
Abstract
Chromatin regulatory complexes localize to specific sites via recognition of posttranslational modifications (PTMs) on N-terminal tails of histone proteins (e.g., methylation, acetylation, and phosphorylation). Molecular recognition of modified histones is mediated by “reader” protein subunits. The recruited complexes govern processes such as gene transcription, DNA replication, and chromatin remodeling. Dysregulation of histone modifications and consequent downstream effects have been associated with a variety of disease states, leading to an interest in developing small-molecule inhibitors of reader proteins. Herein, we describe a generalized time-resolved fluorescence resonance energy transfer (TR-FRET) assay for a panel of methyl-lysine (Kme) reader proteins. These assays are facile, robust, and reproducible. Importantly, this plug-and-play assay can be used for high-throughput screening (HTS) campaigns, generation of structure–activity relationships (SARs), and evaluation of inhibitor selectivity. Successful demonstration of this assay format for compound screening is highlighted with a pilot screen of a focused compound set with CBX2. This assay platform enables the discovery and characterization of chemical probes that can potently and selectively inhibit Kme reader proteins to ultimately accelerate studies of chromatin reader proteins in normal biology and disease states.
Introduction
Chromatin structure and dynamics are governed by an interplay of activities including DNA and histone modifications, incorporation of histone variants and nucleosome remodeling, and noncoding RNAs. 1 Regulation of chromatin states through these mechanisms affects cellular diversity and plasticity without altering the genetic sequence. Posttranslational modifications (PTMs), such as methylation of lysine or arginine residues on the N-terminal tails of histone proteins, are key determinants of chromatin regulation. For example, transcriptionally active gene promoters have enriched levels of histone H3 trimethylation at lysine 4 (H3K4me3), whereas gene promoters that are transcriptionally repressed have enriched levels of histone H3 trimethylation at lysine 9 (H3K9me3) and lysine 27 (H3K27me3). 2 As such, aberrant modification to the PTM landscape has been connected to human diseases and disorders. 1 Because PTMs are inherently reversible, unlike extensive DNA mutations, modulation of chromatin regulation with small-molecule inhibitors offers a promising therapeutic strategy for numerous diseases, including cancer.
Recognition of PTMs by proteins with “reader” domains enables further addition or removal of marks via recruitment of enzyme complexes that ultimately regulate cellular activities including gene transcription, DNA replication and recombination, and chromatin remodeling.
1
Methyl-lysine (Kme) reader proteins recognize lysine residues that are mono-, di-, or trimethylated at the ε-amino position of the lysine side chain using an aromatic cage binding site, typically formed by two to four aromatic residues (
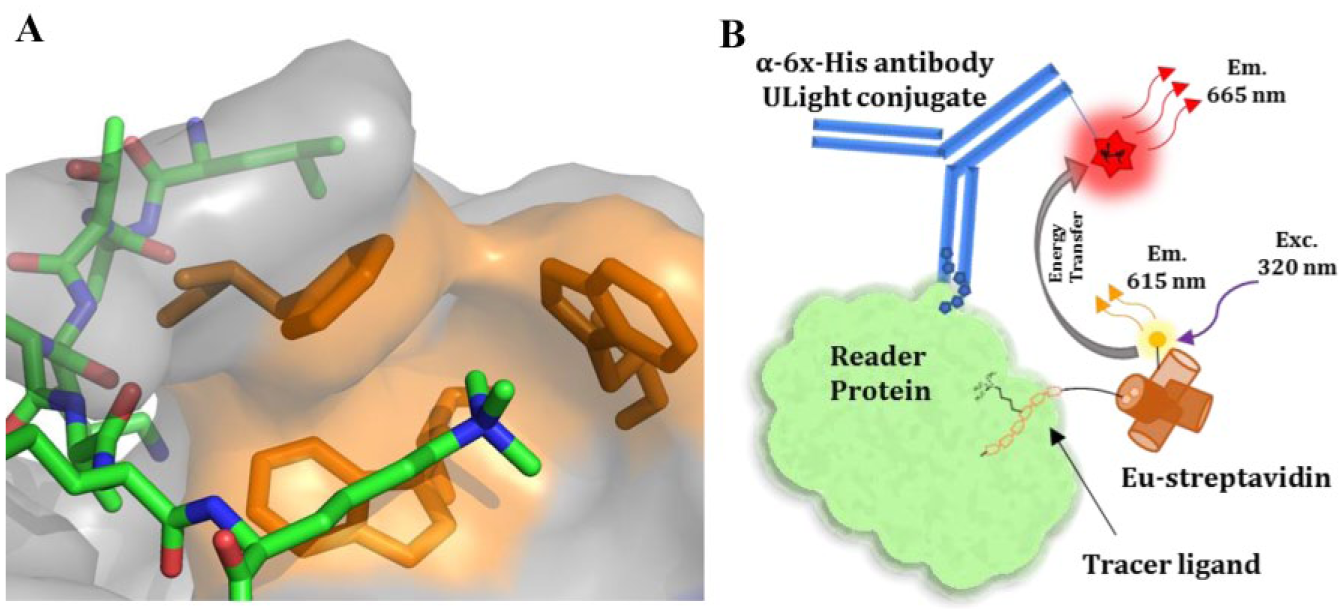
(
In mammals, the chromodomain is a highly conserved Kme binding motif structurally consisting of a three-stranded antiparallel β-sheet that folds against a C-terminal α-helix. 4 The human genome encodes 29 chromodomains in total. Eight members belong to the subfamily of chromodomain-containing chromobox (CBX) proteins that play critical roles in two major transcriptionally repressive complexes: the heterochromatin protein 1 (HP1) proteins consisting of CBX1, -3, and -5 (also known as HP1-β, -γ, and -α, respectively) and the polycomb (Pc) proteins consisting of CBX2, -4, -6, -7, and -8. The HP1 chromodomains are selective for binding to H3K9 methylation, whereas mammalian Pc proteins are reported to be more promiscuous in vitro but are mostly associated with H3K27 methylation in vivo. 5 Although CBX proteins have been associated with numerous biological functions, deciphering the exact role of each homolog is an ongoing endeavor and requires high-affinity and selective inhibitors, also known as chemical probes.6,7
Use of small-molecule chemical probes is a preferred strategy for investigating and validating the biological functions of specific targets within cells. They are particularly useful for mechanistic studies of biological processes relative to traditional genetic approaches in that they enable modulation of a specific component or activity rather than genetically removing an entire protein from the cellular system. A true chemical probe requires extensive assessment of selectivity, mechanism of action, and cellular activity. 7 Chemical probes can also provide starting points for drug discovery campaigns. For example, efforts on acetyl-lysine reader proteins have led to both high-quality chemical probes and molecules suitable for clinical trials. 8 Currently, there are only a limited number of chemical probes available for Kme reader domains, reinforcing a need for new tool molecules. 3
A robust, dependable, and reproducible biochemical interaction assay is essential for implementing hit discovery campaigns using high-throughput screening (HTS). Additionally, the ability to accurately characterize inhibitors of a biomolecular interaction is an essential component of developing structure–activity relationships (SARs) to drive hit optimization efforts. Both primary and secondary screening are fundamental requirements for discovering and developing new chemical probes. Criteria considered when choosing a method to employ for these purposes are cost, sensitivity, speed, ease, and reliability.
The AlphaScreen (Amplified Luminescent Proximity Homogeneous Assay; PerkinElmer, Waltham, MA) format has been widely used for HTS and characterization of reader protein inhibitors.9,10 Since AlphaScreen is a bead-based method, it has the particular advantage of detecting weak interactions by virtue of the avidity effect. However, for analysis of competitor compounds, the avidity effect can be a drawback as it potentially can make competitive compounds appear significantly weaker compared with the dissociation constant. 11 Another commonly used method for reader protein assays is fluorescence polarization (FP) using fluorescently labeled histone peptides. 12 One potential issue with the FP assay format is that it often requires high protein concentrations, which results in a higher tight-binding limit for accurately characterizing potent inhibitors. More so, the results of an FP assay using nanomolar concentrations of fluorescently labeled peptide can be affected considerably by compounds with fluorescent or absorbent properties, leading to fluorescence artifacts. 13 While both of these assay formats have utility for measuring Kme reader protein–peptide interactions and characterizing inhibitors,6,10,12 we sought a generalizable assay platform that avoids some of the pitfalls of these other techniques.
Here, we report establishment of a homogeneous, non-bead-based approach using time-resolved fluorescence resonance energy transfer (TR-FRET) for Kme reader proteins. This plug-and-play assay platform (
Materials and Methods
Materials
The LANCE Eu-W1024 streptavidin conjugate and the LANCE Ultra ULight anti-6X-histidine antibody were obtained from PerkinElmer. Trizma hydrochloride, sodium chloride, and Tween 20 were obtained from Sigma-Aldrich (St. Louis, MO). 1,4-Dithio-
Expression and Purification of Recombinant Chromodomain Proteins
The chromodomains of CBX2 (residues 9–66 of NP_005180), CBX4 (residues 8–65 of NP_003646), CBX5 (residues 18–75 of NP_036249), CBX8 (residues 8–61 of NP_065700), and MPP8 (residues 55–116 of NP_059990) were expressed with N-terminal His tags in pET28 expression vectors. The chromodomain of CBX7 (residues 8–62 of NP_783640) and CDYL2 (residues 1–75 of NP_689555) were expressed with C-terminal His tags in pET30 expression vectors. Proteins were expressed and purified as previously described.6,10
General TR-FRET Assay Conditions
A stock solution of 10× Kme reader buffer (200 mM Tris [pH 7.5], 1500 mM NaCl, and 0.5% Tween 20) was prepared, 0.2 µm filtered, stored at room temperature, and used throughout. Assays were completed using freshly made Kme reader buffer containing 20 mM Tris [pH 7.5], 150 mM NaCl, 0.05% Tween 20, and 2 mM DTT. White, low-volume, flat-bottom, nonbinding, 384-well microplates (Greiner, cat. 784904) were used for assay development and screening with a total assay volume of 10 µL. V-bottom, 384-well polypropylene plates (Greiner, cat. 781280) were used for compound serial dilutions and for transfer of assay mixtures. For compounds with stock solutions in water, serial dilutions were made using Kme reader buffer. For compounds stored in DMSO, serial dilutions were made using DMSO. Following the addition of all assay components, plates were sealed with clear covers, gently mixed on a tabletop shaker for 1 min, centrifuged at 1000g for 2 min, and allowed to equilibrate in a dark space for 1 h before reading. Measurements were taken on an EnVision 2103 Multilabel Plate Reader (PerkinElmer) using an excitation filter at 320 nm and emission filters at 615 and 665 nm. Emission signals (615 and 665 nm) were measured simultaneously using a dual mirror D400/D630 (using a 100 microsecond delay). TR-FRET output signal was expressed as emission ratios of acceptor/donor (665/615 nm) counts. Percent inhibition was calculated on a scale of 0% (i.e., activity with DMSO vehicle only) to 100% (100 µM UNC3866) using full column controls on each plate. The interquartile mean of control wells was used to calculate Z′ values. For dose–response curves, data were fit with a four-parameter nonlinear regression analysis using GraphPad Prism 7.0 or ScreenAble software to obtain IC50 values.
Two-Dimensional Titration of Protein and Tracer Ligand
A 10-point, twofold serial dilution was prepared separately for both the protein and biotinylated tracer ligand in a deep, 384-well polypropylene plate using Kme reader buffer. Solutions were made to three times the final concentration and typically started at 750 nM for protein and 90 nM for biotinylated tracer ligands. Upon addition to the plate, the top final concentrations were typically 250 and 30 nM, respectively. The 10th well of each dilution only contained buffer for background signal information. A 3× solution of the fluorophore conjugates composed of Eu-labeled streptavidin and anti-6X-histidine ULight-labeled antibody was prepared in Kme reader buffer solution and is referred to as the TR-FRET reagent. The protein dilution (3.3 µL) was added to rows in the assay plate followed by addition of 3.3 µL of the peptide dilution to columns across the assay plate. Lastly, 3.3 µL of the TR-FRET reagent was added to all wells.
Z′ and DMSO Tolerance Experiment
Full columns of high signal (no inhibitor, DMSO only) and low signal (100% inhibition with competitor compound; 100 µM UNC3866) were produced to calculate the Z′ factor. 14 To complete a DMSO tolerance test, the high and low signals were obtained using concentrations of 0, 0.5, 1, 2, and 3% DMSO. A 1 mM solution of UNC3866 was prepared using Kme reader buffer. Using an automated multichannel pipette, 1 μL of the inhibitor was added to five columns of the plate to be read for the low signal. Likewise, 1 μL of Kme reader buffer was transferred to five columns for the high signal measurement. Solutions of DMSO (10×) were made using Kme reader buffer to 0, 5, 10, 20, and 30% DMSO. One microliter of the DMSO solutions was transferred to both high- and low-signal columns measurement appropriately. A mixture of protein, biotinylated tracer ligand, and the TR-FRET reagents was made into a single tube. The solution was gently mixed and then aliquoted appropriately to a new column in a deep-well plate. Using an automated multichannel pipette, 8 µL of the mixture was added to the assay plate.
TR-FRET Competition Assay in Dose–Response Format
A 16-point, threefold serial dilution of compound (UNC3866 or UNC4219) was prepared using Kme reader buffer in a deep, 384-well polypropylene plate. Using an automated multichannel pipette, 3.3 µL of the compound dilution was added to three columns of the assay plate. A mixture of protein, biotinylated tracer ligand, and the TR-FRET reagents was made into a single tube. The solution was gently mixed and then aliquoted to a new column in the deep-well plate. Using an automated multichannel pipette, 6.7 µL of mixture was transferred from the deep-well plate to the assay plate.
Compound Screening Using the Optimized TR-FRET Assay and Assay-Ready Plates
For testing compounds in higher throughput, 384-well assay-ready plates were prepared in standard plate format: columns 1 and 2 were used for low-signal controls (100% inhibition with competitor compound), columns 23 and 24 were used for high-signal controls (DMSO only), and columns 3–22 were used for 25 µM single-dose test compounds. Two compound sets maintained in the UNC Center for Integrative Chemical Biology and Drug Discovery (CICBDD) were used: EpiG (960 compounds) and EpiDiamond (64 compounds), where the compounds generally contain Kme mimetic features and known epigenetic-related chemical probes. First, controls were added to a mother plate where columns 1 and 2 were filled with 10 mM stock of UNC3866 in DMSO and columns 23 and 24 were filled with DMSO. Test compounds were dispensed across the mother plate at 100× (10 mM) concentration in columns 3–22 using a TECAN Freedom EVO liquid handling workstation. Using a TTP Labtech Mosquito HTS liquid handling instrument, assay-ready plates were prepared by stamping 100 nL of control compound into columns 1 and 2, 25 nL of compounds from the mother plate into columns 3–22, and 25 nL of DMSO into columns 23 and 24. Protein, biotinylated tracer ligand, and the TR-FRET reagents were added together and gently mixed by pipetting and rocking. Ten microliters was then added to each well of an assay-ready plate using a Multidrop Combi (Thermo Fisher, Waltham, MA). Percent inhibition was calculated on a scale of 0% (i.e., activity with DMSO vehicle only) to 100% (100 µM UNC3866) from the full column controls on each plate.
Dose–Response Follow-Up for Hit Validation
To test hit compounds selected for follow-up in dose–response curves, assay-ready plates were formatted and prepared similar to the single-dose test plates. Columns 3–12 and 13–22 were used for 10-point serial dilutions of test compounds. For mother plate preparations, test compounds were serially diluted in DMSO 3× across the plate at 100× concentration using a TECAN Freedom EVO liquid handling workstation. The top concentration was 10 mM. Using a TTP Labtech Mosquito HTS liquid handling instrument, assay-ready plates were stamped with 100 nL of compound solutions from the mother plate. Protein, biotinylated tracer ligand, and the TR-FRET reagents were added together and then mixed gently by pipetting and rocking. Ten microliters was added to each well of an assay-ready plate using a Multidrop Combi (Thermo Fisher).
Results and Discussion
For target class drug discovery, such as the Kme reader family, development of a screening panel is essential for discovering and evaluating new potent and selective inhibitors and driving SAR studies.
15
TR-FRET is well established as a screening platform for HTS, hit validation, and SAR. The principle of TR-FRET is based on nonradiative transfer of energy between a donor and acceptor fluorophore that results when the fluorophores are within close proximity (∼100 Å) of each other. To establish a general TR-FRET method for Kme reader proteins, we employed biotinylated tracer ligands and 6X-histidine tagged proteins, labeled with Eu-streptavidin (donor) and fluorophore-conjugated anti-6X-histidine antibody (acceptor), respectively (
To determine the conditions for an optimal TR-FRET signal, titrations of both protein and tracer ligand were carried out in a 10 × 10 (two-dimensional [2D] titration) format (
Kme Reader Protein and Tracer Ligand Pairs with Finalized Concentrations Used for TR-FRET Assays, the Z′ Values Obtained Using Those Parameters, and the IC50 Values Obtained for UNC3866.
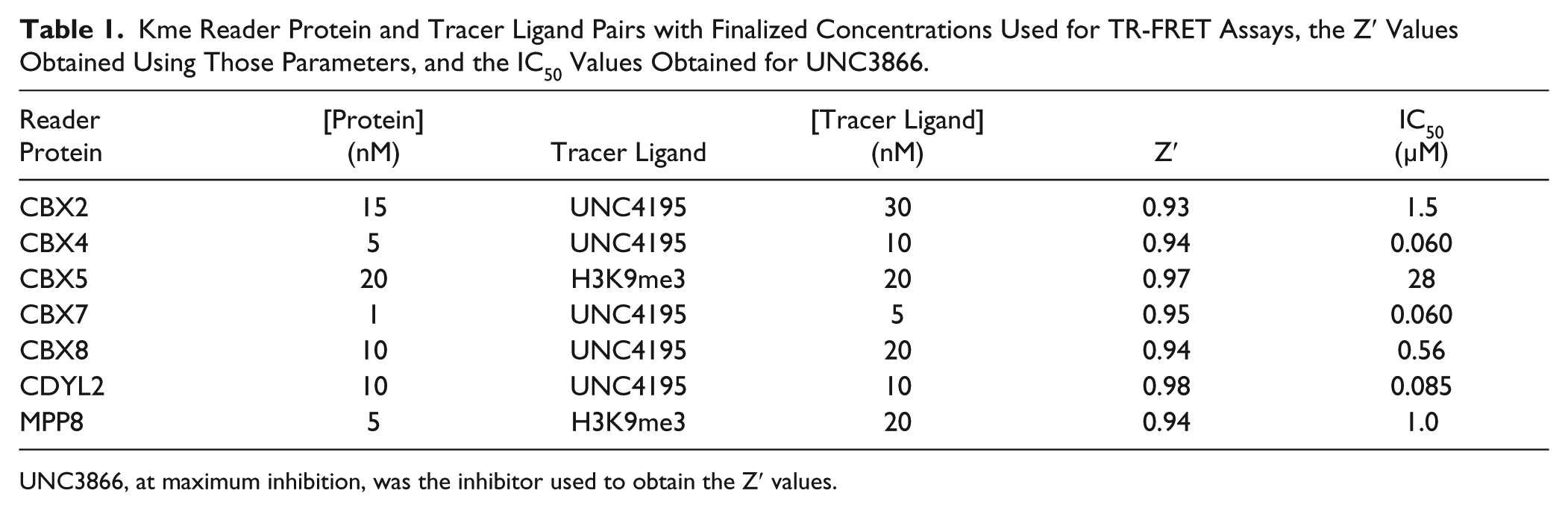
UNC3866, at maximum inhibition, was the inhibitor used to obtain the Z′ values.
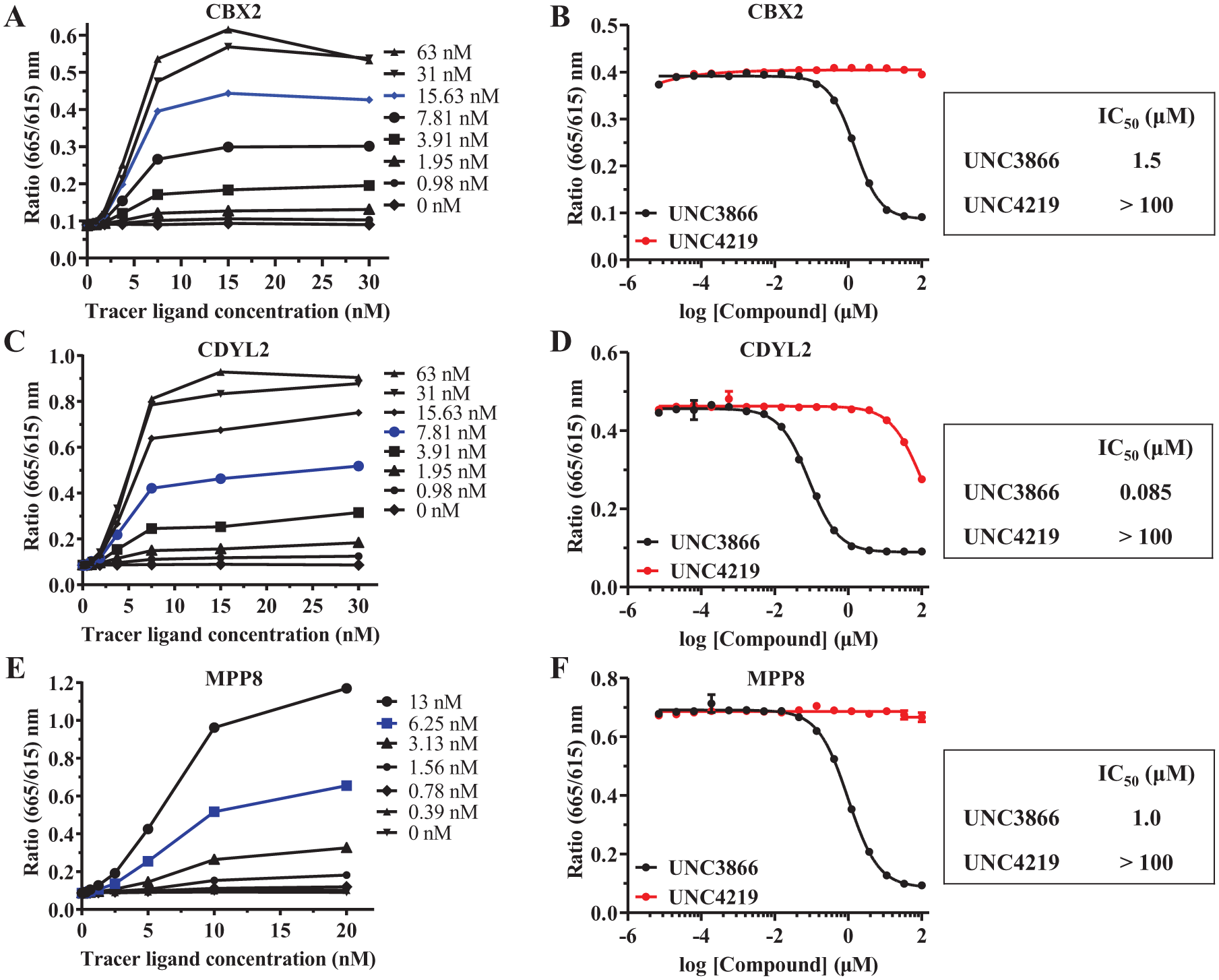
Examples of 2D titrations for CBX2 (
All of the assays we developed performed well, with Z′ values ≥0.9, at 1% DMSO ( Table 1 ). We next completed a DMSO tolerance test to determine the maximum solvent limit. 14 With increasing amounts of DMSO, the maximum TR-FRET signals decreased slightly; however, the Z′ values remained ≥0.9 with up to 3% DMSO ( Suppl. Table S1, Suppl. Fig. S3 ). Therefore, DMSO’s effect on assay performance is practically negligible. For further work using assay-ready plates, an appropriate range of final compound concentrations can be obtained while achieving ≤1% DMSO (v/v) using automation.
To validate these chromodomain TR-FRET binding assays for HTS and demonstrate their utility in finding competitive inhibitors, we performed a series of competition experiments. Assay validation using competitive displacement of the tracer ligand can be completed using the same biotinylated peptide (with the addition of excess free biotin after coupling biotinylated tracer ligand to Eu-labeled streptavidin), a nonbiotinylated analog of the tracer ligand, or a known inhibitor. For this work, we used UNC3866 as the competitive inhibitor for these TR-FRET reader assays (
An additional advantage of this homogeneous TR-FRET assay is a low tight-binding limit achieved by using low (nanomolar) concentrations of protein. This is in contrast to most FP assays for reader proteins where micromolar protein concentrations are typically required to achieve appropriate signal with histone peptides as tracer ligands.6,12 Furthermore, when an assay is developed such that the reagents used are well below the Kd of the ligand, the IC50 value can be directly compared with the dissociation constant Kd or Ki. The relationship of IC50 to Ki is described by a modified Cheng–Prusoff equation where a protein–ligand interaction is reversible, has 1:1 stoichiometry, and the concentration of protein is significantly lower than the tracer ligand concentration ([L]; eq 1). 18 Importantly, the IC50 values obtained with our TR-FRET assays ( Table 1 ) match favorably with Kd values reported previously using isothermal titration calorimetry (ITC).6,10
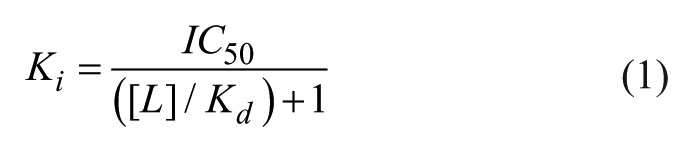
We next completed a focused screening campaign with CBX2 in an attempt to find new chemical moieties as starting points for designing small-molecule inhibitors. Even though this Kme reader has been demonstrated as a therapeutically relevant target for diseases such as advanced prostate cancer, there are currently no reported potent and selective small-molecule inhibitors for CBX2.
19
Our primary screening campaign utilized the EpiG compound sets (maintained in the UNC CICBDD), which includes one set of 960 compounds that are known to bind chromatin modulators or have been produced for chromatin regulatory protein inhibitor efforts and another set of 64 previously described inhibitors for chromatin regulatory proteins. The initial screen of CBX2 with the EpiG sets was completed by testing compounds at a single dose of 25 µM. The screen was completed in duplicate with consistency in high and low signals and screening statistics (Z′ > 0.9) between all plates (
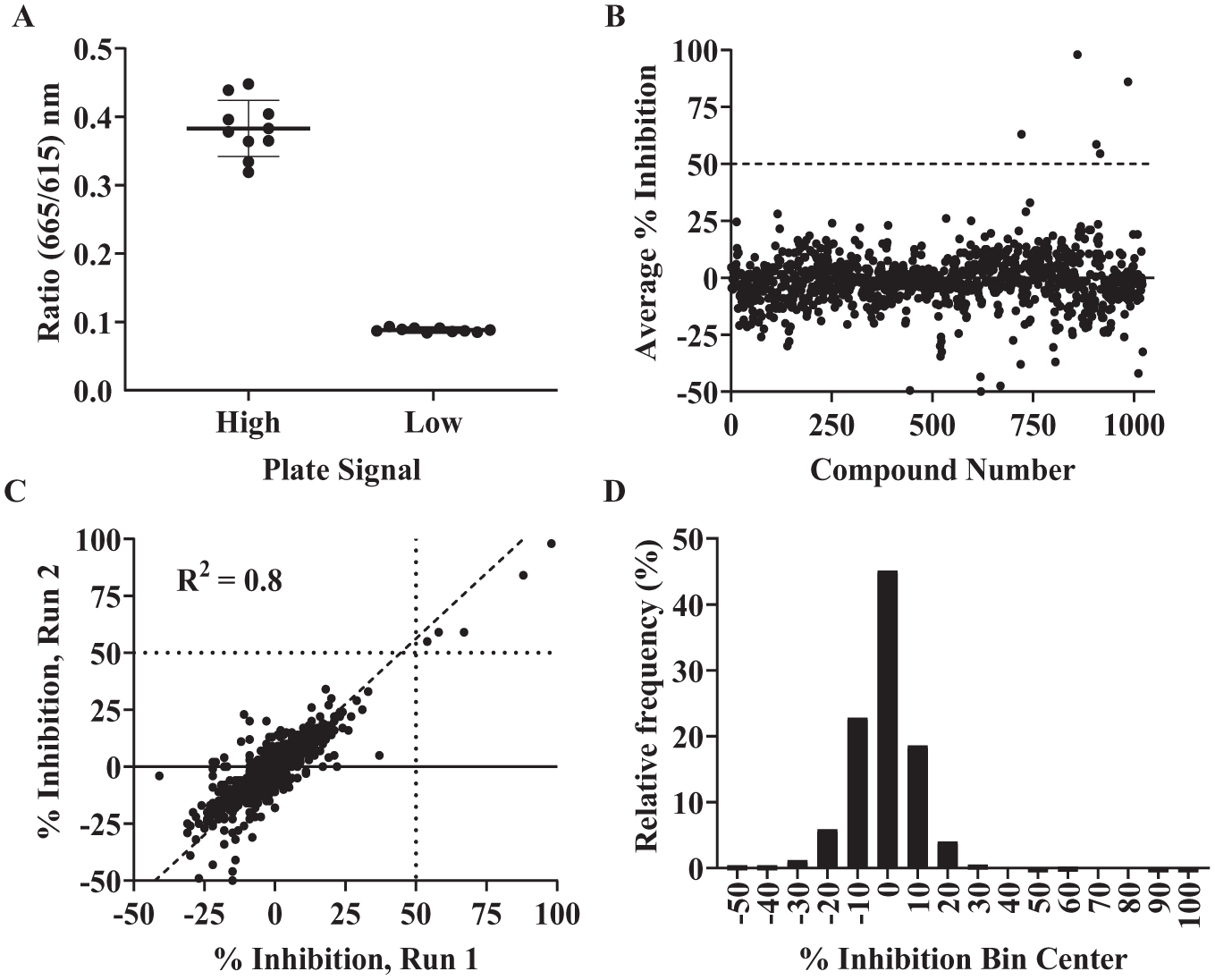
(
As mediators of active and repressed genomic states, chromatin-modifying complexes are important targets to pursue understanding of basic biological function and to conduct drug discovery efforts in numerous therapeutic areas. Producing chemical probes that are selective within families of reader proteins creates opportunities for advancing our understanding of chromatin regulation and also provides information needed to produce more effective therapeutics targeting complex diseases. These efforts are not possible without robust and reliable in vitro assays. The panel of TR-FRET assays reported here will support our efforts to develop novel chemical probes for Kme readers. These assays are appealing because they are suitable for either HTS campaigns or quantitative dose–response analysis. Importantly, for chemical probe progression, these assays enable the generation of SAR and evaluation of inhibitor selectivity. It is expected that this plug-and-play platform can also be applied to other families of Kme reader proteins such as those containing Tudor, PHD, and WD-40 domains.
Supplemental Material
DS_DISC844569 – Supplemental material for A General TR-FRET Assay Platform for High-Throughput Screening and Characterizing Inhibitors of Methyl-Lysine Reader Proteins
Supplemental material, DS_DISC844569 for A General TR-FRET Assay Platform for High-Throughput Screening and Characterizing Inhibitors of Methyl-Lysine Reader Proteins by Justin M. Rectenwald, P. Brian Hardy, Jacqueline L. Norris-Drouin, Stephanie Cholensky, Lindsey I. James, Stephen V. Frye and Kenneth H. Pearce in SLAS Discovery
Footnotes
Acknowledgements
We thank Kaelin Amaya, Brittany Castellanos, and Meenu Immaneni for contributions in the early stages of this project. We also thank Michael Stashko for reviewing experimental data and providing helpful suggestions.
Supplemental material is available online with this article.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Funding for this work was supported by NIH 5R01 GM100919-06, the UNC Eshelman Institute for Innovation, and the UNC Lineberger Comprehensive Care Center.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.