
Abstract
D-dimer is an essential diagnostic index of thrombotic diseases. Since the existing anti-D-dimer antibodies vary in quality and specificity, a search for alternative anti-D-dimer antibodies is required. The present study aimed to screen a novel monoclonal antibody (mAb) against D-dimer using a light-initiated chemiluminescence assay (LiCA). In this work, mice were immunized with antigen prepared from human plasma by enzyme hydrolysis. After screening, a novel mAb, DD 2G11, was obtained. The results of sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) and Western blot analysis indicated that DD 2G11 could be used as a standard marker for D-dimer. The isotype of DD 2G11 was IgG1, the Ka value was 0.646 nM-1, and the Kd value was 50 nM, indicating that the binding affinity to D-dimer was very high. Furthermore, no cross-reactivity between DD 2G11 and other fibrinogen degradation products (FgDPs) was found. Finally, the correlation between DD 2G11 and the reference antibody (commercial antibody) was investigated by analyzing 56 clinical samples using a latex-enhanced turbidimetric immunoassay (LTIA). The R2 value of the linear regression was 0.94538, indicating that DD 2G11 met clinical requirements. In conclusion, the present study provides a more expeditious protocol to screen mAbs and provides a clinically usable mAb against D-dimer.
Keywords
Introduction
D-dimer is a 180 kDa specific degradation product generated through the breakdown of fibrinogen/fibrin clots in the fibrinolytic pathway. 1 D-dimer consists of three chains (α, β, and γ chains) of fibrinogen cross-linked by disulfide bonds. The dimeric structure of D-dimer is held by two covalent, intermolecular isopeptide bonds between the γ chains.2–4 The concentration of plasma D-dimer changes drastically during fibrin production; therefore, it is an ideal blood-native biomarker of coagulation and fibrinolysis and may be used as a measure of normality or abnormality of the coagulation system. 5 In general, <0.5 μg/mL of D-dimer is detectable in healthy individuals. 6 However, the plasma D-dimer level is significantly elevated in patients with disseminated intravascular coagulation (DIC), 7 acute myocardial infarction, 8 cerebral embolism, 9 serious hepatitis, 10 and certain malignant solid tumors. 11 Additionally, the high negative predictive value of plasma D-dimer makes it the gold standard for excluding a deep vein thrombosis (DVT) or pulmonary embolism (PE) diagnosis. 12 Meanwhile, following thrombolytic therapy, D-dimer levels fall rapidly if treatment is effective. 13 From that point of view, D-dimer assays may serve as a useful strategy for monitoring patients undergoing thrombolytic therapy and play an essential role in clinical diagnosis.
The clinical application of D-dimer cannot be separated from its accurate determination. At present, the clinical detection of D-dimer is based on the antigen–antibody reaction. 14 D-dimer molecules are first captured by monoclonal antibodies (mAbs) and then quantified by colorimetric or fluorescent methods. Thus, the accuracy of the test results relies primarily on the antibody used, the method of capture, the instrumentation required, and the calibration standard. 15 In particular, the antibody must target the specific epitopes on the D-domains without interacting with other fibrinogen degradation products (FgDPs) or fibrinogen in the plasma. Studies on the development of mAbs against different specific epitopes of D-dimer have been ongoing since the 1990s. The mAbs prepared by Doh et al. react with D-dimer only and not with other FgDPs. 16 Meanwhile, as techniques have improved, the application of immobilized antibodies has simplified the antibody screening process. 17 Moreover, to reduce immunogenicity, single-chain fragment variable (scFv) antibodies, Fab fragment antibodies, and recombinant antibodies have also been investigated to produce more effective antibodies. 18 However, there remain issues with the quantitative determination of D-dimer. One of these is that the results of D-dimer assays using these antibodies are not necessarily consistent due to differences in the reactivity and analytical sensitivity of the antibodies. 19 Also, it is difficult to develop an antibody that is suitable for use in a clinical immune turbidimetric assay. 20 Thus, research should focus on seeking efficient antibody screening methods and the preparation of novel anti-D-dimer antibodies, to ultimately establish better immunological detection methods to meet clinical needs.
The traditional screening method for mAbs is the enzyme-linked immunosorbent assay (ELISA), which requires multiple washing steps. Errors and hook effects are easily induced when high-affinity clones are screened. Thus, alternative screening methods are in demand. Notably, a luminescent oxygen channeling immunoassay (LOCI)-based liquid proximity assay, the light-initiated chemiluminescence assay (LiCA), was first reported in 1994 and applied to antibody screening recently. 21 When a light-sensitive bead (coated with captured antibody) approaches a chemiluminescent bead (coated with detection antibody), the biological interaction between them generates a detectable amplified optical signal. 22 This technique takes approximately half the time of an ELISA, avoids the washing steps, and exhibits excellent sensitivity, precision, accuracy, and linearity for a broad range of targets. 23 It has already been widely applied in protein–protein interaction assays, protease assays, and kinase assays, and has recently been used in the high-throughput screening (HTS) of antibodies.24,25 Additionally, in separate experiments, the authors of the present study have successfully screened high-specificity anti-HBsAg antibodies using LiCA, which can fully meet clinical requirements. The present study aimed to screen a novel mAb against D-dimer using LiCA, which may lead to the development of novel D-dimer diagnostic reagents.
Materials and Methods
Reagents
Anticoagulant human plasma from 50 healthy volunteers (5 mL per person) and 56 patients with suspected DIC (5 mL per person) was kindly provided by the Affiliated Hospital of Jilin Medical University (Jilin, China) in March 2017. DIC was diagnosed according to the DIC criteria. 26 Plasma were stored at −20 °C. Thrombin, plasmin, aprotinin, CaCl2, citric acid, EDTA, Tris, NaCl, barbital, glycine, paraffin oil, and Tween 20 were all obtained from Sigma-Aldrich (Merck KGaA, Darmstadt, Germany). Easy II Protein Quantitative Kit (BCA kit, TransGen Biotech Ltd, Beijing, China, cat. DQ111-01), Innovance D-Dimer reagents (Siemens Healthineers, New York, NY, cat. OPBP03), horseradish peroxidase-labeled goat anti-mouse IgG H&L (HRP-IgG, cat. ab6789), anti-β-actin antibody (cat. ab8226), and D-dimer antigen (cat. ab35949) were purchased from Abcam (Cambridge, MA). Anti-D-dimer antibody DD 1D10 (positive control) was kept in our laboratory. Fibrinogen was purchased from Sigma-Aldrich (Merck KGaA, cat. F3879). D and E fragments and FgDP mixture were purchased from Biomean Tech Co., Ltd (Chongqing, China, cat. FAP-H004). Dulbecco’s modified Eagle’s medium (DMEM) and fetal bovine serum (FBS) were purchased from Gibco (Invitrogen, Carlsbad, CA).
This study was performed in accordance with institutional ethical guidelines and was approved by the Affiliated Hospital of Jilin Medical University in Jilin, China. Informed consent for the collection of plasma was obtained from all subjects.
Preparation and Identification of D-Dimer Antigen
D-dimer was prepared from human plasma as reported previously. 16 Ten milliliters of human plasma was incubated with 250 µL of 200 U/mL thrombin and 1 mL of 0.2 M CaCl2 at 37 °C for 6 h to form cross-linked fibrin clots. After centrifuging at 10,000g for 15 min, the precipitate was sequentially washed with washing solution (citric acid 0.001 M, EDTA 0.04 M, NaCl 0.16 M; pH 7.4 ± 0.1) three times and double distilled (DD) water overnight. Then, the precipitate was ground into powder with liquid nitrogen. Following resuspension with Tris-buffered saline (TBS; Tris 0.05 M, NaCl 0.16 M; pH 7.4 ± 0.1), the solution was digested with 20 mU/mL plasmin at 37 °C for 16 h. The reaction was terminated with 2–10 μg/mL aprotinin. After centrifuging at 10,000g for 15 min, the supernatant was concentrated using an ultrafiltration centrifuge tube with a 30 kDa molecular weight cutoff membrane and was further analyzed by nonreducing and reducing sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE), Western blotting, and indirect ELISA. D-dimer antigen purchased from Abcam served as a positive control.
Preparation of D-Dimer-Specific mAbs
Female BALB/c mice (6 weeks old, weighing 18–22 g) were purchased from Jilin University (Changchun, China). Animals were housed under conditions of controlled temperature (23 ± 1 °C) and maintained on a 12 h light/dark cycle (lights on 7:00-18:00 h) with food and tap water available ad libitum. The experimental protocol was approved by the Lab Animal Centre of Jilin University (license number SCXK-(JI) 2017-0004). Five female BALB/c mice (6 weeks old) were immunized with 5 mg/kg of purified human D-dimer every 14 days for 4 weeks by subcutaneous injection. For the first injection 5 mg/kg D-dimer was administered in equal Freund’s complete adjuvant (Sigma-Aldrich; Merck KGaA). The second and third injections were immunizations with 5 mg/kg D-dimer in equal Freund’s incomplete adjuvant (Sigma-Aldrich; Merck KGaA). Three days after the last injection, 5 μL blood samples were collected from the tail vein and drawn into an Eppendorf tube, keeping a recommended ratio of blood to saline solution at 1:9. After centrifuging at 2000g for 10 min, the supernatants were determined using indirect ELISA. Mice with a sera titer above 1:16 × 104 were selected for cell fusion. Hybridoma cells producing D-dimer-specific mAbs were prepared according to the previous protocol. 27 The positive mice were euthanized by cervical dislocation 3 days after the last injection, and spleen cells were harvested aseptically. A total of 1 × 108 spleen cells were suspended with DMEM and then mixed with 2 × 107 mouse myeloma cells (Sp2-Ag0/14, ATCC). In a 37 °C water bath, 1 mL of preheated 50% polyethylene glycol 4000 (Sigma-Aldrich; Merck KGaA) was slowly added for 1 min, and then 3 mL of DMEM for 3 min. After centrifuging at 800g for 5 min, the precipitates were suspended with DMEM and plated into 96-well cell plates that had been paved with feeding cells. The plates were placed in a 5% CO2 incubator at 37 °C for culture. On the 5th and 10th days after the fusion, half of the medium was replaced with 20% HAT medium (Invitrogen, Thermo Fisher Scientific, Waltham, MA); on the 12th and 15th days after the fusion, the medium was replaced with 20% HT medium (Invitrogen). When the fusion cells grew to more than one-fourth of the well bottoms, the supernatants of fused cells were assayed by LiCA for the presence of anti-D-dimer antibodies.
Screening of Anti-D-Dimer Antibodies Using LiCA
The primary screening procedures were performed as follows. First, D-dimer was biotinylated according to the protocol of EZ-Link Sulfo-NHS-SS-Biotinylation Kit (Thermo Fisher Scientific, cat. 21445). Briefly, 1 mL of a 2 mg/mL D-dimer was incubated with ~27 μL of 10 mM Sulfo-NHS-SS-Biotin on ice for 2 h. The mixture was dialyzed against phosphate-buffered saline (PBS) buffer using an 8 kDa molecular weight cutoff dialysis bag to remove excess biotin reagent. Afterward, 1 μg/mL biotinylated D-dimer, a mixture of 25 μL culture supernatants of fusion cells, and 80 μg/mL goat anti-mouse antibody-conjugated luminous beads (acceptor beads, Bo Yang Biotech Co., Ltd, Shanghai, China, cat. A1807) were added to the same well in a 96-well plate at 25 μL each. Meanwhile, a mixture of 25 μL of 0.1 M PBS (pH 7.4 ± 0.1) + DMEM, 25 μL of 1 μg/mL biotinylated D-dimer, and 25 μL of 80 μg/mL goat anti-mouse antibody-conjugated luminous beads served as the negative control. The serum of D-dimer-immunized mice served as the positive control. The microplate was placed in an HT LiCA high-throughput chemiluminescence analyzer (Bo Yang Biotech). Two hundred microliters of 20 μg/mL streptavidin-coated photosensitive beads (donor beads, Bo Yang Biotech, cat. A1808) were added to each well by the analyzer automatically using LiCA Master HT Edition 2.0 software. Following the reaction at 37 °C for 35 min, the fluorescent signals of the negative control and the samples were obtained with excitation at 450 nm and reading emission at 620 nm. The sample/negative values (S/N values) were calculated as follows: value of sample/value of negative control. All experiments were repeated six times. Samples with an S/N value ≥2 were identified as positive hybridoma cells, which were also called positive clones.
Indirect ELISA Assay
The affinity of mAbs to the D-dimer and the antigenicity of the prepared D-dimer were determined by indirect ELISA as described previously. 28 Ninety-six-well plates were coated with 5 μg/mL purified D-dimer or other antigens (fibrinogen, D and E fragments, and FgDP mixture) in coating buffer (0.2 M carbonate-bicarbonate buffer, pH 9.2) with 100 μL in each well at 4 °C overnight. After plate coating, the plates were washed twice with 200 μL of PBS containing 0.1% Tween 20 per well. Then, 200 μL of blocking solution (2% BSA/PBS) was added to each well and incubated at 37 °C for 1 h. The plates were washed three times and incubated with 100 μL of hybridoma cell culture supernatants or positive antibody DD 1D10 at 37 °C for 1 h. PBS (0.1 M; pH 7.4 ± 0.1) in DMEM served as the negative control and DMEM served as the blank control. Afterward, the plates were washed three times and incubated with a 1:2000 dilution of HRP-IgG at 37 °C for 40 min. Ortho-phenylenediamine (OPD; Sigma-Aldrich; Merck KGaA) in citrate phosphate buffer, pH 5.5 (0.5 mg/mL), containing H2O2 (1 µL/mL, added just prior to use) was used for the color reaction. The reaction was stopped by adding 100 μL of 2 M sulfuric acid. The absorbance at 490 nm was read with a Sunrise microplate reader (TECAN, Salzburg, Austria). All experiments were repeated six times.
Secondary Screening of Monoclonal Antibodies
The LiCA positive clones were further screened for secondary screening. During the secondary screening, the culture supernatant of positive clones was incubated with a high D-dimer value of human plasma from patients suspected to have DIC (D-dimer concentration: 30–50 μg/mL) or a low D-dimer value of human plasma from healthy volunteers (D-dimer concentration: <0.55 μg/mL) at 37 °C for 30 min at 25 μL each. Then, 25 μL of the mixture, 25 μL of 1 μg/mL biotinylated D-dimer, and 25 μL of 80 μg/mL goat anti-mouse antibody-conjugated luminous beads were added to the well in a 96-well plate. A mixture of 25 μL of 0.1 M PBS (pH 7.4 ± 0.1) + DMEM, 25 μL of 1 μg/mL biotinylated D-dimer, and 25 μL of 80 μg/mL goat anti-mouse antibody-conjugated luminous beads served as the negative control. A mixture of 25 μL of 3 μg/mL DD 1D10, 25 μL of 1 μg/mL biotinylated D-dimer, and 25 μL of 80 μg/mL goat anti-mouse antibody-conjugated luminous beads served as the positive control. The plate was placed in an HT LiCA high-throughput chemiluminescence analyzer (Bo Yang Biotech). The remaining steps were the same as for the primary screening. All experiments were repeated six times. The intensive positive mAbs should be the ones with decreased S/N values when the high-value D-dimer plasma is added and increased S/N values when the low value D-dimer plasma is added.
Purification and Identification of Monoclonal Antibodies
After the second screening, four intensive candidate positive cell lines, which can secrete high-affinity antibody, were obtained. The selected positive hybridoma cells were cloned (isolate a single cell from a mixture of hybridoma cells) at least twice by limited dilution. Hybridomas to be cloned are diluted to 0.8 cells per well and cultured overnight at 37 °C, 5% CO2 in air, and 98% humidity in a CO2 incubator. The cloning procedure was repeated until a stable and single hybridoma cell line was established. The cloned cell lines were massively cultured in high-glucose DMEM containing 10% FBS. Then, the hybridomas were expanded in ascites fluid according to the manufacturer’s instructions and mAbs were purified as follows. 27 Before the hybridoma injection, several adult female BALB/c mice were intraperitoneally injected with sterile paraffin oil (0.5 mL per mouse) 7 days prior to inoculation with 1 × 106 hybridoma cells per mouse. The fresh ascites fluid was collected from the mouse abdominal cavity using a needle and was dripped into a sterile 15 mL centrifuge tube. After centrifuging at 1500g for 10 min, the supernatant was diluted with an equal amount of barbital buffer solution (4 mM barbital, 0.16 M NaCl, 0.8 mM MgSO4·7H2O, 0.3 mM CaCl2; pH 7.2 ± 0.1). Ten milliliters of diluted ascites fluid was mixed with 150 mg of silicon dioxide and incubated at 200 rpm for 30 min at room temperature in a shaker. After centrifuging at 10,000g for 30 min, the supernatant was filtered using a 0.45 μm filter membrane. Subsequently, the sample was subjected to an ÄKTA prime FPLC purification system (GE Healthcare Bio-Sciences, Pittsburgh, PA) equipped with a Protein A/Protein G column (10 mL; Thermo Fisher Scientific, cat. 89962). Briefly, 10 mM PBS (pH 7.4 ± 0.1) served as the column equilibration buffer, which was driven by a pump at a flow rate of 6 mL/min. Following equilibration using ~50 mL of equilibration buffer, 10 mL of ascites fluid samples was subjected to the column manually. The column was sequentially washed with the equilibration buffer for a while until the baseline of the purification system was flat (~10 mL of equilibration buffer). Then, the column was eluted with 0.1 M glycine-HCl buffer (pH 2.7 ± 0.1). The eluted mAb fractions (30 mL each) were collected using a fraction collector (GE Healthcare Bio-Sciences) and detected with a UV analyzer at 280 nm (Waters 150, Millipore, Billerica, MA). The elution peaks were pooled (~50 mL) and neutralized using 1 M Tris-HCl (pH 9.0 ± 0.1). The eluted mAbs were fully dialyzed against 10 mM PBS at 4 °C for 48 h using an 8 kDa molecular weight cutoff dialysis bag. The immunoglobulin isotyping of the antibodies was identified using mouse mAb isotyping reagents (ELISA/Ouchterlony Double Diffusion, Sigma-Aldrich; Merck KGaA, cat. ISO2-1KT), according to the manufacturer’s instructions. Briefly, the antibody isotyping identification was a capture ELISA-based method, using IgG1, IgG2a, IgG2b, IgG3, IgM, and IgA isotype-specific antibodies as primary antibodies, purified mAbs as antigens, and peroxidase-labeled rabbit anti-mouse IgG antibody as second antibodies. The high optical density (450 nm) suggests the right antibody isotype. Finally, purified mAbs were analyzed by reduced SDS-PAGE, Western blotting, and a BCA kit.
SDS-PAGE and Western Blotting
Purified D-dimer was analyzed in SDS-PAGE under nonreducing conditions (absence of DTT, presence of SDS/denaturing) and reducing conditions. Specifically, D-dimer was mixed with 5× nonreducing loading buffer (Sangon Biotech Co., Ltd, Shanghai, China, cat. C516031) or 5× protein loading dye (Sangon Biotech) and loaded onto 10% SDS-PAGE. Purified mAbs were analyzed using reduced SDS-PAGE. Western blot analysis was performed as described by Towbin et al. 29 The protein concentration of purified D-dimer was determined via a BCA kit. Sixty milligrams of total protein was mixed with 5× protein loading dye and boiled at 98 °C for 5 min. Samples were loaded onto 10% SDS-PAGE under reducing conditions. Proteins were electrophoretically transferred to 0.22 μm polyvinylidene fluoride (PVDF) membranes with a Western blot transfer system (Bio-Rad Laboratories, Inc., Hercules, CA) for 2 h at 300 mV. The PVDF membranes were washed with TBST (Tris-buffered saline containing 0.5% Tween 20) and blocked with 5% BSA for 2 h at 37 °C. Thereafter, the membranes were washed with TBST five times and incubated with mAbs overnight at 4 °C, and then washed with TBST four times and incubated with HRP-labeled goat anti-mouse IgG (1:5000) for 1 h at 37 °C. Proteins of interest were visualized with enhanced chemiluminescence in Gel Doc EZ Image Lab (Bio-Rad Laboratories). β-Actin served as a loading control and was detected using the anti-β-actin mAb (Abcam, cat. ab8226).
Monoclonal Antibody Affinity Constant Determination by ELISA
The affinity constant was calculated according to a previous method. 30 A total of two different concentrations of D-dimer (2 and 0.5 μg/mL) were coated in a 96-well plate and assigned [Ag] and [Ag′], respectively. The mAbs were serially diluted to 60, 6, 0.6, 0.06, 0.006, and 0.0006 μg/mL and added to the plate. The process was performed as previously described for the indirect ELISA. According to the S-shaped curve of OD490 values versus D-dimer concentration, the antibody concentrations (mol/L) at the corresponding half maximum OD490 value at two antigen concentrations were extrapolated and set as [Ab] and [Ab′], respectively. The affinity constant was determined using the following equation:
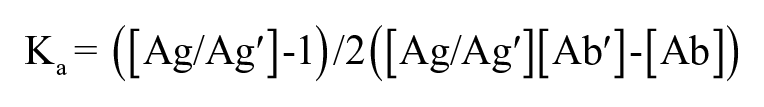
Monoclonal Antibody Dissociation Rate Analysis Determination by SPR
The dissociation equilibrium constant (Kd) was analyzed by surface plasmon resonance (SPR) technology using a Biacore T200 system (Biacore AB, Piscataway, NJ). The D-dimer was immobilized onto CM5 biosensor chips (Biacore AB) to 400–480 response units (RU) using an amine coupling kit. After the D-dimer chip was ready, increasing concentrations of the four antibodies, prepared in HBS-EP buffer (10 mM HEPES, pH 7.4, 150 mM NaCl, 3 mM EDTA, 0.005% surfactant P20), were injected over the chip at 50 μL/min for 250 or 300 s, after each injection. HBS-EP buffer was passed over the chip for 250 s to monitor the dissociation phase. Equilibrium binding responses were plotted against the concentrations of analyte, and from the fitted saturation binding curves, the Kd values were derived.
Measurement of D-Dimer Level in Clinical Human Plasma by LTIA
To evaluate the clinical value of screened mAbs against D-dimer, D-dimer concentrations in 56 individuals were detected by latex-enhanced turbidimetric immunoassay (LTIA). LTIA was performed as follows: latex particles (<1 μm diameter) coated with 0.5 mg/mL mAbs were activated by 1-ethyl-3-[3-dimethylaminopropyl] carbodiimide hydrochloride (EDC) and N-hydroxysulfosuccinimide (NHS) and incubated for 30 min at room temperature. The solution was blocked with 50 μL of 1% BSA/PBS and ultrasonically dispersed for further analysis. Meanwhile, anticoagulant-treated blood was collected and centrifuged at 2500g for 15 min at 4 °C to separate the serum samples. The mixture of serum and latex particles was subjected to a Sysmex CA-7000 Coagulation Analyzer (Dade-Behring, Marburg, Germany) to evaluate the OD570 values. Innovance D-Dimer reagents (Siemens Healthineers) served as the reference antibody.
Results
Preparation and Characterization of D-Dimer
D-dimer was prepared via hydrolyzation of human plasma using thrombin and plasmin. The natural human D-dimer is a dimer, the dimeric structure of which is held by two covalent, intermolecular isopeptide bonds between the γ chains.2–4 Therefore, the obtained antigen was identified using nonreducing SDS-PAGE, reducing SDS-PAGE, Western blotting, and indirect ELISA. D-dimer antigen purchased from Abcam served as the control, and antibody DD 1D10 was used as the primary antibody. DD 1D10 is a specific anti-D-dimer antibody obtained previously, with an antibody titer of 1:256 × 104. As presented in
Figure 1A
, a clear band of 180 kDa was noted under the nonreducing condition, which is the intact protein of D-dimer. Also, a clear band of 70 kDa (γ chain of D-dimer) and two bands around 75 kDa (α chain of D-dimer) and 30 kDa (β chain of D-dimer) were noted under the reducing condition. Meanwhile, the purified D-dimer exhibited no inferiority in reactivity toward the anti-D-dimer antibody DD 1D10, compared with the commercial D-dimer, in Western blotting and indirect ELISA analysis (
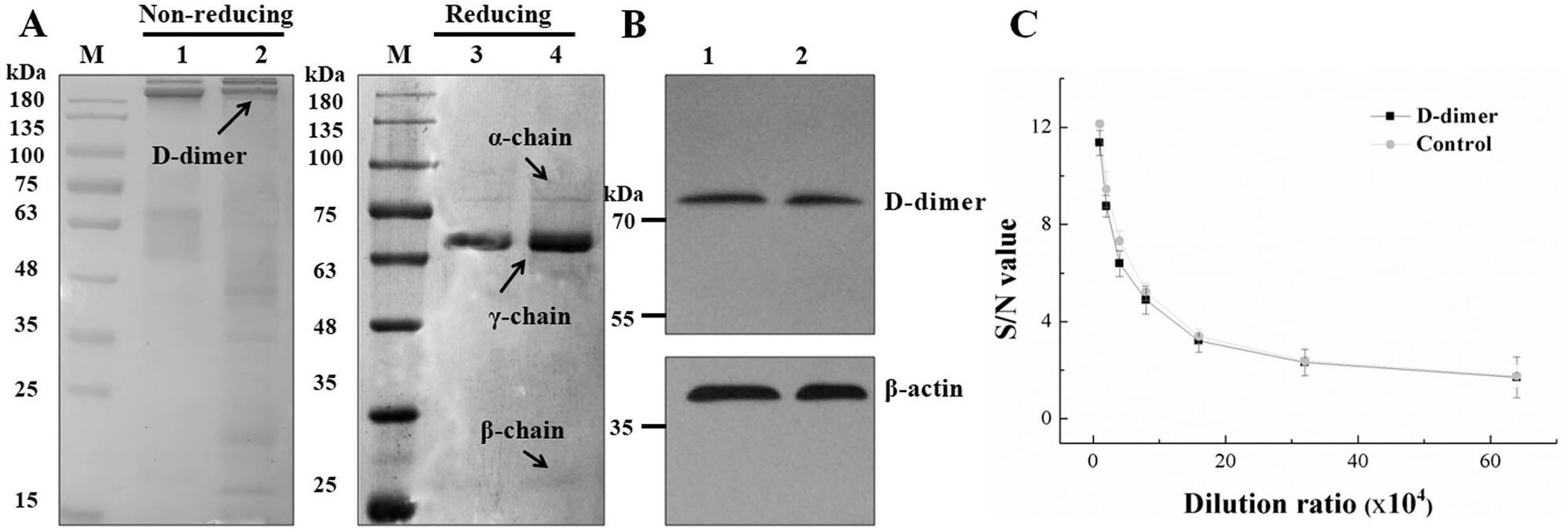
Characterization of D-dimer. D-dimer was prepared via hydrolyzation of human plasma using thrombin and plasmin. The hydrolysate was further analyzed by (
Screening of Anti-D-Dimer mAbs Using LiCA
Following cell fusion, more than 500 culture supernatants were numbered and tested by LiCA. Culture supernatants of hybridoma cells with an S/N value ≥2 were identified as positive clones during the preliminary screening. The results are presented in Figure 2 . A total of 62 clones of hybridomas producing IgGs that recognize the D-dimer antigen were obtained using LiCA. In the second screening, the addition of high-value D-dimer plasma can compete with the biotin-labeled D-dimer and prevent approaches of acceptor beads and donor beads, which lead to a significant decrease of S/N value. Meanwhile, the addition of low-value D-dimer plasma does not cause a significant change of S/N value compared with no plasma added, which indicates that plasma has no effect on the detection. After secondary screening and cloning, four intense positive mAbs were obtained and named DD 2G11, DD 3H4, DD 4E6, and DD 5B9 ( Table 1 ).
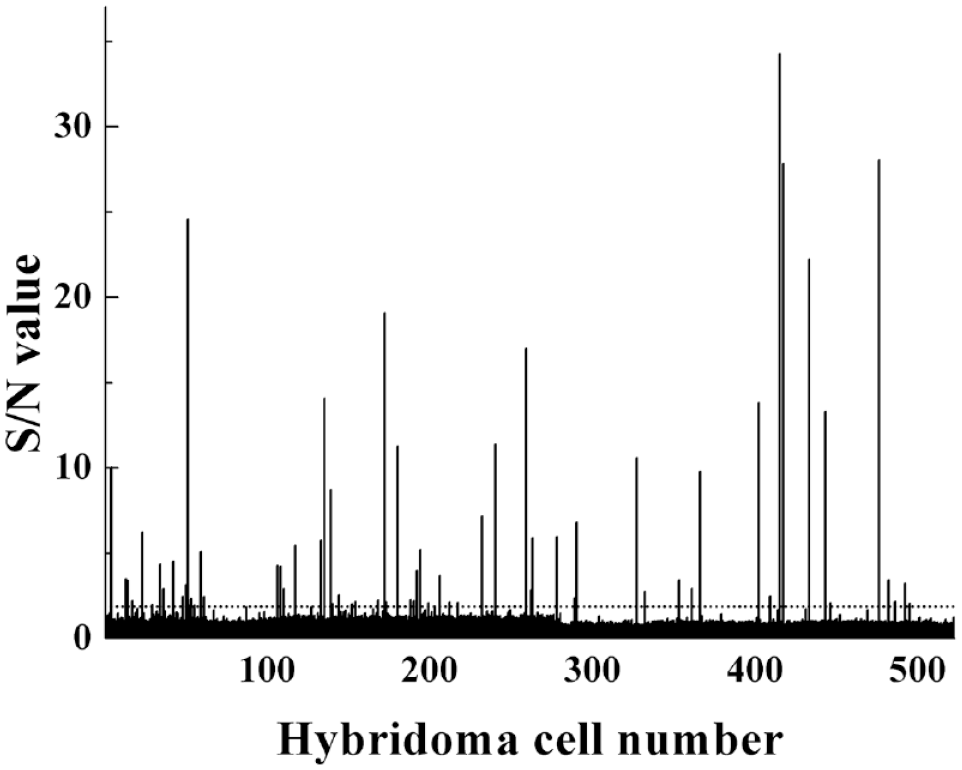
After cell fusion, more than 500 hybridoma cells were obtained and numbered (x axis). The culture supernatants of hybridoma cells were tested by LiCA for the presence of anti-D-dimer antibodies. Draw a horizontal dotted line at the S/N value 2. Hybridoma cells with an S/N value ≥2 (above the dotted lines) were selected as positive clones.
S/N Values of Four Intensive Positive mAbs Obtained after Secondary Screening and Cloning Determined by LiCA.
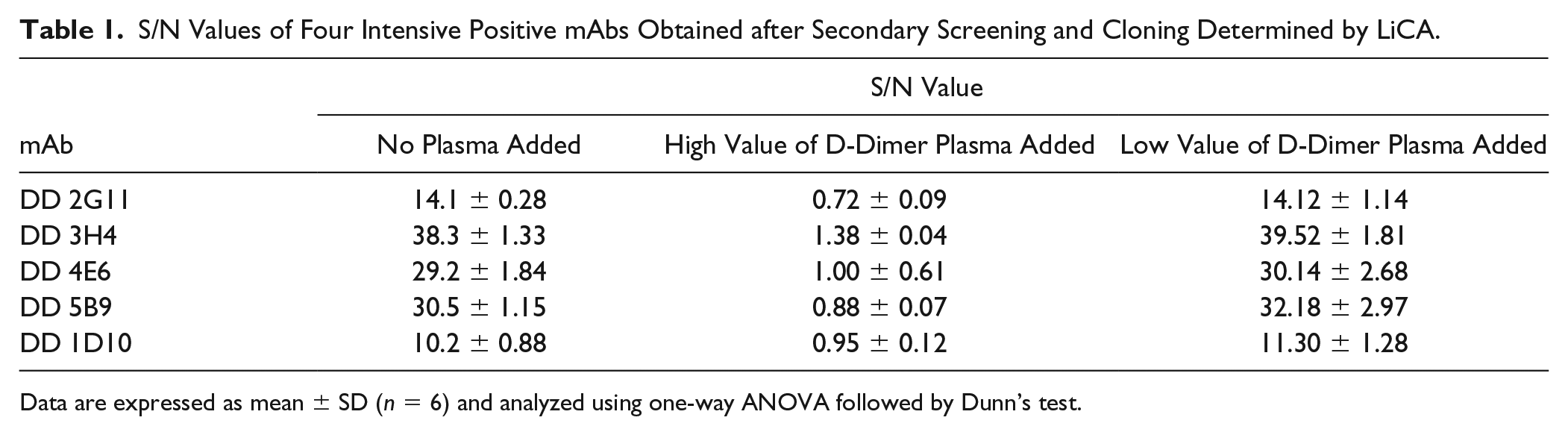
Data are expressed as mean ± SD (n = 6) and analyzed using one-way ANOVA followed by Dunn’s test.
Production and Purification of mAbs
Following reduced SDS-PAGE identification of purified ascites, two clear bands around 55 kDa (heavy chain) and 25 kDa (light chain) were noted, and the contaminating proteins were few. The Western blotting results (
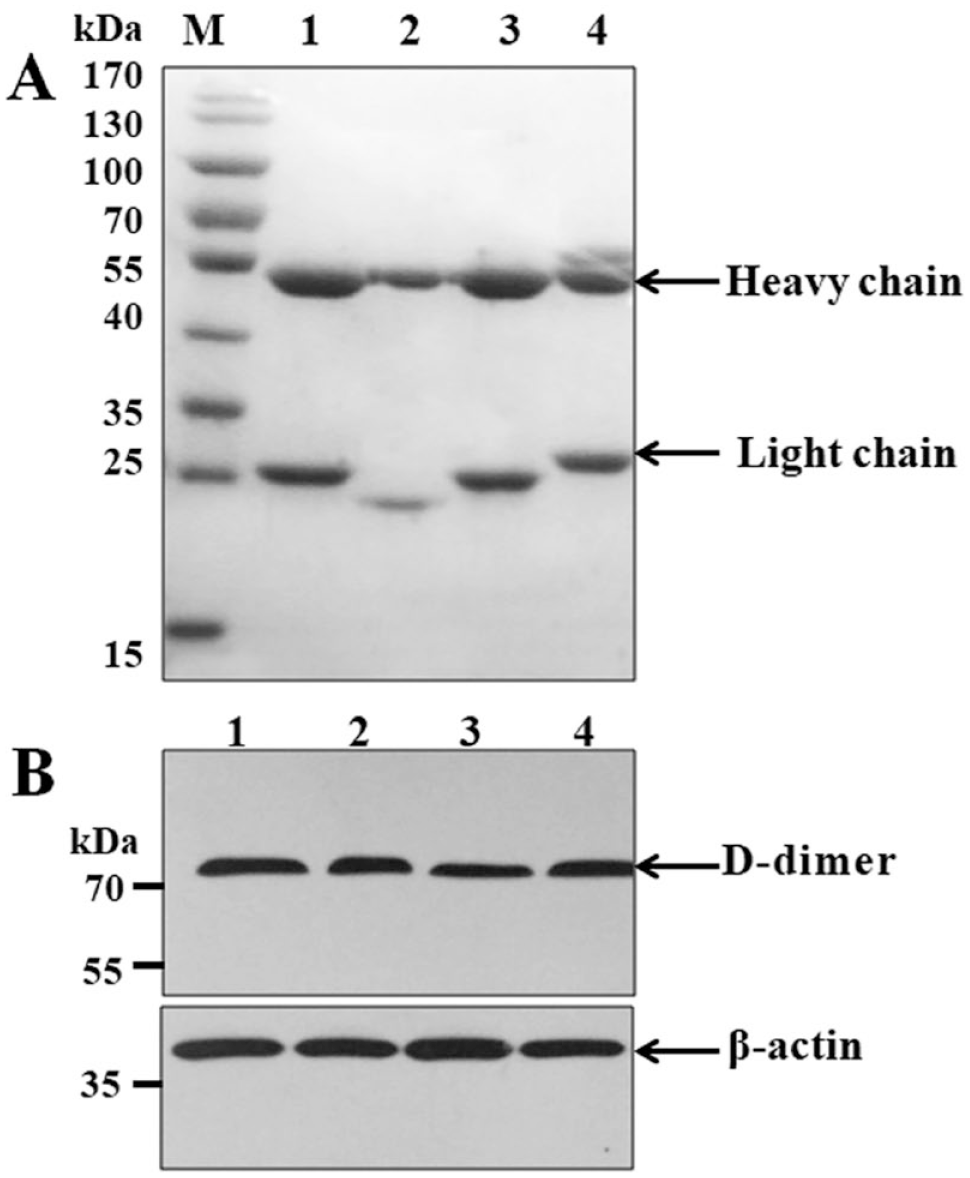
Characterization of anti-D-dimer antibodies. (
Result of the mAb Isotypes, Protein Concentrations, Affinity Constants, and Titer.
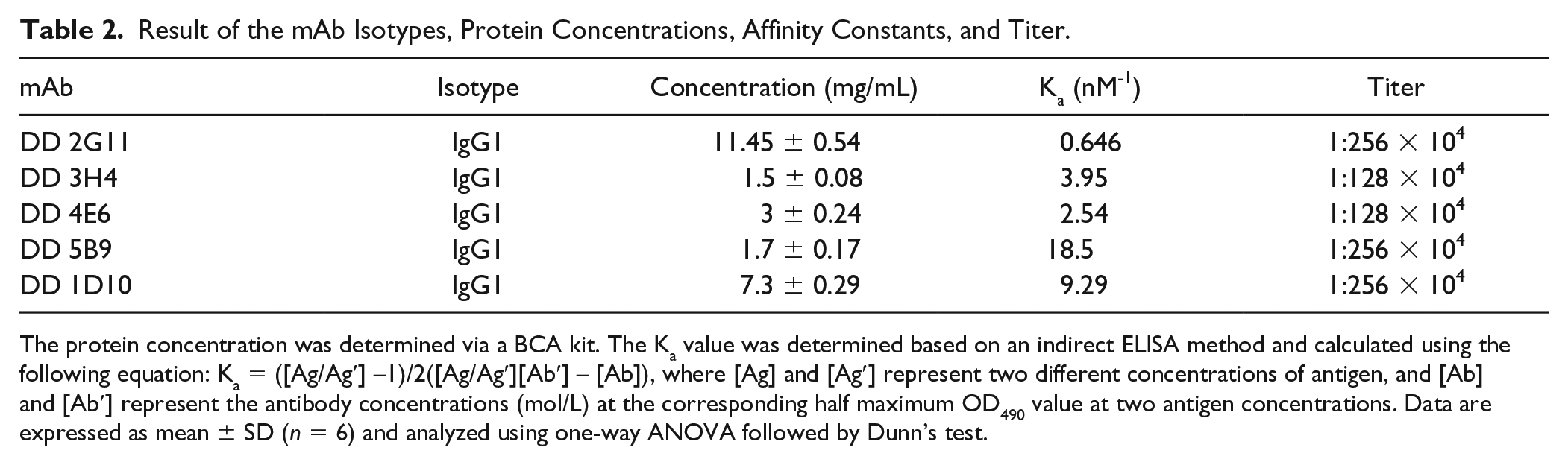
The protein concentration was determined via a BCA kit. The Ka value was determined based on an indirect ELISA method and calculated using the following equation: Ka = ([Ag/Ag′] –1)/2([Ag/Ag′][Ab′] – [Ab]), where [Ag] and [Ag′] represent two different concentrations of antigen, and [Ab] and [Ab′] represent the antibody concentrations (mol/L) at the corresponding half maximum OD490 value at two antigen concentrations. Data are expressed as mean ± SD (n = 6) and analyzed using one-way ANOVA followed by Dunn’s test.
Isotypes and Affinity Constants of mAbs
The affinity constants and isotypes of four clones are presented in Table 2 . The results indicate that the isotypes of the four clones were all IgG1. The Ka values of DD 2G11, DD 3H4, DD 4E6, and DD 5B9 were calculated to be 0.646, 3.95, 2.54, and 18.5 nM-1 respectively, which were all lower than that of DD 1D10 (9.29 nM-1). Affinity measurements by SPR are shown in Figure 4 . The Kd values of DD 2G11, DD 3H4, DD 4E6 and DD 5B9 were 50, 322, 394, and 113 nM, respectively, which indicates that they had high affinity to D-dimer.
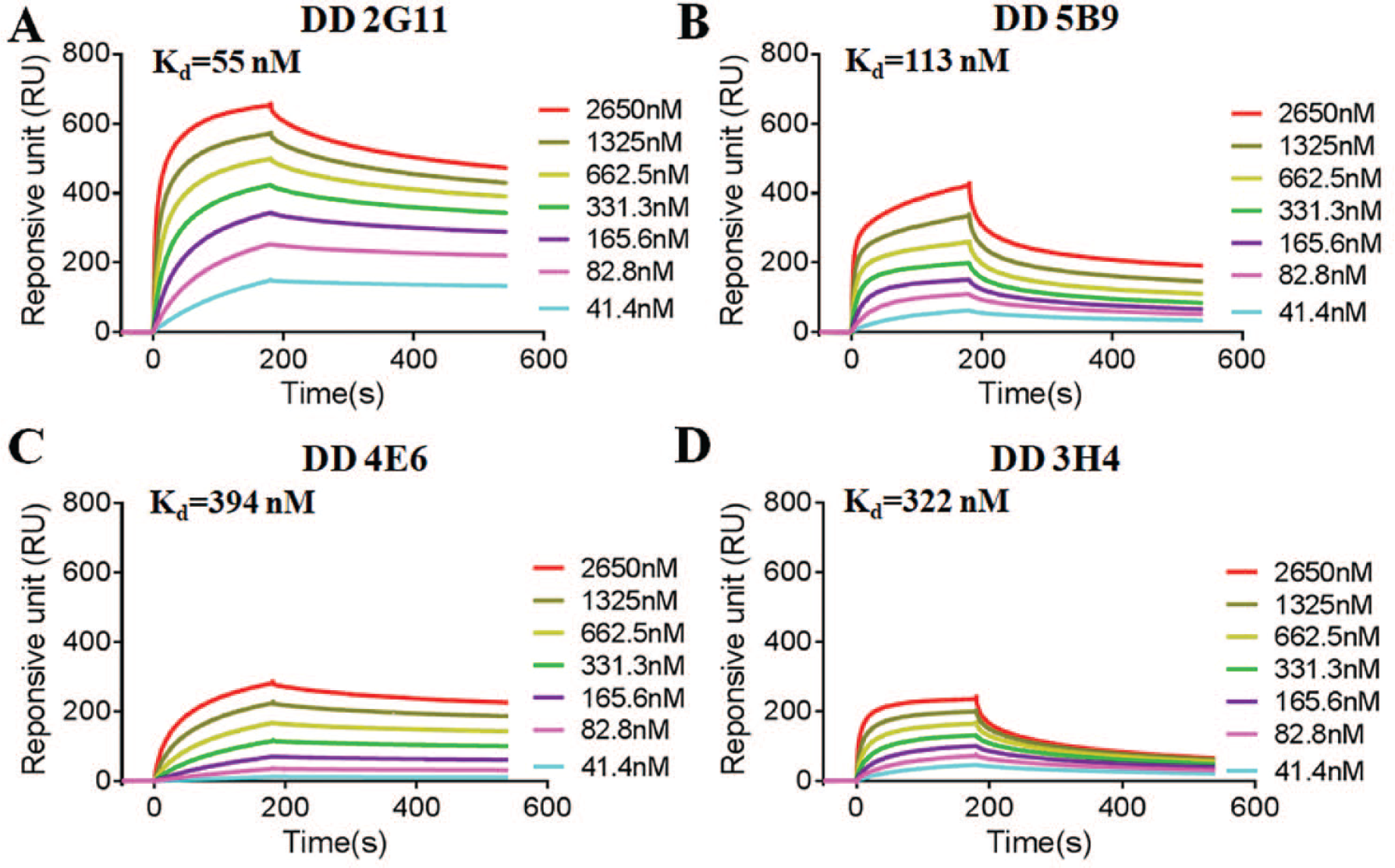
Dissociation equilibrium constant analysis by SPR for the binding of D-dimer to the four mAbs: (
Correlation between Four mAbs and Reference Antibody Based on LTIA
Comparison of four mAbs and the reference antibody using the LTIA method was performed on 56 blood samples. The reference antibody was a commercial anti-D-dimer antibody that is used in the majority of medical testing institutions. Furthermore, it is very suitable for clinical LTIA.31,32 The R2 value of linear regression between DD 2G11 and the reference antibody was 0.94538, which demonstrated that DD 2G11 had the highest correlation with the reference antibody (
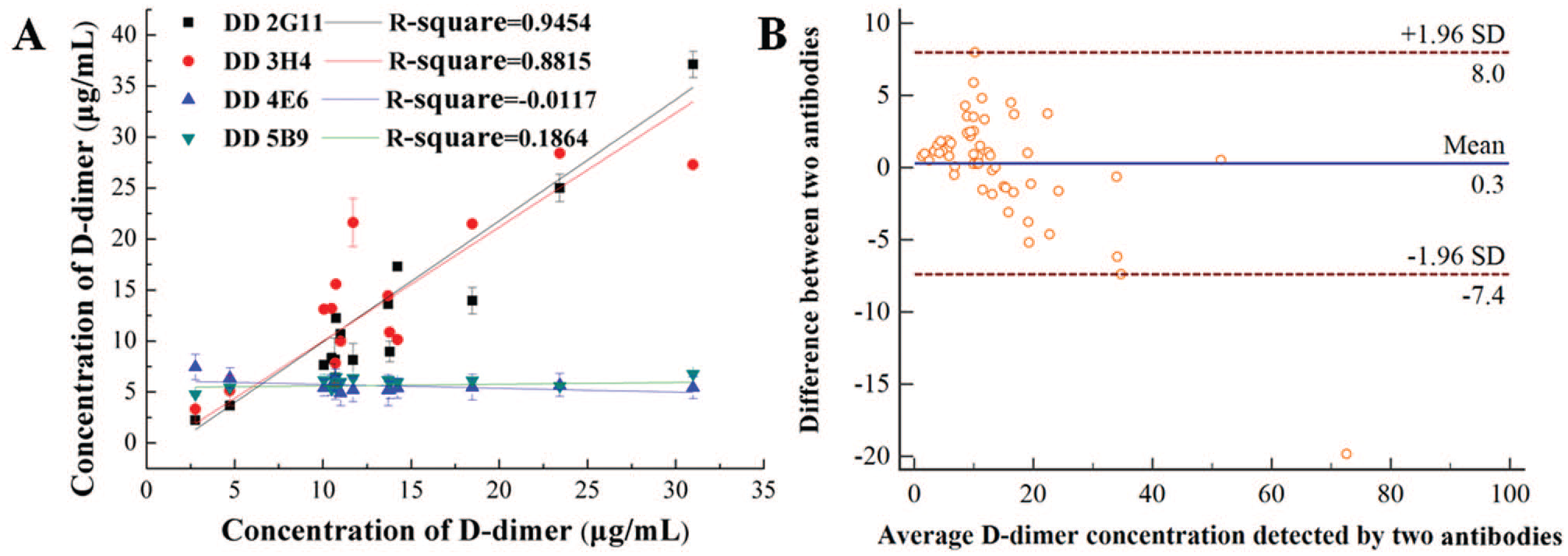
Correlation between four mAbs and Innovance D-dimer reagents (reference antibody) for detecting D-dimer by LTIA. (
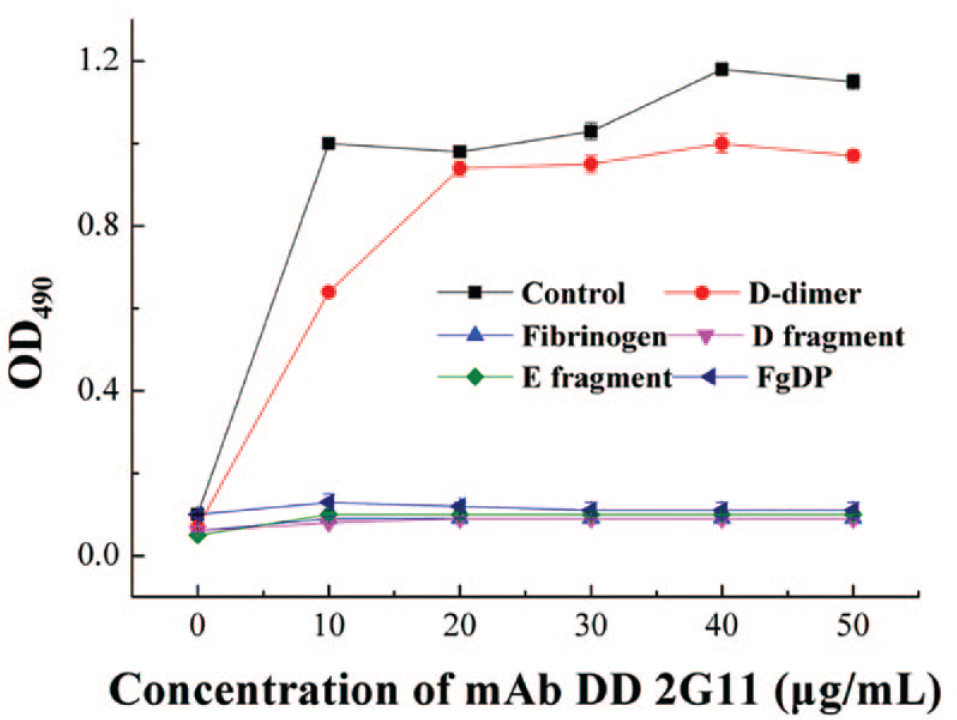
Specificity analysis of mAb DD 2G11 to various antigens (D-dimer, fibrinogen, D and E fragments, and FgDP mixture) using indirect ELISA. The commercial D-dimer served as the control.
Discussion
The assessment of D-dimer may provide clinical information for diagnosis and to guide the treatment of DIC, DVT, and PE. Although ~30 different assays are currently available for the clinical detection of D-dimer, none of them are independent from the application of specific mAbs. Therefore, screening for high-affinity mAbs against D-dimer is of importance.
In the present study, mAbs against D-dimer were successfully developed using an HTS method, LiCA. The LiCA technology provides an easy and reliable way to screen mAbs. Due to its high sensitivity of chemiluminescence, it has a lower detectable limit (LDL) compared with ELISA. Therefore, there was a greater opportunity to select positive clones. Moreover, this technology takes approximately half the time of ELISA and avoids the washing steps. It offers advantages in saving time and in its automatic operation, which make it more efficient and suitable for HTS and miniaturized screening compared with traditional screening methods. However, due to the short history of this technique, it has not been used in screening mAbs, which highlights the success of the present study.
Although previous studies have reported a number of mAbs reacting to D-dimer, the present study confirmed that the reactivity of DD 2G11 is consistent with the commercial antibody in the following aspects: (1) The antigen used for immunization was prepared from human plasma, which may contribute to the high immunogenicity of the antigen and generate specific antibodies. At the same time, our preparation technology of human D-dimer is the same as that of the commercial D-dimer. And Western blot and indirect ELISA analysis data show that the D-dimer purified from plasma had no inferiority in the reactivity compared with the commercial D-dimer. (2) In this study, four positive hybridoma clones were generated that produced mAbs of high affinity and no cross-reactivity with other FgDPs, possibly due to the nature of the IgG1-type antibody. Meanwhile, the ELISA results demonstrated that the mAbs were able to recognize folded D-dimer. Among the four mAbs, DD 2G11 exhibited the best performance in antibody titer, affinity, and protein concentration. (3) Data from the clinical LTIA test indicated that DD 2G11 had the highest correlation with the reference antibody and could meet clinical requirements. However, the details of this antibody remain limited at this time. It is important to note that the linear epitopes of DD 2G11 remain unknown, which is important for further studies.
Supplemental Material
supplementary_materials – Supplemental material for Generation of a Monoclonal Antibody against D-Dimer Using HTS-Based LiCA
Supplemental material, supplementary_materials for Generation of a Monoclonal Antibody against D-Dimer Using HTS-Based LiCA by Yuan Dong, Hanjin Hou, An Chen, Wei Ma, Moli Yin, Fanwei Meng, Chuanmin Hu, Huiyan Wang and Jianhui Cai in SLAS Discovery
Footnotes
Acknowledgements
The authors thank Xiuqiong Pu (Army Medical University) and Song Xu (Army Medical University) for their excellent technical assistance.
Supplemental material is available online with this article.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the “Research and Development of Industrial Technology” Program of Jilin Province, PR China (grant nos. 20170204005YY and 20180623045TC), National Natural Science Foundation of China (no. 11604120), and National Training Program of Innovation and Entrepreneurship for Undergraduates (no. 201913706059).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.