening each target using deconvoluted pools from our library as well as two additional pools of oligos with different sequences from independent libraries. Using these methods, we found that in cases where two or more siRNA reagents confirmed, we had strong enrichment in processes involved in the PARP pathway such as double-strand break repair and response to DNA damage stimulus. Overall, the two strategies used to probe potential off-target effects produced comparable results with nearly identical confirmation rates.
To further elucidate the roles of these confirmed genes in the PARPi pathway, a high-content multiplexed imaging assay was performed to measure cleaved caspase-3, phosphohistone H2A.X, phosphohistone H3, Rad51, and cell cycle profiles. Three distinct clusters of genes were revealed when the assay results were profiled. Imaging analysis parameters that distinctively separated these clusters included cell viability, PH2A.X foci formation, Rad51 levels, and 2N/4N DNA content analysis. The three clusters shared similarities in that they were sensitive to PARPi treatment, had an increased number of PH2A.X foci, and showed cell cycle arrest at the G2/M phase. The main differences among the three clusters resulted from the Rad51 assay. Each cluster showed distinct phenotypes with regard to Rad51 protein, indicating that Rad51 might be a key modulator of cellular response to PARPi treatment.
A multiplexed primary screening strategy identified biologically relevant genes involved in the PARPi pathway. The multiplexed viability/apoptosis assay allowed us to identify both weak and strong PARPi enhancers, effectively casting the widest net to capture biologically relevant genes. Our strategy of confirming the hits with additional siRNA reagents enabled us to rank these genes based on the probability of their siRNA activity of being on target. The addition of high-content imaging assays allowed us to multiplex a variety of readouts to determine similarities and differences between groups of genes to both mechanisms of action and specific profiles. These methods have generated a well-validated set of PARPi enhancers with different mechanisms of action that can be used as biomarkers for tumor profiling and PARPi therapy patient selection.
Footnotes
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.