
Abstract
Many diseases, such as diabetes, autoimmune diseases, cancer, and neurological disorders, are caused by a dysregulation of a complex interplay of genes. Genome-wide association studies have identified thousands of disease-linked polymorphisms in the human population. However, detailing the causative gene expression or functional changes underlying those associations has been elusive in many cases. Functional genomics is an emerging field of research that aims to deconvolute the link between genotype and phenotype by making use of large -omic data sets and next-generation gene and epigenome editing tools to perturb genes of interest. Here we review how functional genomic tools can be used to better understand the biological interplay between genes, improve disease modeling, and identify novel drug targets. Incorporation of functional genomic capabilities into conventional drug development pipelines is predicted to expedite the development of first-in-class therapeutics.
Introduction
A major challenge facing pharmaceutical research and drug development is the high attrition rate of therapies in clinical development. This has led to the enormously high costs for bringing new drugs to market, with a recent estimate of about $2.5 billion per new drug approval. 1 When assessing the driving factors behind high attrition rates, it was reported that the majority of drug failures are due to toxicity and lack of efficacy. 2 Further exemplifying the reduction in R&D productivity, many new drugs that do gain regulatory approval have limited commercial success due to a failure to significantly differentiate from the standard of care.1,2 There has also been a reduction in both the proportion of late-stage targets in the pipeline that are classified as first-in-class and the percentage of approvals considered first-in-class. 1 This decline in innovation is highlighted by annual peak pharmaceutical sales decreasing by almost 50% in recent years.1,3 Although 2018 is considered a blockbuster year for the number of drug approvals by the Food and Drug Administration (FDA), a large portion of these approvals are for orphan or rare oncology indications, where despite the clinical impact of these drugs, the commercial potential is projected to be minimal. 4 A recent analysis examining the drug discovery process over the last 60 years highlights that despite tremendous improvements in technology, pharmaceutical research has been plagued with deficiencies in reproducibility and efficiency. 5 Pulling together the high attrition rates, reduced differentiation, and issues with reproducibility, a clear question emerges for drug discovery: How can biotech and pharmaceutical companies identify and de-risk new first-in-class drug targets that will successfully translate into the clinic?
Companies are approaching this problem by expanding the toolsets used to identify new targets, with greater emphasis on human genetics and functional genomic technologies as a means to improve understanding of the molecular mechanisms driving disease.2,6 Functional genomics is a broad term that covers the investigation of biochemical, cellular, or physiological properties of gene products to understanding the relationship between genotype and phenotype. 7 Functional genomics is used to better understand various processes related to genomic sequence, gene expression, and encoded protein function, including the study of coding and noncoding transcription, protein translation, and interactions between proteins, DNA, and RNA species. Here we review the recent history and cutting-edge tools for functional genomics and outline how these approaches are being used to improve the drug development process.
Tools for Functional Genomics
The first robust tool for making site-specific perturbations to the transcriptome was RNA interference (RNAi).
8
RNAi is a multistep process that results in targeted degradation and subsequent repression of specific mRNA transcripts (
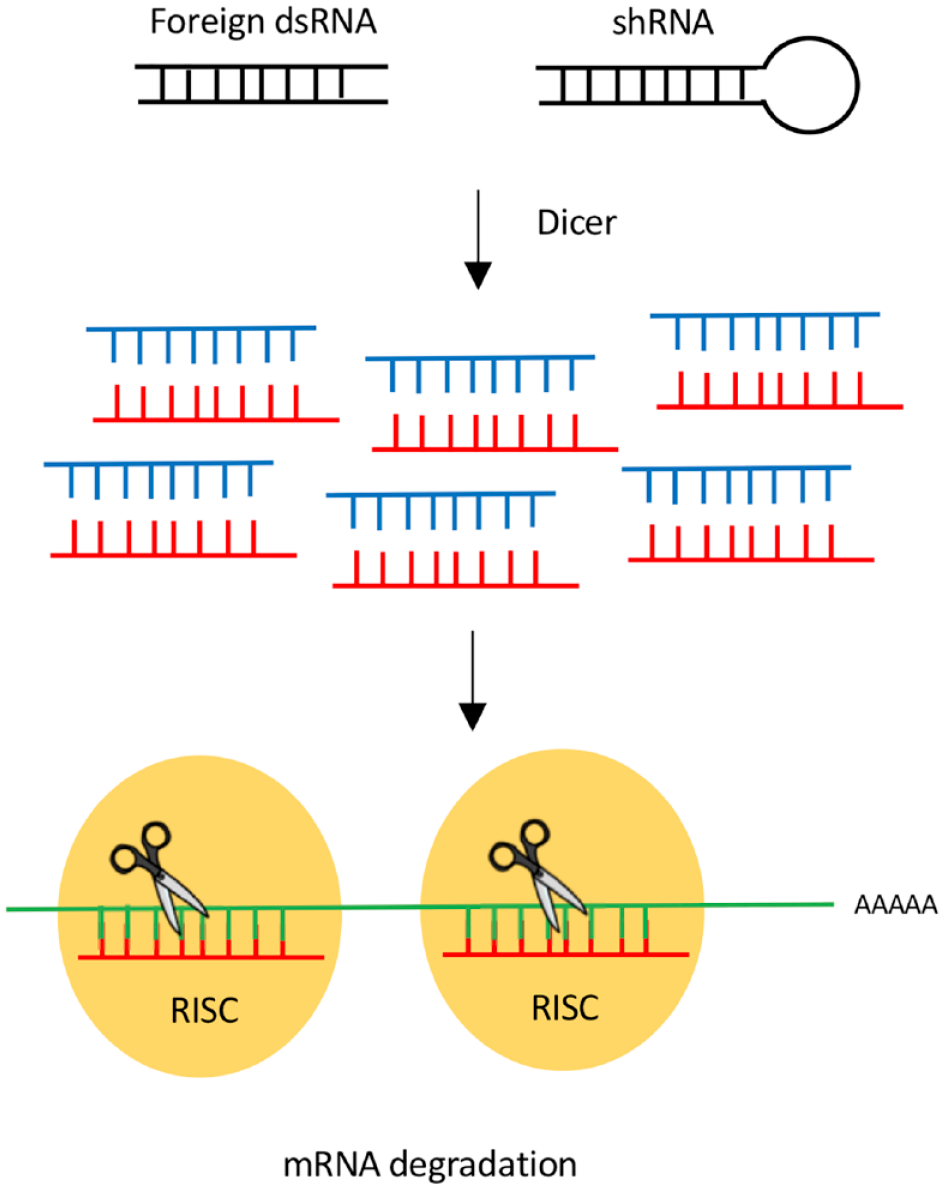
Mechanism of RNAi. Short dsRNA can either be directly delivered to cells or be expressed from a plasmid as shRNAs. The dicer enzyme processes the dsRNA into ssRNA. ssRNA associates with the RISC complex. Through sequence homology, the RISC complex binds and subsequently degrades the targeted mRNA sequence.
Recent advances in genome- and epigenome editing tools now allow researchers to readily make site-specific perturbations to the transcriptome, genome, and epigenome. While a reduction in mammalian gene expression has been facilitated through RNAi-based technologies for the last 18 years,
8
gene editing platforms provide expanded functionality, including, but not limited to, gene repression. Gene editing enzymes induce a site-specific double-stranded break at the loci of interest (
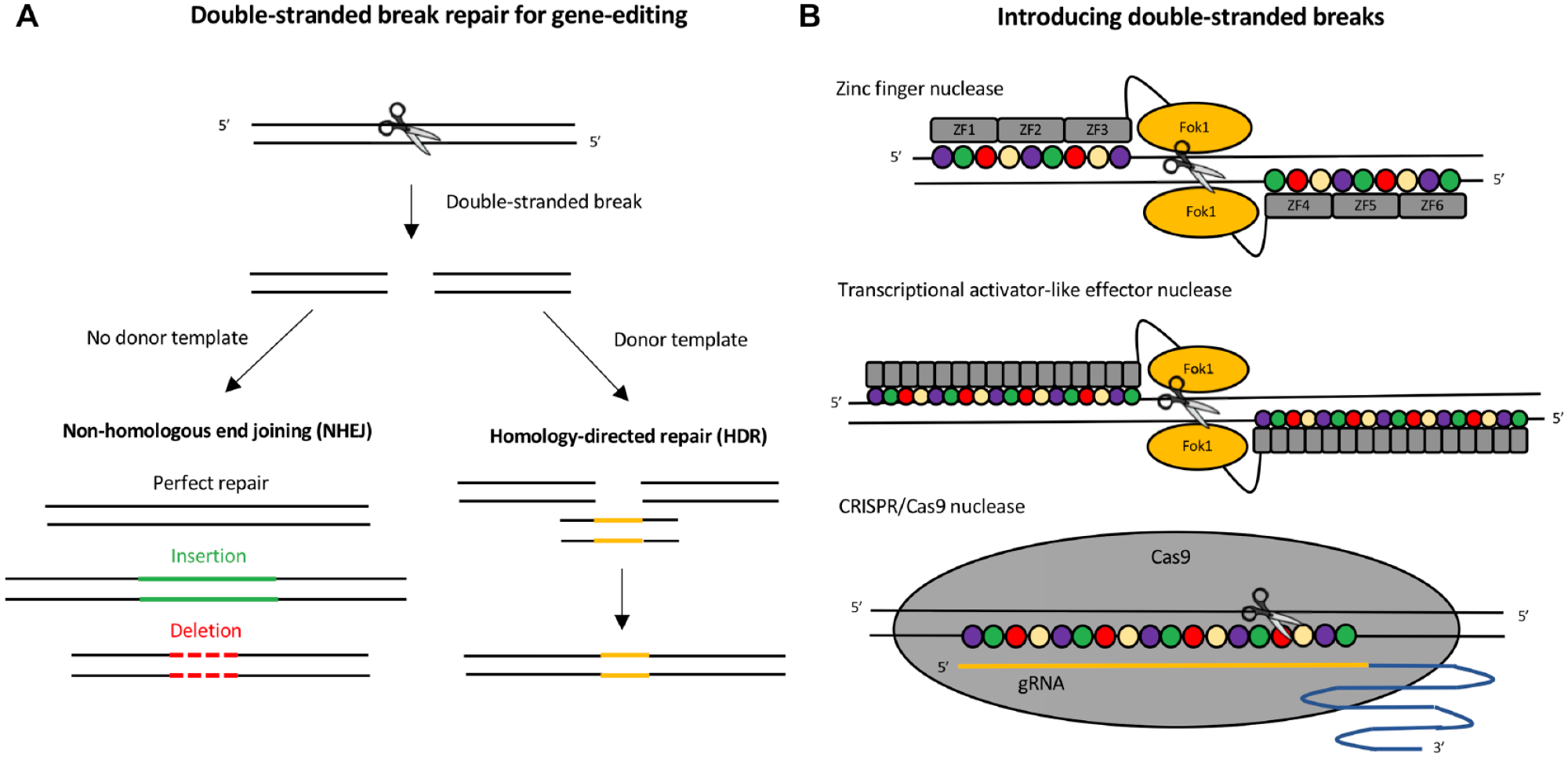
Induction and manipulation of DNA double-stranded break repair for genome editing applications. (
While the principle of utilizing double-stranded DNA break repair for gene editing applications has been around since the 1990s,21–23 the first gene editing tools were quite complex and not accessible to most research laboratories. Before the discovery of clustered regularly interspaced short palindromic repeats and associated endonucleases (CRISPR/Cas9), the most widely used platforms for gene editing were zinc finger nucleases (ZFNs)
24
and transcription activator-like effector nucleases (TALENs)25–28 (
The field of gene editing became far more accessible to the general scientific community with the discovery of CRISPR/Cas9 for gene editing in mammalian cells.30,31 In contrast to earlier platforms, the specificity of the Cas9 nuclease is conferred by a RNA–DNA interaction (
In contrast to gene editing, where permanent changes are made to the DNA sequence, epigenome editing involves modifying the chromatin structure and proteins recruited to a specific locus to influence gene expression. Recent advances have led to the development of engineered epigenome editing tools capable of modulating both gene expression and chromatin state (
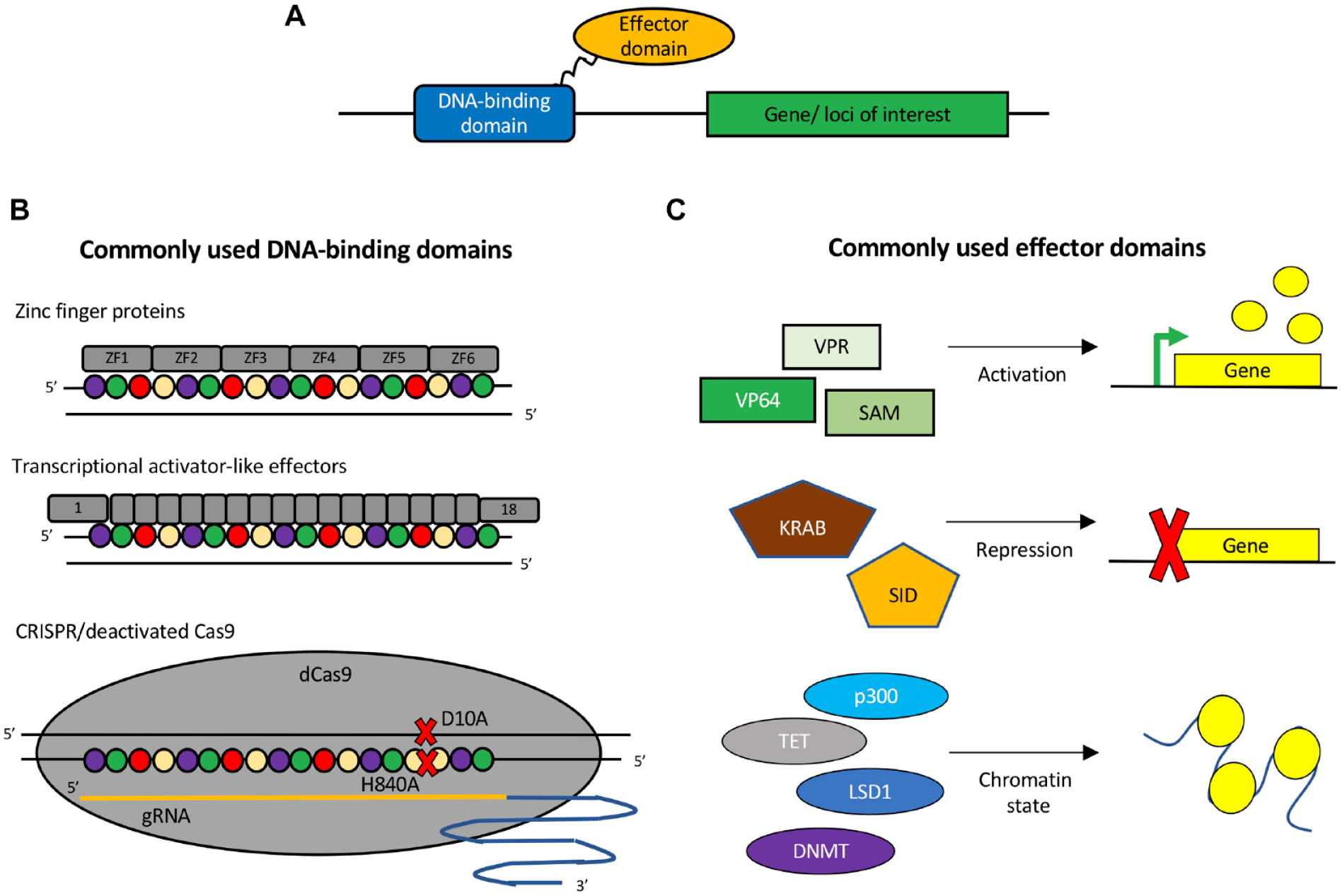
Tools for epigenome editing. (
The use of dCas9 fused to epigenetic regulators has vastly increased the flexibility and applicability of CRISPR/Cas9, particularly from a drug discovery perspective. Without altering the underlying DNA sequence, this CRISPR/Cas9 platform enables researchers to disable (CRISPR-knockout [CRISPR-KO]), turn on (CRISPR activation [CRISPRa]), or decrease (CRISPR inhibition [CRISPRi]) gene expression from single or multiple genomic loci. The platform versatility provides new avenues for studying and modeling both monogenic and complex diseases. Furthermore, the CRISPR/Cas9 platform allows for cost-effective high-throughput screening on endogenous gene regulation.
Improved Disease Understanding: Genetic Variants and Human Health
Functional genomic tools are critical for mechanistically linking genetic variation to health. For decades, geneticists have used candidate gene approaches to elucidate the function of individual genes associated with rare hereditary disorders. While the study of human monogenic disorders has provided many drug targets, these diseases are typically rare.44,45 Following the conclusion of the Human Genome Project in 2001–200346,47 (
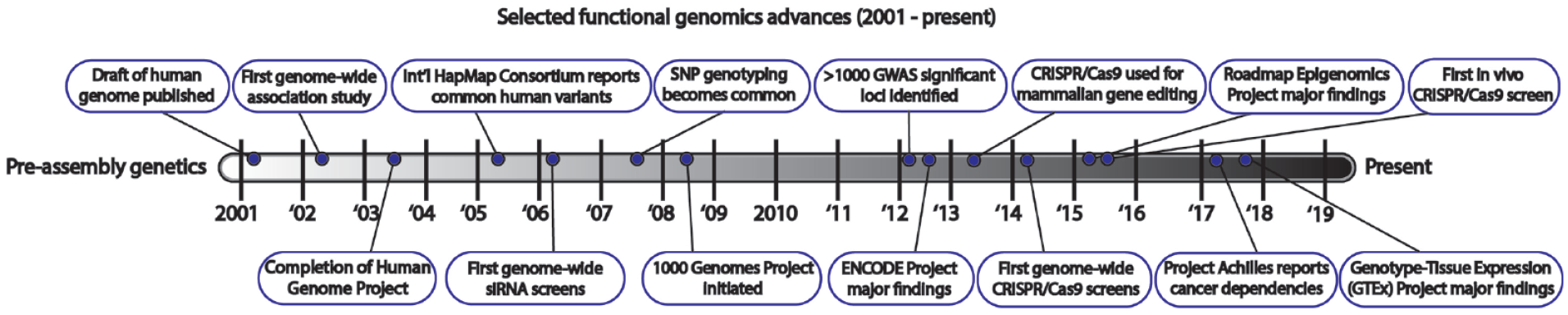
Timeline of select functional genomic advances in the post-human genome assembly era.
GWAS efforts have found that common human traits or diseases are usually highly polygenic, with individual genetic variants explaining little of the overall trait variance or the risk of developing disease. 54 Interestingly, these same techniques have uncovered cases of rare variants with strong effect sizes, either substantially increasing risk or conferring protection from disease, but in small patient populations or individual families. 55 As functional genomic techniques become more accessible to researchers, we will gain further understanding of the molecular mechanisms behind these observations. The relatively small number of well-annotated GWAS loci indicate that rare variants with strong effects may represent the extreme, where a disease-linked gene exhibits complete loss or gain of function. For example, there are more than 100 loci associated with obesity and type 2 diabetes, with many alleles contributing modest risk, and rare loss-of-function variants in the adenylate cyclase 3 (ADCY3) gene confer a severe obesity phenotype.56,57 This finding led to the pursuit of ADCY3 enhancers as antiobesity medicines.58,59 In another example, variants that reduce function of the immune receptor encoded by the triggering receptor expressed on myeloid cells 2 (TREM2) gene are associated with an increased risk of Alzheimer’s disease, 60 while complete loss of TREM2 signaling has been shown to cause rare Nasu–Hakola disease that includes progressive early-onset dementia. 61 Together, these findings indicate that enhancing TREM2 signaling is a potential therapeutic strategy in neurodegenerative disease and therefore is an active area of research.
As compared with exonic variants that clearly modify the function of a particular gene, the majority of GWAS discoveries fall in the noncoding region of the human genome in putative gene regulatory elements. These putative regulatory elements are often defined with active enhancer histone marks that may be cell type specific.62–64 A major challenge of modern functional genomics is how to mechanistically link specific noncoding variants with gene regulation and the associated disease processes. To date, functional genomic consortia, such as ENCODE and Roadmap Epigenomics, have struggled to do this at scale due to the myriad possible gene regulatory mechanisms involved. 65 One scalable approach is the identification of expression quantitative trait loci (eQTLs), the systematic association of genetic variants with variation in gene expression levels. As large microarray and RNA-seq data sets that sample across many individuals have become available, eQTL mapping efforts have identified thousands of cis- and trans-acting loci across diverse tissue types for most human genes.66,67 Similar quantitative association techniques have been applied to smaller data sets to study protein abundance, DNA methylation status, and chromatin states.68–70 eQTL approaches help establish a link between disease-associated variants and particular genes or genomic features, but alone often fall short of detailing molecular mechanisms indicating proteins or pathways suitable for pharmacological intervention.
The availability of high-throughput sequencing has facilitated widespread use of genome-wide biochemical assays to characterize the genomic landscape surrounding disease variants. Long-range chromatin interactions can be studied genome-wide using techniques such as “Hi-C,” which captures proximal genomic regions in a sequencing library by dilute ligation reactions. These tools have helped confirm that the human genome is organized into hundreds of kilobase to megabase size topologically associating domains (TADs), often bounded by insulating CCCTC binding factor (CTCF) protein binding sites.71,72 TADs are thought to provide a microenvironment for gene and gene regulatory element sequences to move around and establish long-range contacts. Enhancer looping within TADs reinforces basal promoter activity and explains why sets of genes located within the same TAD are often co-regulated or developmentally linked.73,74 In addition, functional genomic consortia have discovered millions of putative gene regulatory elements that are cell or tissue dependent.64,75 These regulatory elements are defined by transcription factor binding, active histone modifications, and increased local chromatin accessibility. The mapping of the human genomic regulatory landscape has set the stage for interrogation of molecular mechanisms underlying disease-associated loci.
Individual loci with multiple disease-associated single-nucleotide polymorphisms (SNPs) in linkage disequilibrium may indicate altered transcription factor binding sites, perturbation of noncoding RNAs, splicing changes, disruption of local chromatin structure, or altered enhancer looping.76–79 There has been an increased focus on using functional genomic tools to deconvolute complex GWAS loci. Miller et al. provide an early example of integrating modern functional genomic techniques and analyses to connect regulatory variants to gene function in the context of coronary artery disease. 80 By integrating genomic, epigenomic, and transcriptomic profiling of cells and tissues, the authors describe how particular regulatory variants influence disease gene expression profiles. However, even after noncoding variants are connected to the regulation of a particular gene, it still may be unclear how the encoded protein or RNA from that gene influences key disease biology. Fortunately, the toolbox to fill this gap is also expanding, enabled by improved human genomic annotation, high-throughput sequencing, proteomics, and bioinformatic insights. Claussnitzer et al. provide an impressive example of leveraging many of these advances to understand the connection between a particular noncoding SNP and obesity risk. 77 The authors demonstrate that SNPs disrupting the binding site of ARID5B, a transcriptional repressor, results in increased expression of IRX3 and IRX5 genes and a shift from energy-dissipating beige adipocytes to energy-storing white adipocytes associated with obesity.
In addition to inherited disease risk, de novo or somatic mutation has emerged as a secondary source of genetic variation underlying disease. Cancers are increasingly classified by a molecular taxonomy of their mutation burden and driver genes. 81 This work has improved patient outcomes by facilitating the development of specific mutation targeted therapies. 82 While certain acquired somatic mutations have been linked to oncogenic pathways for a number of years, there is a recent accumulation of evidence that somatic mutation is involved in other disease types as well, such as neurological and autoimmune conditions.83,84 For instance, some focal epilepsies appear to be driven by somatic mutations impacting a localized lineage of neurons and glia in the brain. 85 As another example, a subset of autoimmune diseases may be linked to somatic mutation generating autoantigens that are recognized by the adaptive immune system as foreign.86,87 Somatic mutations may explain the apparent tissue specificity, late onset, or unusual presentation of particular conditions.
Functional genomic tools are increasingly being used to investigate both somatic and heritable mutation-driven disease in various cell and animal models. In particular, CRISPR/Cas9-based genome engineering has emerged as the tool of choice to introduce mutations into endogenous genomic loci, including at particular developmental time points.88–90 Functional genomic tools such as these are increasingly being used to better study these complex mutation-associated phenotypes and rapidly improving the way we model and study disease.
Applications of Functional Genomics in Disease Modeling
Historically, researchers have relied on model organisms to study human disease. While these studies have made important contributions toward understanding key pathways, many animal models are unable to fully recapitulate complex human disease biology. For example, a single rodent model of Alzheimer’s disease is unable to exhibit the full spectrum of human disease pathologies, including the accumulation of amyloid beta, tau tangles, and extensive neuronal loss. 90 This is likely due to differences in genetic drivers as well as neural network development, connectivity, and complexity between model organisms and humans. 91 Furthermore, it can be difficult or cost-prohibitive to produce the multiplicity of animal models that would demonstrate the diversity of a given human disease phenotype. For example, there are multiple mutations associated with Alzheimer’s disease with different severity, time of onset, and pathologies. It is currently unrealistic and impractical for most researchers to construct and/or study multiple animal models simultaneously to get a holistic evaluation of disease biology.
For next-generation therapies including antibody, RNA, or gene editing approaches, it is important that the therapies are evaluated in a human background because these reagents may not exhibit the same specificity toward the gene, transcript, or protein in another species. While the genomes of nonhuman primates and humans are 92% conserved,92,93 small changes in the genome, transcriptome, and proteome can greatly affect efficacy, off-target effects, and subsequent toxicity. With the advent of induced pluripotent stem cell (iPSC) technology, in principle, scientists can engineer virtually any cell type/tissue of interest from an unlimited cell source. However, in practice, these engineered tissues are often lacking in some of the transcriptomic, epigenomic, and phenotypic hallmarks of mature tissue. Some protocols achieve a mature cell phenotype or tissue formation, but the cultures are often impure or the long timescales required are not amenable for routine use. These limitations increase the cost and lengthen the timelines of conventional drug discovery where candidate therapeutics are screened in iPSC models.
By pairing in vivo animal model studies with robust human-derived in vitro cell models, scientists will gain a more complete understanding of human disease biology, therefore enabling the development of effective therapeutics. Functional genomic tools have had a large impact on the quality and efficiency of generating novel animal models, though these tools are only starting to address some of the current limitations of human-derived in vitro disease modeling. In this section, we discuss the current status of disease models and how functional genomic tools are being used to improve animal model generation, model the genetic variants of human disease, improve the quality of iPSC-derived disease models, and recapitulate mature tissue transcriptomic and epigenetic profiles.
Generating Novel Animal Models
Functional genomic tools have greatly expedited the process of generating novel animal models. As of 2005, most transgenic mice were generated through the injection of genetically modified mouse embryonic stem (mES) cells into wild-type mouse blastocysts. 94 Through homologous recombination, a stable mES cell line is generated containing the desired genetic mutation. Upon injection into the blastocyst, the mES cells contribute to the germline of the animal. The blastocytes are then implanted into a host mother. The resulting chimeric animals are bred to generate a homozygous model with the desired genetic modification. 94 In best-case scenarios, these methods take about 1–1.5 years to generate a new transgenic strain. 95
Historically, ES cells were required for generating transgenic animals because gene targeting technologies were not efficient enough to directly induce genetic modifications in mouse embryos. Conventional gene targeting technologies relied on the delivery of donor DNA constructs where the desired mutation is straddled between two DNA sequences that have homology to the target genomic site. With initial studies only achieving a targeting efficiency of ~1/1000 cells, a large number of ES clones needed to be screened for the desired mutation before being injected into the blastocysts for transgenic animal generation. 94 As the field of gene editing progressed, it was found that the rate of HDR can be greatly increased by inducing a targeted double-stranded break at the desired integration site. Depending on the desired edit, gene editing tools including ZFNs, TALENs, and CRISPR/Cas9 can be used to edit ES cells at efficiencies of more than 80%. 96 Currently, these methods are used for generating transgenic animals requiring complex genetic manipulations, for example, when knocking in large DNA segments. In recent years, alternative methods have been developed that further accelerate the process of genome modification by directly injecting DNA or mRNA of site-specific nucleases into single-cell embryos to induce a targeted double-stranded break.96–100 These protocols make use of pronuclear injections or electroporation of gene editing components directly into the embryos. Similar to ES-derived transgenic methods, direct injections still generally give rise to chimeric animals that are then bred to generate a stable mouse strain. Direct injection of editing tools into embryos skips the laborious process of generating stable ES lines and therefore greatly reduces the timeline for generating a transgenic strain to an average of 6–12 months.100,101
For many genetic diseases, there are multiple mutations associated with the disease phenotype. Highly efficient gene editing protocols now allow for multiple genetic mutations to be generated simultaneously. It has been reported that up to five mutations can be simultaneously introduced into mouse ES cells or two mutations directly in mouse embryos. 99 Therefore, rather than sequentially generating compound mutation models or cross-breeding multiple single-mutation strains, these models can be generated in a single project. More robust animal model generation has led to the ability to test new compounds in multiple genetic backgrounds, which will help to determine which mutations are responders to a given treatment, aiding in patient population selection for the resulting therapeutic. In addition, the use of CRISPR/Cas9 technology has allowed the study of disease processes in animal models that were previously out of reach. For example, focal cortical dysplasia is caused by somatic cells that acquire mutations in the brain, leading to dysregulated signaling and epilepsy. This has been difficult to model previously as the mutations only affect a portion of cells in the brain, but with CRISPR/Cas9, researchers can create animals with brain mosaics, thereby mimicking the disease.102,103
Controlling for Genetic Variability
Genetic variation between individuals is one limitation of modeling human disease using primary cells isolated from patients. On average, the human genome varies by about 20 million bases between unrelated individuals (or 0.6% of the 3.2 billion bases).
104
For complex human diseases, this makes it difficult to experimentally deconvolute the causative sequences or transcriptomic profiles linked to disease phenotypes from passive variation. By studying a panel of mutations or disease states in the same genetic background, it is much easier to link the causative mutation to disease outcome. CRISPR/Cas9 has made it possible to create multiple disease models in the same isogenic background. However, while gene editing is a robust method in most immortalized cells, it can be quite difficult to induce high rates of gene editing in primary cell models, therefore necessitating clonal isolation to obtain a pure population of cells containing the desired edit. This is where the fields of iPSC cultures and gene editing come together for the generation of isogenic disease models. For most diseases, the disease phenotype presents in terminally differentiated cells with limited proliferative capacity, making clonal isolation impossible. Therefore, isogenic disease models must be created in the iPSC stem-cell-like state, before being differentiated into the desired cell type. By creating panels of iPSC isogenic disease models, research labs can now study multiple genetic disease backgrounds in parallel and more easily determine causative relationships between genotype and phenotype105–107 (
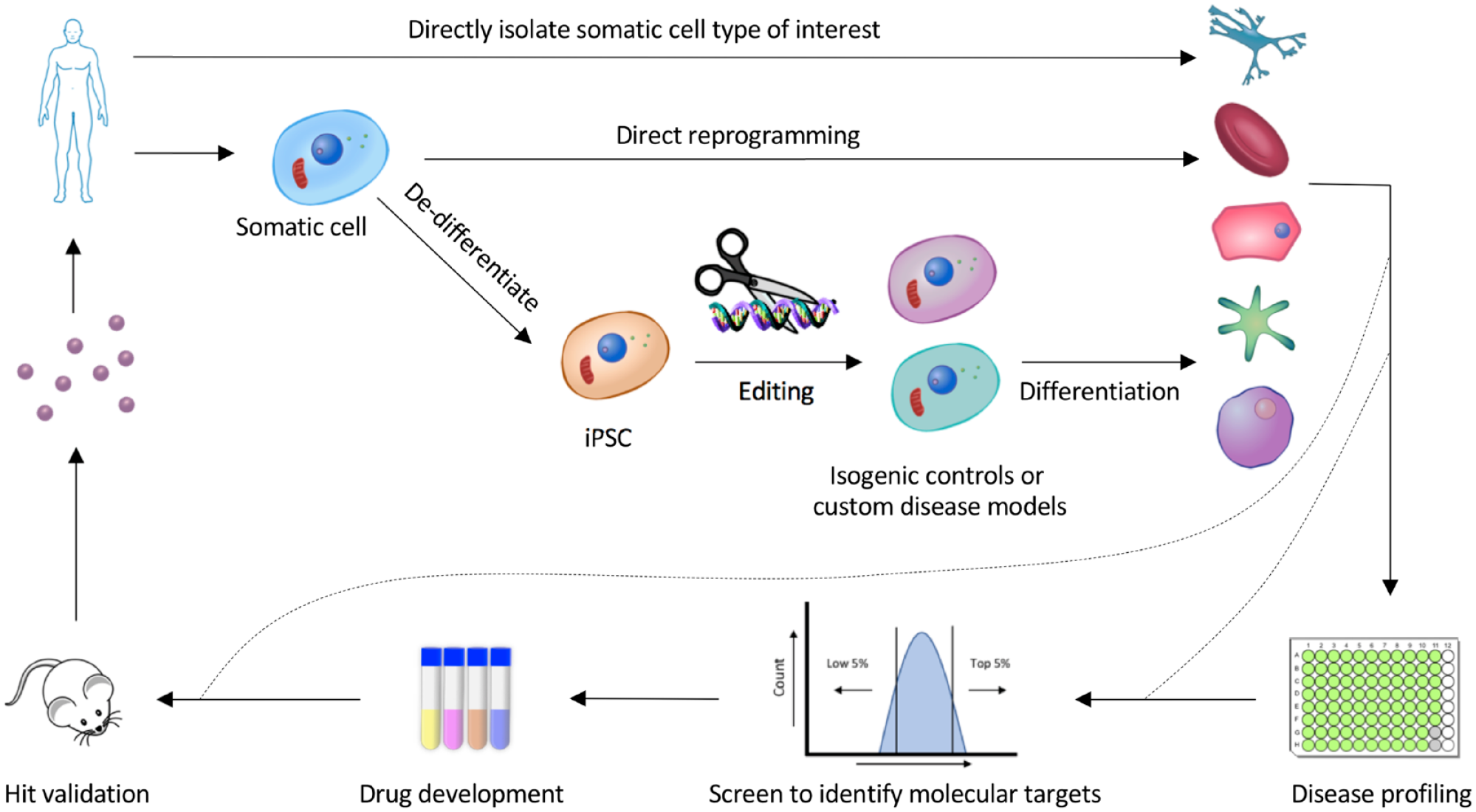
Functional genomic tools contribute to robust disease modeling for drug discovery.
One application of isogenic disease models is experimentally validating potential causal disease SNPs identified through GWAS. Loci identified through GWAS tend to have multiple SNPs within a short distance of one another, all in linkage disequilibrium. For example, there are at least 14 SNPs in the GRM3 locus reported to be associated with schizophrenia. Following meta-analysis of the available data, a significant association with schizophrenia was found for three of these SNPs, 108 yet it is unclear which of these three are causative versus merely tightly associated with the disease allele. However, using CRISPR/Cas9 three iPSC-derived neuronal lines can be generated to model the three GRM3 SNPs. The effect of these mutations on the transcriptomic profile of these cells compared with the healthy isogenic control can then be experimentally determined. Since the resulting differences in GRM3 expression are thought to be subtle, 109 isogenic controls would be necessary to reduce baseline variability and gain statistical power.
Another application of isogenic disease modeling is to identify the genes and pathways that are associated with disease-causing mutations to identify new drug targets. By comparing the transcriptomic profile of neurons derived from Parkinson’s disease patients and corrected isogenic controls, downregulation of the transcription factor MEF2 was identified as a mechanistic driver of mitochondrial damage implicated in Parkinson’s disease. 110 Furthermore, by screening for compounds that increase MEF2 transcription, the compound isoxazole was identified and shown to have protective effects against mitochondria-induced damage. 110 This example demonstrates that isogenic controls help increase the probability of identifying genes implicated in complex disease and how that information can be used to identify new candidate small-molecule therapeutics.
Improving the Quality of iPSC-Derived Disease Models
Functional genomic tools currently allow for relatively simple generation of multiple iPSC-derived disease models. However, the utility of iPSC-based disease models for drug discovery is currently limited by efficiency and the long time frames of current reprogramming methods. The final cultures usually contain a mix of cell types in addition to the target cell type, making downstream data deconvolution difficult. Furthermore, even the best protocols generally produce cultures more closely resembling the fetal or neonatal cellular state rather than the desired mature adult state. For example, neurons derived from human iPSCs fire action potentials as early as 3 weeks postdifferentiation; however, the properties of these early action potentials are relatively immature. Allowing maturation out to day 55, there is a significant improvement characterized by increased sodium and potassium current amplitudes, action potential amplitude, and action potential threshold. 111 However, the increased time and costs associated with long differentiations are prohibitive for use in drug screening and are thereby generally used solely as a tool for validation.
Defined iPSC differentiation protocols aim to mimic the stages of natural development, where stem cells gradually move from a pluripotent state to a multipotent state and finally into a unipotent terminally differentiated cell type. For example, during neuronal differentiation iPSCs first transition into neural progenitors, and then can be further differentiated into excitatory cortical neurons, inhibitory cortical neurons, midbrain dopaminergic neurons, or motor neurons, depending on the stimuli provided. Inefficiencies at each stage of differentiation drastically decrease the purity and maturity of the final cell product. For many lineages, key markers of the different stages of development have been well characterized through lineage-tracing studies in mice.112,113 Using this information, researchers can purify iPSC cultures at each stage of development based on known markers. Using functional genomic tools, this approach has been used to successfully generate iPSC-derived chondrocytes 114 and skeletal muscle progenitor cells. 115 In these studies, CRISPR/Cas9 was used to knock in endogenous reporters for COL2A 114 and Myf5/Pax7, 115 respectively, to purify the desired cell types from a mixed cell population. The resulting cell cultures are more uniform compared with cultures that do not undergo a purification step. Robust and reproducible differentiation protocols are required in order to successfully use iPSC-derived cell cultures for drug development.
Epigenome editing tools have been successfully used to reprogram cells into a variety of cell types, including iPSCs,116–118 myocytes,119,120 and neurons,119,121,122 demonstrating that these synthetic factors are potent enough to drive changes in cell phenotypes. Transcription factor-driven reprogramming and defined reprogramming protocols share the same general limitations; cultures are impure and long times are required to achieve functional maturity. However, the adaptation of CRISPR/Cas9-based transcription factors for high-throughput screening enables systematic identification of the optimal factors required to improve current reprogramming protocols. With the central hypothesis that current reprogramming protocols are failing to induce the expression of necessary genes to drive sufficient reprogramming, high-throughput genetic screens can be used to identify these missing factors. This approach has been successfully used to identify a combination of ZF-based transcription factors that are able to replace the master transcription factor Oct4 for inducing reprogramming into iPSCs. 123 CRISPR/Cas9-based synthetic screens are just starting to be used to identify the necessary genes responsible for controlling differentiation or reprogramming. In the first published example, a genome-wide knockout screen was used to uncover a set of kinases that inhibit the transition of iPSCs into definitive endoderm. Pharmacological inhibition of these kinases leads to an improved generation of definitive endoderm and subsequent differentiation into pancreatic and lung progenitor cells. 124 By screening on differentiation markers, libraries of gRNAs targeting promoters of genes could be used to identify proteins that enhance existing reprogramming protocols. 125 Discovery of novel reprogramming factors would therefore help improve culture quality and maturity, enabling drug discovery in more mature and disease-relevant cell types. Once robust and pure single-lineage cultures can be made, this will increase the ease and availability of multitissue organoids that allow for examination of more complex disease biology for drug development.126–128
Generating Human Disease Models That Recapitulate Mature Transcriptomic and Epigenomic Profiles
DNase-seq/ATAC-seq and RNA-seq allow researchers to comprehensively assess the chromatin and transcriptomic profiles, respectively, of cells and tissues. Application of these assays to iPSC-derived disease models has shown that while iPSC differentiation protocols produce cells that exhibit some of the phenotypic qualities of the desired tissue, there are differences in the transcriptomic 129 and epigenetic (unpublished data) profiles of these cells compared with mature adult tissues. Reprogrammed cells tend to exhibit “epigenetic” memory, meaning that iPSCs derived from one lineage tend to retain epigenetic marks from the parent cell type. 130 This epigenetic memory in iPSCs inherited from the parental cell type influences the differentiation capacity and likely the epigenetic profile of the final cell product. For example, iPSCs derived from nonhematopoietic cells (such as fibroblasts) have a reduced capacity to differentiate into blood-forming cells. 131
To efficiently identify new disease targets and drugs, it is important to develop human therapeutics in the context of disease models that accurately reflect the epigenetic and transcriptomic profiles of the relevant tissues. Most iPSC-derived neuronal protocols produce cells that are fetal in nature, meaning that they may not accurately model advanced neurological disorders associated with aging. 132 One way to partially overcome the immature epigenetic nature of iPSC-derived models is the direct reprogramming of an adult cell type into the desired cell type. By bypassing the pluripotent state, direct reprogramming allows for retention of the epigenetic marks that have accumulated in the parental somatic cell. It has been shown that the epigenetic methylation signatures associated with aging are well conserved when adult fibroblasts are directly differentiated into neurons (correlation of 0.91). 133 However, even these cells likely will not exhibit the full transcriptomic and epigenetic profile of adult tissue. 134 One limitation of this approach is that cells must be directly reprogrammed for each experiment, as somatic cells used for direct differentiation generally have limited proliferation capacity.
Functional genomic tools can induce site-specific genetic and/or epigenetic changes that alter chromatin conformation, transcriptomic profiles, and protein expression. Using a variety of the modular DNA binding platforms discussed previously, different effector domains can be localized to a specific genomic locus to induce changes in DNA sequence or chromatin structure, and thereby influence gene expression profiles.41,135 Should reprogrammed cell models lack the desired epigenetic signature, these tools can be used to induce the correct epigenetic mark. For example, fragile X syndrome is characterized by a CGG expansion in the 5′ UTR of the gene that promotes methylation and gene silencing of the fragile mental retardation protein (FMRP). An increased number of CGG repeats is generally associated with increased methylation and a more deleterious phenotype. Targeting a CRISPR/Cas9-based demethylase to the locus induces normal levels of FMRP expression and alleviates the phenotype. 136 Conversely, this also suggests that by using epigenome editing tools, researchers can model the disease by inducing methylation of the promoter rather than needing to generate multiple model cell lines, each with a different number of CGG repeats to model the spectrum of disease. Tools such as these will be instrumental in providing the understanding of disease biology needed to drive the next generation of therapies.
Functional Genomic Screening for Drug Discovery
High-throughput screening is a critical part of drug discovery. Pharmaceutical R&D has traditionally relied on one of two different pharmacological screening approaches: target-based screens and phenotypic screens. Target-based screens require screening of large chemical libraries for activity toward a known disease-associated target. These screens can be done utilizing high-throughput array-based methods, often screening thousands or even millions of compounds for a known target. Phenotypic screening allows for unbiased evaluation of chemical matter looking for an effect on the phenotype(s) of interest. In recent years, there has been increased focus on phenotypic screens as there has been evidence that such targets lead to more successful outcomes in the clinic.
4
However, phenotypic screening is still fraught with difficulty, predominantly in the target identification stage, which can be both lengthy and costly, as well as potentially unsuccessful. Regardless of target versus phenotype based, pharmacological screens are currently unable to probe the entire set of potential cellular drug targets. There are estimated to be 500–700 unique protein targets currently included in the FDA-approved drug list, though there are approximately 20,000 genes in the human genome, highlighting a lack of robust chemical matter available for targeting the majority of human genes.137,138 Genetic screens offer the potential to perturb every gene and ask if that perturbation influences the target or phenotype of interest. Additionally, genetic screens provide a new way to capitalize on phenotypic screening while avoiding the drawback of target deconvolution (
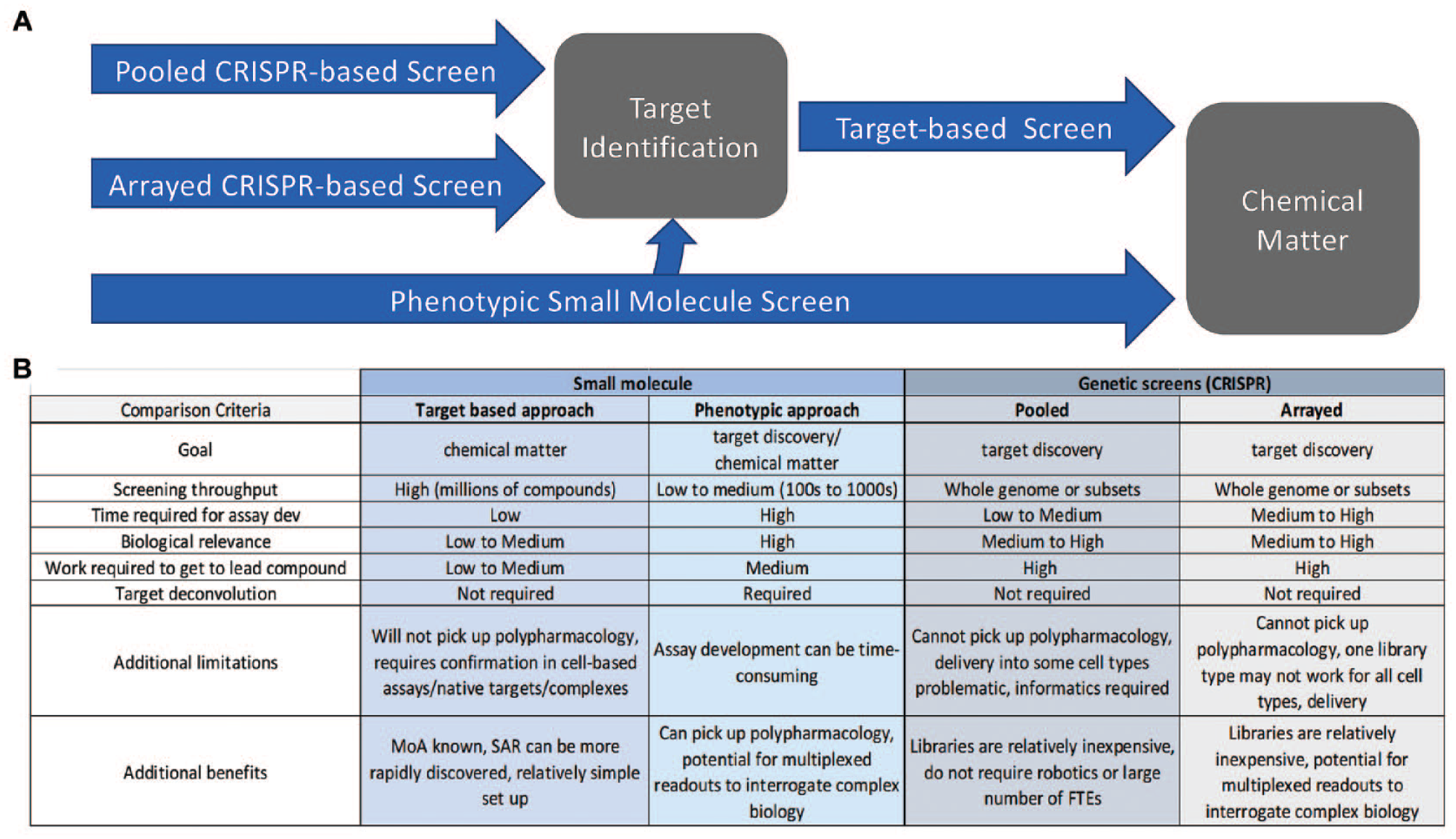
Comparison of screening paradigms. (
The increased throughput of functional genetic screens in recent years has allowed for unbiased, genome-scale screens to answer fundamental biological questions. This technology initially focused on gene essentiality with clear applications in oncology, but has since expanded through interrogation of increasingly complex phenotypes. RNAi was first used to manipulate mammalian gene expression in 2001. 8 This technology enabled modulating the transcriptome with simple antisense oligonucleotides to understand the biological effects of genes. It was not long before the use of RNAi was commonplace and large-scale arrayed and pooled screening became possible in mammalian cells with siRNA and shRNA libraries.
The advent of CRISPR/Cas9-based tools for high-throughput functional genomic screens has transformed genetic screening methods. From essentiality screens focused on genes that contribute to cellular viability to more intricate screens identifying drug response or complex phenotypes, CRISPR/Cas9 tools have opened new avenues in drug discovery. Prior to the use of CRISPR/Cas9 in mammalian cells, high-throughput genetic screens were limited by the lack of specificity and effectiveness of shRNA and siRNA mechanisms. 139 While RNAi was a great advance, the issues of incomplete knockdown and off-target effects limited its broader utility for high-throughput screening.15–17 CRISPR/Cas9 tools have allowed increased specificity in genomic and epigenomic editing in mammalian cells by acting at the level of DNA rather than RNA. The requirement of complementarity of both the gRNA and target DNA, combined with the need for a protospacer adjacent motif (PAM) sequence, has allowed for better specificity of gene targeting.140,141 Direct cutting of target DNA with the Cas9 enzyme has allowed for site-specific induction of indels and subsequent gene knockout, avoiding potential issues with incomplete knockdown that can be seen with RNAi. The combined specificity and complete gene knockout with CRISPR/Cas9 has led to fewer false positives and more reproducible hit identification compared with RNAi methods.142,143 Additionally, the ease of use over other DNA editing technologies such as ZFNs and TALENs has led to the rapid adoption of CRISPR/Cas9 tools in drug discovery.
Implementing CRISPR/Cas9 Functional Genomic Screens
CRISPR/Cas9 screening can be performed in either an arrayed format or, more commonly, in a pooled format. In a pooled screen, a large number of cells are transduced with a pooled library of gRNAs packaged in a lentiviral delivery system that can be combined with a variety of Cas9 effectors to achieve knockout, activation, or inhibition (
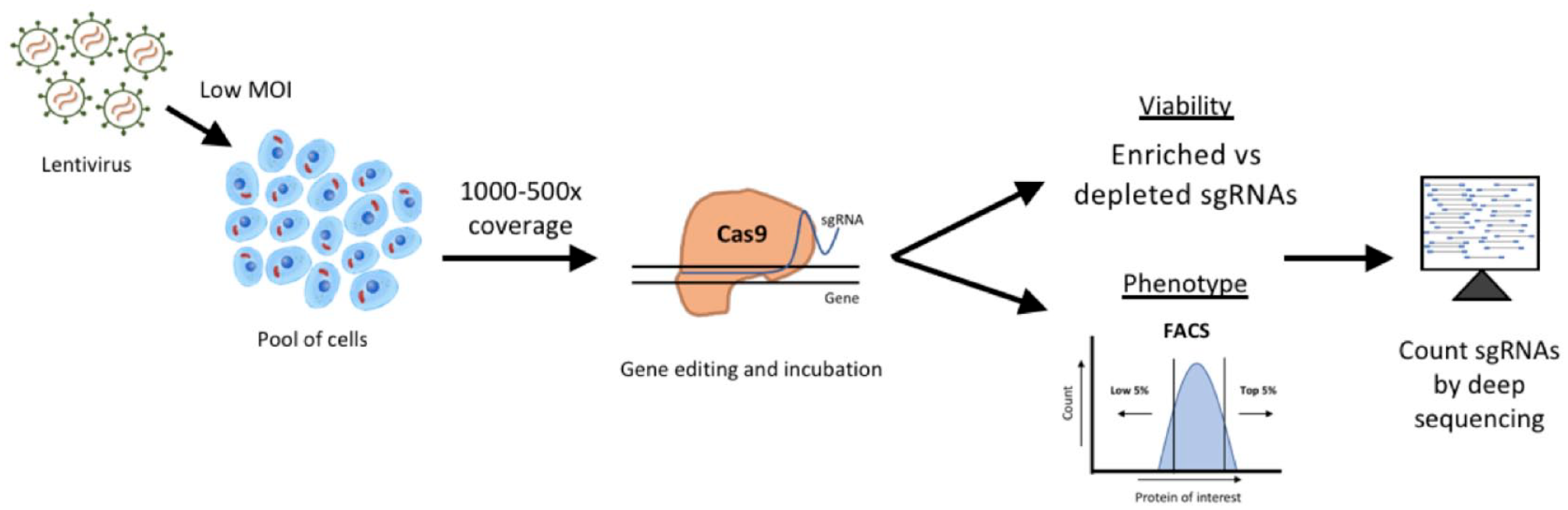
Representative pooled CRISPR screening workflow. Pooled CRISPR screening is typically performed by transducing a large pool of cells with gRNA-containing lentivirus. It is important to maintain the desired gRNA coverage throughout the screen (typically at least 500×), which can mean maintaining a minimum of 40–50 million cells per replicate in genome-wide screens. Virus is given to cells at a low MOI (typically around 0.2) to ensure there is only one gRNA per cell. For an MOI of 0.2, five times the number of cells must be transduced with the viral gRNA library to ensure coverage is maintained after antibiotic selection. In a proliferation screen, cells are collected at time points along the way and finally at the endpoint to monitor changes in gRNA abundance over time. In a phenotypic assay, cells may be stained with an antibody for a particular marker and sorted using FACS for abundance of the marker at the endpoint of the screen. The final phase of the screen involves isolating genomic DNA from all collected samples and PCR amplifying the guide-containing region with barcoded primers. PCR products are then sequenced by next-generation sequencing and the abundance of gRNAs can be compared across conditions or time points.
On/Off-Target Effects of CRISPR/Cas9
As CRISPR/Cas9 technology has developed, a continuing point of discussion has been related to understanding and improving the on- and off-target effects (i.e., efficiency and specificity) of both the gRNAs and Cas9 itself. Initial genome-wide CRISPR/Cas9 screening papers used the “Genome Scale CRISPR Knock-Out” (GeCKO) library of gRNAs, which were selected to minimize off-target effects using a metric that includes the number of predicted off-targets in the genome and the type of mutations (distance from protospacer-adjacent motif and clustering of mismatches). 149 Early studies established that mismatches closer to the PAM are more important for proper DNA binding compared with distal mutations. 30 The first high-throughput screens and other focused studies have since uncovered other important features of gRNAs that are critical for specificity. For instance, several studies have explored the sequence bias of gRNAs in genome-wide libraries by measuring the frequency of bases at each position in high- and low-performing gRNAs.150,151 Other studies have examined the effects of consecutive mismatches. 152 Next-generation libraries make use of more complex gRNA design algorithms and training data for improved specificity and on-target activity. 148 To test the off-target effects of the CRISPR/Cas9 system over the course of several weeks in a pooled screen, Wang et al. conducted an experiment using a two-vector system where Cas9 and a gRNA toward AAVS1 were constitutively expressed for 2 weeks. In this experiment, there was 97% cutting efficiency at the predicted AAVS1 site after 2 weeks compared with <2.5% cleavage at 13 predicted off-target sites. 150 These data gave promise to the specificity and low rate of off-target effects in pooled CRISPR/Cas9 screens over the length of an experiment. gRNA design has continued to be optimized with updated algorithms powered by gRNA cutting efficiency and specificity data, leading to cleaner, more reproducible screens for target discovery.148,153
Pooled CRISPR-Based Screening for Drug Discovery
Whole-genome CRISPR/Cas9 screening libraries can now be purchased or made for a relatively low cost, and a pooled screen can be performed by one person in only a matter of weeks. Besides the ease of use, the other major advantage of CRISPR/Cas9 pooled screening lies in target identification, avoiding the need for target deconvolution that is often faced by small-molecule phenotypic screens. An unbiased CRISPR pooled screen can target every gene in the genome, allowing the possibility of discovering novel targets and disease biology.
One of the prominent applications of CRISPR/Cas9-based pooled screening to date has been uncovering essential genes and genes that regulate cellular proliferation. Comparison of essential genes across tissue types and individual mutations has revealed the ability to define context-specific dependencies on certain genes. 154 For example, the loss of the tumor suppressor retinoblastoma protein (Rb) is a common occurrence in many cancers; however, identifying cellular vulnerabilities in Rb mutant patients has eluded researchers. CRISPR/Cas9 knockout screening of a pooled gRNA library in Rb mutant small-cell lung cancer (SCLC) cells showed that loss of Rb made these cells uniquely reliant on Aurora B kinase (AURKB) compared with wild-type cells. 155 Furthermore, the dependence of Rb null cells on AURKB was confirmed in xenograft models with AURKB inhibitors. These results indicate a potential therapeutic avenue for SCLC patients harboring an Rb mutation and highlight the use of pooled genetic screening for drug discovery.
Understanding the mechanism of action is critical to the successful development of a drug candidate. A clear example of the application of functional genomic tools in this area is the use of high-throughput genetic screens performed in combination with drug treatment. Screens such as these can be used to understand the mechanism of action of a compound with unknown biology or to uncover genes that confer intrinsic or acquired resistance to a particular drug. Drug resistance is a major obstacle in the clinic, particularly in cancer therapy, that can arise through a wide variety of mechanisms. The use of CRISPR/Cas9 screening has uncovered mechanisms of drug resistance pointing to key genes and pathways that dictate the response to individual compounds. 156 Early evidence for the power of pooled CRISPR/Cas9 screens in drug resistance was shown in a proof-of-principle study using a near-genome-wide gRNA library to identify resistance to 6-thioguanine (6-TG), a nucleotide analog that damages DNA. 150 In this screen, cells were transduced with the gRNA library followed by treatment with a lethal dose of 6-TG. Cells that survived the treatment were then sequenced to identify gRNAs that were enriched in this population. As expected, genes known to be involved in DNA mismatch repair were identified as top hits, validating this approach to identify drug resistance and mechanism of action. Later work by Anderson et al. used pooled CRISPR/Cas9 screening with a targeted gRNA library across multiple KRAS mutant cell lines to identify drug sensitizers. 157 By using low-dose small-molecule inhibitors (~IC25), these screens could identify drug combinations that could promote primary drug action and delay drug resistance, in this case to MEK/ERK inhibitors, in KRAS mutant cancers. 157
Pooled CRISPR/Cas9 screening has also been used in vivo. 158 In the first pooled screen performed in vivo, a genome-wide lentiviral library of gRNAs were given to cells in vitro and then transplanted into mice in a xenograft model, and lung metastases were sequenced to determine mediators of metastatic disease. 159 In another example, a small library of gRNAs were packaged into AAV and delivered directly to the brains of immunocompetent mice to uncover the role of genes that are frequently found mutated in glioblastoma multiforme (GBM) patients. 160 This screen identified several driver mutations and co-occurring mutations in GBM in vivo models that correlated with genomic data seen in patients. Future studies will continue to show the utility of pooled CRISPR/Cas9 screens in vivo for target discovery, particularly for specific phenotypes that cannot be reliably reproduced in vitro.
Gain-of-Function Screens
While much focus has been on the manipulation of genes by decreasing their expression, screening using the overexpression of genes can be important in certain contexts. The overexpression of genes for gain-of-function screens has been possible through cDNA expression vectors 161 and later CRISPR/Cas9 activation (CRISPRa) screens. 140 Overexpression allows for a positive manipulation of genes to understand biological activity that occurs when the gene is present, in contrast to loss-of-function studies. One benefit of overexpression is avoiding potential variables of cellular compensation and redundancy that occur with gene knockdown or knockout. The caveats to cDNA overexpression are expressing the gene off of an exogenous plasmid, out of the cellular context, and thereby achieving potentially supraphysiological protein expression, which may alter function and localization. Alternatively, CRISPRa allows for targeted overexpression from endogenous loci to activate gene expression from endogenous promoters, or enhancers, of a gene and in this way can regulate a gene in a manner, and to a level, that may be more physiologically relevant. Furthermore, activation of endogenous promoters can lead to expression of multiple gene splice variants, 162 something that is currently not possible with a single cDNA construct.
Phenotypic Genetic Screening
As functional genomic screening has evolved, more complex screens have significantly expanded the range of biology that can be interrogated. The use of fluorescence-activated cell sorting (FACS) has allowed for studies to be performed using pooled gRNA or shRNA libraries at a genome-wide scale, followed by sorting cells based on the abundance of a protein of interest.163–168 FACS-based pooled genomic screens can be applied to a wide variety of disease states by screening on changes in the abundance of a particular protein of interest. For example, to screen on regulators of autophagy, a lentiviral genome-wide gRNA library was delivered to H4 neuroglioma cells stably expressing green fluorescent protein (GFP)-tagged p62, a well-known substrate, and marker of autophagic activity. 164 After 7 days, cells were sorted on the upper and lower quartile of GFP protein levels, followed by high-throughput sequencing, to determine changes in gRNA abundance between p62 high and low populations. Regulators were identified as gRNAs that changed abundance in the p62 low or high population, signifying active degradation or accumulation of p62, and thus altered autophagy. mTOR is a known regulator of autophagy and, accordingly, the majority of negative regulators identified in the screen were positive regulators of the mTOR pathway, such as Rheb and Raptor, as well as mTOR itself. In addition, several potentially novel regulators of autophagy were identified, showing the utility of such screens in identifying candidate drug targets. 164
In addition to pooled FACS-based screens, genomic perturbations can be assayed using arrayed methods. Arrayed-based screens are done in plate format and thus are more labor-intensive and may require automation depending on the size/type of screen. However, arrayed screens can be used to study specific cellular phenotypes that would not otherwise be possible in a pooled format, such as screening on an image or kinetic-based phenotype.169,170 The ability to complex multiple endpoints into the same screen also allows much more information to be gathered about how the probed gene influences the cell phenotype. Therefore, array-based screens are an attractive option for specific phenotypic outputs, especially for targeted gRNA or RNAi libraries that could potentially be combined with chemical screens.
Moving forward, the ability to analyze and compare the scale of data being generated by genome-wide screens has become increasingly important. Project Achilles was initiated by the Broad Institute to aid in the effort to compare these screens by compiling close to 1000 cell lines screened with RNAi and CRISPR/Cas9 knockout libraries to enable analysis across screens and identification of cellular dependencies across cell lines.171–175 In this data portal, screen data sets can be analyzed in combination with gene copy number and expression data in a publicly available data set to examine unique and context-specific genetic vulnerabilities.172–175 As genome-wide functional screens become increasingly popular, it will be critical to comprehensively analyze these data sets to gain a deep biological understanding to uncover new drug targets and therapeutic avenues.
Future of Functional Genomics in Drug Discovery
In the short history of the functional genomic field, progress has been rapid (
To address the issues with translatability in drug discovery, many pharmaceutical companies are moving toward examining patient samples to better understand the molecular mechanisms driving disease and identify genetic biomarkers of therapeutic response. 176 This precision medicine approach has been used successfully in the clinic, particularly in oncology. One such example is the clinical benefit seen with the use of a poly (ADP-ribose) polymerase (PARP) inhibitor, olaparib, as a monotherapy in metastatic breast and advanced ovarian cancer patients with BRCA mutations that have received prior chemotherapy.177,178 The use of targeted therapies, such as olaparib, demonstrates the benefits of identifying mechanistically distinct patient populations that dictate the clinical response to a given therapy. Similar approaches are being explored in other diseases, such as epilepsy, where genetic evaluation has demonstrated that refractory epilepsies can be caused by different underlying mechanisms that can lead to variation in clinical response. 179 These examples demonstrate the value of having increased information about the molecular mechanism of disease and response to therapy.
An issue faced in many diseases is the difficulty in accessing disease tissue and obtaining enough genetic material in relevant patient samples for testing; this is particularly true for neurological disorders. New low-cell-input profiling techniques, such as single-cell RNA-seq, 180 ATAC-seq, 181 and CUT&RUN, are opening up new opportunities in this area. By being able to extract more information from small amounts of sample, scientists can more broadly apply these functional genomic techniques. Additionally, as technical hurdles limiting genome editing efficiencies in primary tissue are overcome, it may become possible to use CRISPR/Cas9 tools to conduct drug discovery campaigns directly in patient samples and to examine relevant phenotypes or endpoints in single-cell format. Improved barcoding technologies that use proteins as barcodes may allow for more direct links of cellular perturbation to phenotypes compared with conventional DNA barcoding. 182
Generally, genetic screens are used to modulate a single target per cell. However, modulating a single gene at a time may not be suitable for the screening of complex polygenic diseases. Future studies will likely examine combinatorial effects of genetic modifications. This could potentially be addressed by combinatorial CRISPR/Cas screens, either by increasing the number of gRNAs introduced per cell in pooled or arrayed screens, or by screens performed in various isogenic disease cell lines to identify phenotype modifying genes. As methods become more refined and robust, complex combinatorial screens will likely lead to the discovery of novel biological pathways and interactions, subsequently expanding the number of future drug targets.
Conclusion
Discovering and developing new medicines is a difficult and high-risk process. Functional genomic tools provide an avenue to gain a comprehensive understanding of human disease biology and enable drug development. With genomic and epigenomic tools, endogenous regulatory networks can be directly probed and clearly linked to phenotypic disease outcomes. As functional genomic technologies continue to develop, they will increasingly be implemented into conventional drug discovery pipelines, aiding the efforts to develop novel therapeutics.
Footnotes
Declaration of Conflicting Interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: All authors are employed by UCB Pharma or Element Genomics, a wholly owned subsidiary of UCB Pharma.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.