
Abstract
Regulators of G protein signaling (RGS) proteins serve as critical regulatory nodes to limit the lifetime and extent of signaling via G protein–coupled receptors (GPCRs). Previously, approaches to pharmacologically inhibit RGS activity have mostly focused on the inhibition of GTPase activity by interrupting the interaction of RGS proteins with the G proteins they regulate. However, several RGS proteins are also regulated by association with binding partners. A notable example is the mammalian RGS7 protein, which has prominent roles in metabolic control, vision, reward, and actions of opioid analgesics. In vivo, RGS7 exists in complex with the binding partners type 5 G protein β subunit (Gβ5) and R7 binding protein (R7BP), which control its stability and activity, respectively. Targeting the whole RGS7/Gβ5/R7BP protein complex affords the opportunity to allosterically tune opioid receptor signaling following opioid engagement while potentially bypassing undesirable side effects. Hence, we implemented a novel strategy to pharmacologically target the interaction between RGS7/Gβ5 and R7BP. To do so, we searched for protein complex inhibitors using a time-resolved fluorescence resonance energy transfer (FRET)–based high-throughput screening (HTS) assay that measures compound-mediated alterations in the FRET signal between RGS7/Gβ5 and R7BP. We performed two HTS campaigns, each screening ~100,000 compounds from the Scripps Drug Discovery Library (SDDL). Each screen yielded more than 100 inhibitors, which will be described herein.
Introduction
G protein–coupled receptors (GPCRs) are pivotal cell signal transducers that influence nearly every aspect of human physiology. GPCR signaling is initiated by hormones, peptides, cleavage of an internal ligand, and myriad other ligands. The ligand-bound receptors generate intracellular responses through activation of heterotrimeric G proteins composed of Gα and Gβγ subunits. GPCRs facilitate the exchange of guanosine diphosphate (GDP) for guanosine triphosphate (GTP) on Gα, which promotes its dissociation from Gβγ and allows interaction with downstream effectors modulating their activity. 1 The Gα proteins can intrinsically hydrolyze GTP to GDP to terminate signaling. However, the rate at which this occurs is too slow to account for the dynamic signaling kinetics of physiological processes. 2 Hence, Gα proteins rely on regulators of G protein signaling (RGS) proteins to accelerate the return of the G protein to its inactive, heterotrimeric state where Gα is bound to GDP.3,4 As such, RGS proteins function as GTPase-activating proteins (GAPs), increasing the rate of GTP hydrolysis on the Gα subunits to moderate the extent and lifetime of GPCR signaling ( Fig. 1A ).3,5,6
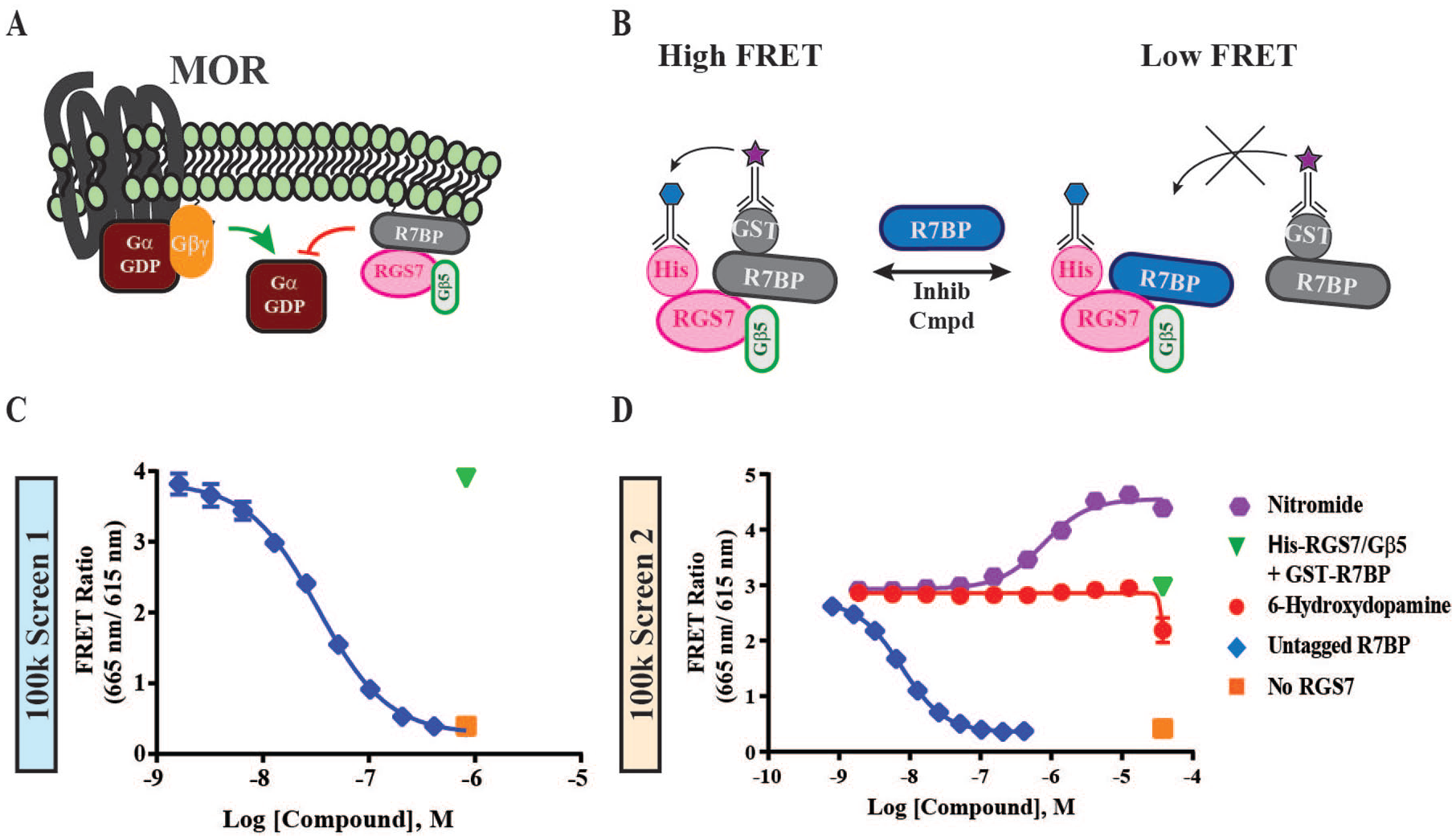
Assay design to identify inhibitors of the RGS7/Gβ5/R7BP complex. (
The 30 known RGS proteins are grouped based on their sequence similarity and domain composition.7,8 The R7 family (RGS6, RGS7, RGS9, and RGS11) comprises GAPs with selectivity for the Gi/o type of G proteins. 9 In addition to the conserved RGS (RH) domain, the R7 family contains a Gγ-like domain that forms an obligate heterodimer with Gβ5.10,11 R7 RGS proteins also feature a DEP/DHEX domain that mediates the interaction with several proteins, including the small transmembrane protein RGS9 anchor protein (R9AP), orphan receptors GPR158/GPR179, and the palmitoylated membrane-anchoring protein RGS7 binding protein (R7BP).12–14 These proteins function as adaptors for R7 RGS proteins to regulate their protein levels, to anchor them to the plasma membrane, and to augment their GAP activity.15–17
This study focuses on pharmacologically targeting RGS7, the prototypic member of this family. RGS7 has been shown to be a powerful negative regulator of mu opioid receptor (MOR) signaling. 18 Studies in knockout mice demonstrate that elimination of RGS7 augments the antinociceptive effects of opioid analgesics and increases their rewarding effects. 19 Interestingly, R7BP knockout mice also exhibit enhanced morphine-induced analgesia and reward but, surprisingly, do not develop opioid tolerance. 20 Taken together, the RGS7/Gβ5/R7BP complex endogenously dampens opioid signaling, and its modulation by R7BP also plays a role in the development of tolerance. Thus, modulation of the RGS7/Gβ5 interaction with R7BP, rather than general suppression of the GAP activity of RGS7, could serve to increase opioid analgesic sensitivity while suppressing tolerance side effects. Such a profile could prove useful for enhancing safety margins in pain management.
Our high-throughput screening (HTS) approach aimed to find small molecules that could induce conformational changes in the RGS7-Gβ5-R7BP complex. Recently, we developed a biochemical-based time-resolved fluorescence resonance energy transfer (TR-FRET) detection assay to monitor the RGS7-Gβ5 interaction with R7BP. 21 In the present report, we extend the initial assay development studies to two HTS campaigns screening more than 200,000 compounds selected from the SDDL. 22 These screens, as designed, measured the small-molecule–mediated loss of FRET signal between RGS7/Gβ5 and R7BP and identified several potential modulators with desired activity.
Materials and Methods
Reagents
The anti-GST-Tb and anti-His-d2 antibodies were purchased from CisBio US, Inc. (Bedford, MA). 6-hydroxydopamine HCl and nitromide were from Sigma Aldrich (St. Louis, MO). R7BP, GST-R7BP, and His-RGS7/Gβ5 were expressed and purified as described. 21
Primary and Secondary Screening Assays
The primary ultra-high-throughput screening (uHTS) campaigns were performed in a 1536-well format testing one compound per well using a fully automated HTS robotic platform. 22 The assay allows simultaneous screening for both inhibitors and activators, which had separate controls. Controls for high FRET inhibition consisted of the His6-RGS7/Gβ5 and GST-R7BP FRET complex with the addition of 825 nM untagged R7BP for the first HTS campaign or 37.5 µM 6-hydroxydopamine for the second HTS campaign. Low FRET inhibition controls were the FRET complex in the presence of buffer. For the first screen, no activating compounds were available as a positive control for enhanced FRET signal relative to the buffer control. However, in the second screen, 12.5 µM nitromide, an activating compound identified in the pilot HTS screening with an EC50 of approximately of 0.76 ± 0.10 µM, was used. 21 Anti-GST-Tb and anti-His-d2 antibodies were reconstituted in freshly prepared assay buffer (20 mM HEPES, pH 8.0, 200 mM NaCl, 1 mM dithiothreitol, and 0.1% [w/v] bovine serum albumin). Antibodies were combined in a 1:1 ratio, and 1 µL of the antibody mixture was added to each well using the Aurora Flying Reagent Dispenser at a final concentration of 0.8 nM (anti-GST-Tb) and 6.7 nM (anti-His-d2). One microliter of untagged R7BP (825 nM final concentration) or 1 µL of 6-hydroxydopamine (37.5 µM final concentration) was added to the high FRET inhibition control wells, and 1 µL of buffer was added to all other wells. Compounds or DMSO controls were transferred into appropriate wells using the GNF pintool transfer station (Genomics Novartis Foundation, San Diego, CA) using 30 nL pintools (V&P Scientific, San Diego, CA). Finally, 1.5 µL each of GST-R7BP and His-RGS7/Gβ5 was dispensed into each well at a final concentration of 3.9 nM and 15.5 nM, respectively. Compounds were screened at 5 µM nominal concentration and were incubated with the proteins for 16 to 18 h at 25 °C, a time point that was empirically determined to yield the largest assay window. 21 The final DMSO concentration did not exceed 0.52% (v/v). The next day, the TR-FRET signal was measured on the PerkinElmer ViewLux uHTS microplate imager using the LANCE/DELFIA mirror modules UV2 (TRF) 320 excitation filter, the APC 665 emission filter, and the Europium 615 emission filter 2. The terbium (Tb) donor fluorophore was excited at 340 nm with 615 nm emission (donor emission), and 665 nm (d2 acceptor emission) was acquired.
For the secondary (confirmation) screening assays, compound hits were cherry-picked and rescreened at the same concentration and in the same manner as the primary HTS, albeit in triplicate.
Data Analysis
Changes in the TR-FRET signal were determined by dividing the 665 nm signal by the 615 nm signal and normalizing each compound as the percentage inhibition of controls. All raw data were incorporated into the Scripps database. The percentage response of each compound was calculated by subtracting the median response of all compounds tested from the response of the individual test well and dividing that by the median response from all compounds subtracted from the median response of the inhibitory or activating controls. The control for maximum inhibition was set as the FRET signal obtained in the presence of 825 nM unlabeled R7BP or 35 µM 6-hydroxydopamine for the first and second screen, respectively. In the second HTS campaign, the percentage activity of compounds that enhanced the FRET signal was calculated using 12.5 µM nitromide as a control. The negative activity control for both assays was the FRET pair (His-RGS7/Gβ5 with GST-R7BP) incubated with buffer.
The signal-to-background ratio and the Z′ for the inhibitory compound screening was calculated by comparing the high-inhibition control (unlabeled R7BP or 6-hydroxydopamine) to the low FRET inhibition control (DMSO) within each plate. Twenty-four control wells were included per plate to establish Z′ and the signal-to-background (S:B) value. Z′ = 0.5 was considered the minimum passable value, and any plate that did not achieve that value was rescheduled until it did so. 23
The percentage activity and cutoff values were calculated as described. 24 Briefly, four values were calculated: (1) the average percentage response of the high FRET controls + 3 SD, (2) the average percentage response of the low FRET controls + 3 SD, (3) the average percentage response of all compounds between the high and low controls, and (4) 3 SD of the average percentage response for the tested compounds calculated in (3). The sum of the average response of the tested compounds (3) plus 3 SD (4) yielded the activity cutoff parameter.
Compounds were analyzed for pan-assay interference structures (PAINs) and for promiscuity across primary screening assays performed at Scripps Research (Jupiter, FL), using the SDDL as described.24,25
Results
Implementation of an Ultra-High-Throughput TR-FRET Assay to Identify Modulators of the RGS7/Gβ5/R7BP Complex
Our screening approach is capable of assaying for modulators that either disrupt or enhance the protein-protein interaction of RGS7-Gβ5 with R7BP. 21 The association of RGS7/Gβ5 with R7BP enhances the GAP activity of RGS7 toward its endogenous substrates Gαi and Gαo. Thus, inhibiting this interaction will likely moderate, rather than completely negate, RGS catalytic activity.15,16,26 A milder influence on the RGS GAP activity could prove valuable for tuning GPCR signaling downstream of ligand-mediated activation instead of agonizing the receptor itself, a strategy conceptually similar to the development of positive allosteric modulators for GPCRs ( Fig. 1A ). Before implementation, we performed extensive pilot experiments measuring the feasibility and reproducibility of the current HTS strategy, including a screen of the Library of Pharmacologically Active Compounds (LOPAC) 1280 compound library and of the MicroSource Discovery Spectrum Collection 2320 compound library. 21
To implement the assay, we used fluorophore-conjugated antibodies that recognize epitope tags on each protein ( Fig. 1B ). In solution, the His-RGS7-Gβ5 and GST-R7BP coupled to the TR-FRET antibodies form a FRET pair. In the presence of an increasing amount of competing, untagged R7BP, this FRET signal decreased ( Fig. 1C,D ). Similarly, incubation with inhibitory small molecules could result in a dose-dependent loss of protein-protein interaction, translated as a loss of FRET signal.
Each of the two 100,000 compound screens employed a subset of the SDDL. The first screen comprised 100,829 compounds. The compounds chosen were bioactive, druglike compounds that included, among others, the Food and Drug Administration–approved library, the Prestwick, Tocris, and Cayman collections. This 100k set also comprised diversity sets that were enriched for analogs of potential ligands with biological properties reported in the literature. For example, 27 compounds of interest were selected from the published data (PubChem AID 1837) that had demonstrated good RGS activity profiles and were free of known screening artifacts and chemical reactivity. These scaffolds were then used to mine for similar compounds in the SDDL deck that exhibited a Tanimoto similarity score >80. This enrichment approach identified compound plates that contained >10 of these analog compounds in the field of 1280 to be screened. Post-HTS analysis demonstrated an average hit rate of 0.5% from this diversity set. The second screen was comprised of 101,317 compounds and enriched for central nervous system/blood-brain barrier (CNS/BBB)–compliant properties and included randomly selected compounds but excluded any compounds from the first screen as well as from the LOPAC library, which we screened previously. 21 The selection criteria for CNS/BBB-compliant compound properties were as follows: 2 to 10 H-bond acceptors, 0 to 5 H-bond [HB] donors, 175 to 360 molecular weight, 1 to 6.6 LogP, 40 to 62Å topological polar surface area (TPSA), and zero to five rotatable bonds. These criteria were based on accepted literature values for leadlike CNS compounds.27–29 For both HTS campaigns, we used His-RGS7/Gβ5 and GST-R7BP incubated with buffer as the no-inhibition (high FRET) control. In the first 100k screen, unlabeled R7BP was incubated with the His-RGS7-Gβ5 and GST-R7BP as the high complex inhibition (low FRET) control; it inhibited the complex with an IC50 of 32 ± 1.10 nM ( Fig. 1C ). Indeed, untagged R7BP inhibited the FRET ratio to such a degree that incubation without His-RGS7/Gβ5 yields a similar FRET ratio ( Fig. 1D ). For the second HTS campaign, we used 6-hydroxydopamine (37.5 µM) in place of the untagged R7BP as the low FRET (high complex inhibition) control ( Fig. 1D ). 6-hydroxydopamine was identified as an inhibitor of RGS7/Gβ5 binding to R7BP in the initial LOPAC screening campaign and was used in lieu of untagged R7BP to increase reproducibility across plates and because of ease of use. 21 In the initial dose-response studies, 6-hydroxydopamine inhibited complex formation with an IC50 of 7.65 ± 3.74 µM. However, once miniaturized, the IC50 was greater than 10 µM ( Fig. 1D ).
The primary purpose of this screening effort was to identify compounds that could inhibit complex formation. However, we also found that some compounds enhanced, rather than inhibited, the emitted FRET signal. In the first HTS campaign, we lacked positive controls for compounds that could enhance the FRET signal. However, this was circumvented by using nitromide, a small molecule that enhanced the FRET signal with an EC50 of 0.76 ± 0.10 µM in the second 100k screen ( Fig. 1D ). Nitromide, like 6-hydroxydopamine, was identified through previous pilot screening of the LOPAC library. 21
Assay Optimization to a 1536-Well Format
The primary biochemical TR-FRET assay with recombinant RGS7/Gβ5/R7BP complex was optimized into a 1536-well format for the two 100k screening campaigns. Given the scope and number of compounds in the SDDL, we miniaturized the assay into 1536-well format at a final volume of 5 µL per well. Optimization involved assessing the effects of miniaturization on key assay parameters, such as demonstration of consistent IC50s using positive controls, maintenance of the assay S:B and assay signal window (Z′), DMSO tolerance, and assay robustness by pilot screening against the LOPAC library. The assay was successfully optimized for robotic HTS automation, having demonstrated good stability, DMSO tolerance (~1%), and reproducibility with an acceptable Z′ score that was >0.5, as presented below.
Primary Screening Results
Knowing that compounds tested could either enhance or inhibit the emitted FRET signal, we analyzed the primary screening data to determine in which direction the TR-FRET signal was altered. To set up the assay, antibodies were first mixed 1:1 in each well. This was supplemented with either the untagged R7BP/6-hydroxydopamine (high-inhibition control) or buffer alone (no inhibition control). Each compound was then pintooled into a single well, and finally, His-RGS7-Gβ5 and GST-R7BP were added last ( Fig. 2A ). All data were normalized as percentage inhibition or percentage activation relative to the median positive controls and the median compound response. Unlabeled R7BP (HTS screen 1) or 6-hydroxydopamine (HTS screen 2) were set as 100% inhibition, whereas DMSO control was set as 0% inhibition ( Fig. 2B,C ). For the activating compound mode, the first screen did not have a positive control. However, for the second HTS effort, nitromide was used as the positive control ( Fig. 2D ).
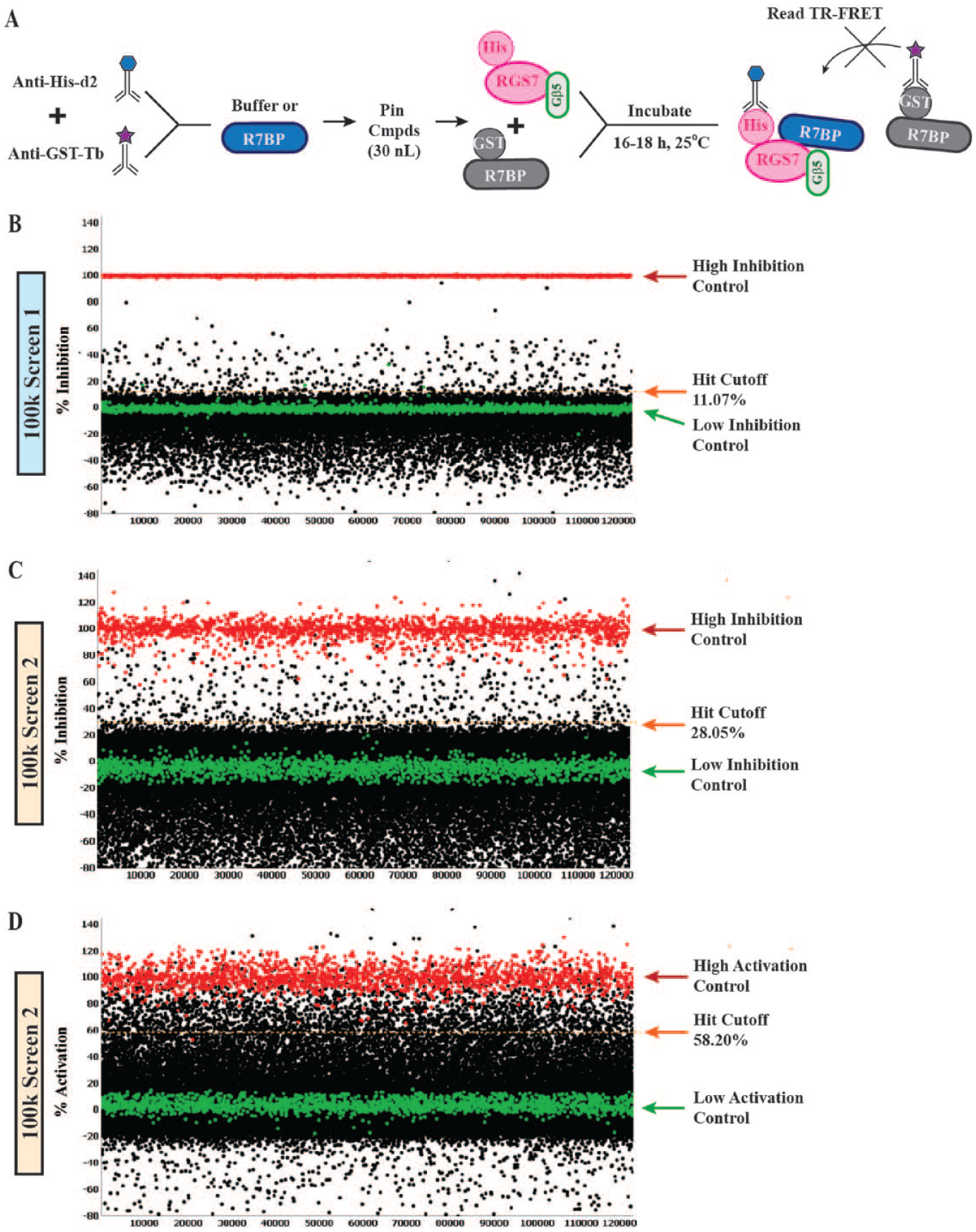
Primary screening for inhibitory and activating compounds. (
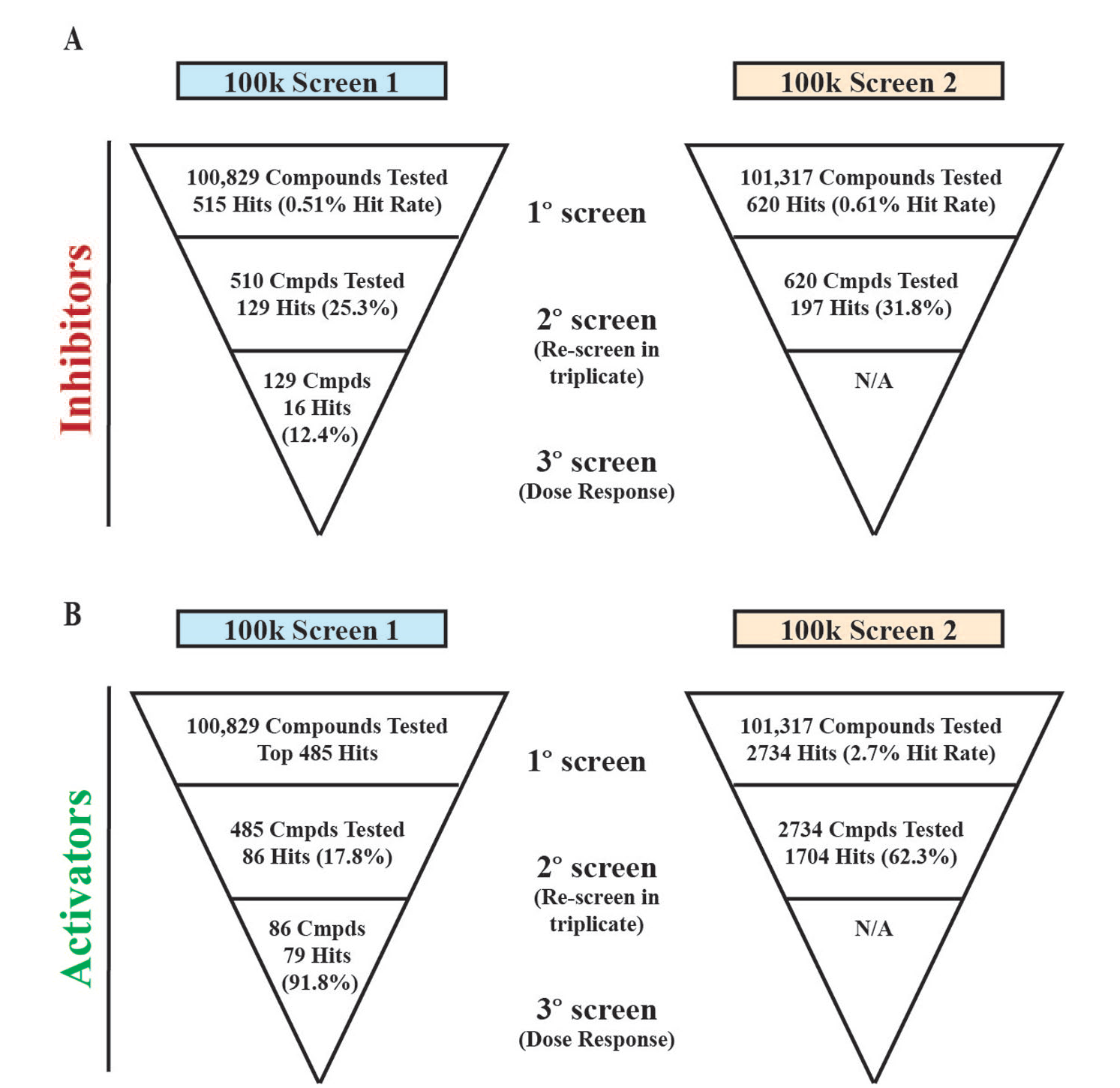
For the first inhibitory mode 100k HTS campaign, the hit cutoff was 11.07% inhibition. The average Z′ for the assay was 0.94 ± 0.03, the average Z score was 0.77 ± 0.06, and the average S:B was 7.40 ± 0.40. Of the 100,829 compounds tested, 515 compounds inhibited the FRET complex greater than 11.07% and were moved to secondary confirmation assays.
With no positive control available for the activator mode portion of the screening for the first 100k HTS, we relied on the Z′ and S:B from the inhibitory screening mode to maintain day-to-day robustness. The average S:B was 8.25 ± 0.01, and the average Z′ was 0.93 ± 0.03. The hit cutoff was calculated at 45.03% of the maximum FRET signal in the absence of any test compounds. The top 485 compounds that enhanced the FRET signal of the protein complex were chosen for confirmatory screening ( Fig. 2B ).
For the second campaign, the analysis of the inhibitory compounds yielded a 28.05% hit cutoff. The average Z′ for the assay was 0.67 ± 0.08, the average Z score was 0.24 ± 0.15, and the average S:B was 2.04 ± 0.12. From the 101,317 compounds tested, 620 compounds showed greater than 28.05% inhibition ( Fig. 2C ).
For the analysis of the activating compounds for the second 100k HTS campaign, nitromide was used as a positive control to determine both the hit cutoff and the Z′ score. The data yielded a Z′ of 0.64 ± 0.07, the average Z score was 0.21 ± 0.18, and the average S:B was 1.52 ± 0.02. This mode yielded 2734 hits that exceeded the 58.20% activity cutoff ( Fig. 2D ).
Secondary Confirmation Screening
Compounds from the primary screens that fulfilled the cutoff criteria as RGS7/Gβ5/R7BP inhibitors were cherry-picked and rescreened in triplicate to confirm their activity ( Fig. 3A–C ). Similar high-inhibition and low-inhibition controls were performed for each assay as in the cognate primary screens. In addition, the same cutoff criteria as determined in the primary screens were applied to the secondary screen.
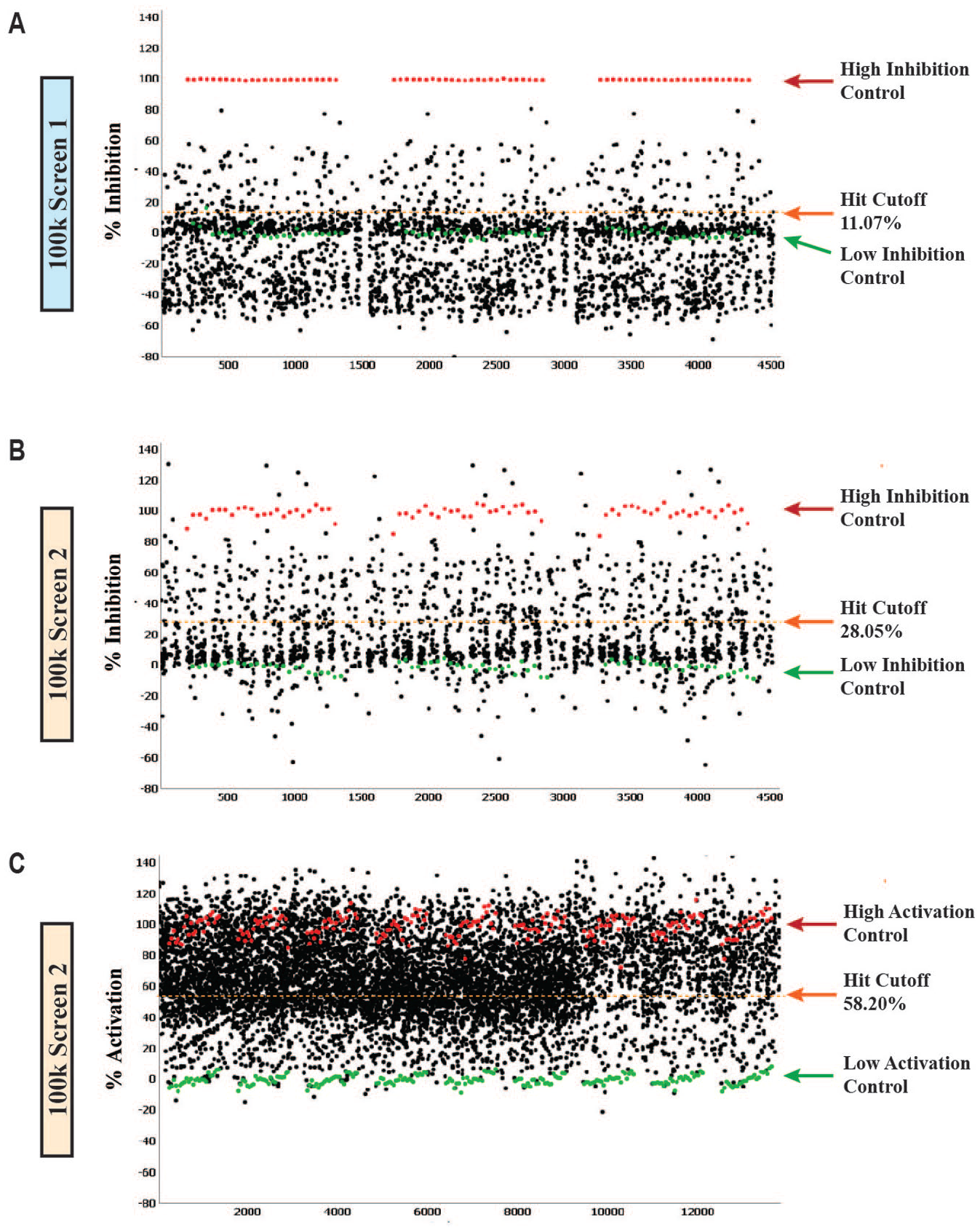
Confirmation screening for inhibitory and activating compounds of the RGS7/Gβ5/R7BP complex All data from the confirmation assay plates were performed in triplicate. Scatter plots including high and low controls are displayed. The same cutoff criteria from the primary screening were applied to the confirmation screening. (
The confirmation screen for the first 100k HTS had an average Z′ of 0.90 ± 0.03 and an average S:B of 8.21 ± 0.05. Of the 515 inhibitory hits tested, 129 hits reproducibly inhibited the complex formation ( Fig. 3A ). These 129 compounds were moved to tertiary screening to determine the potency of the inhibitors. On the other side, of the 485 activating hits tested in triplicate, only 86 hits enhanced FRET activity greater than 45.03% ( Fig. 3A ).
Similarly, for the second 100k screen, 620 inhibitory hits from the primary screen were tested in triplicate at the same concentration, 5.17 µM. This confirmation screen had an average Z′ of 0.78 ± 0.02 and an average S:B of 2.16 ± 0.02, and applying the primary cutoff of 28.05% yielded 197 compound hits ( Fig. 3B ).
A total of 2734 activating hits from the second primary HTS campaign were retested and exhibited excellent assay quality. The average Z′ was 0.70 ± 0.05, and the average S:B was 1.52 ± 0.01. Of the 2734 compounds, 1704 increased the FRET signal greater than 58.20% ( Fig. 3C ).
Tertiary Screening Results
To narrow down the potentially useful inhibitors from the confirmation screen, we subjected compound hits to another round of validation by measuring the compound potency. Only hits from the first HTS campaign were analyzed; hits from the second HTS campaign did not undergo the same tertiary screening. The 129 inhibitory compounds identified from the primary screen were assayed across a range of doses to yield concentration response curves. Compounds that demonstrated IC50s lower than 10 µM were considered hits. Of the 129 tested inhibitory compounds, only 16 satisfied this criterion ( Suppl. Fig. S1 ). 1-[2-(2-oxopyrrolidin-1-yl)-2-sulfanylideneethanethioyl]pyrrolidin-2-one (SR-01000319258-1; Fig. 4 ) was the most potent compound identified, with an IC50 of 173 nM and a maximum inhibition of 54.2 ± 0.84%. However, this compound was also a hit in 15 of 69 primary assays in which it was previously screened at Scripps Research and was also a hit in 1 of 3 bioassays reported in the PubChem database. Despite this optimal potency for an initial hit, it is likely too nonselective for continued study.
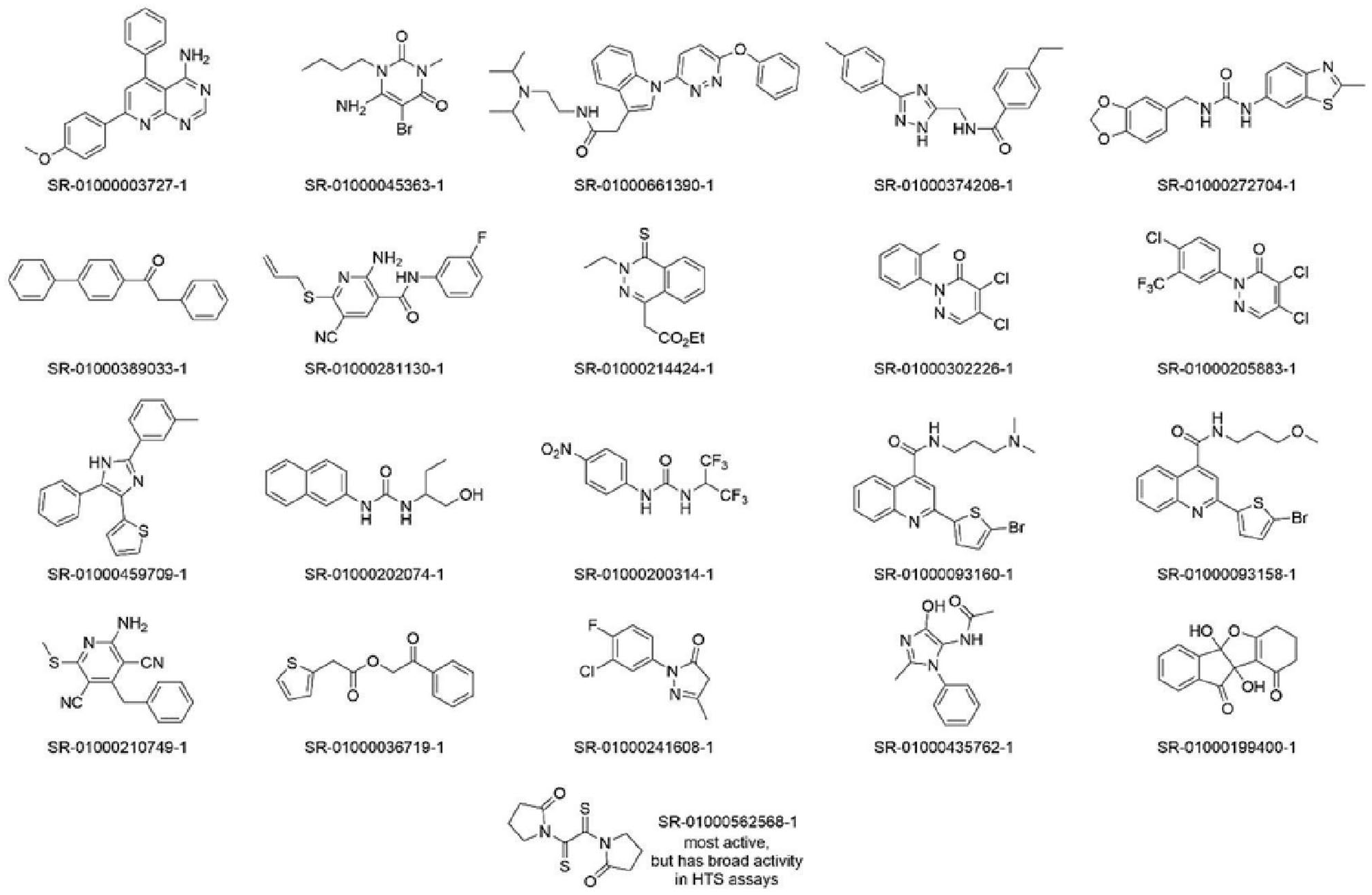
Structures of top inhibitory hits from both 100k screens. The top row represents hits from the first 100k screen, whereas the next three rows are hits from the second 100k screen. The bottom compound was the most potent compound from the first screen but showed promiscuity in other assays.
The next most active inhibitory compound, 7-(4-methoxyphenyl)-5-phenylpyrido[2,3-d]pyrimidin-4-amine (SR-01000003727-1; Fig. 4 ), although less potent than the top hit with an IC50 of 2.0 µM, was a full inhibitor (maximum inhibition of 98.96 ± 0.41%). It also has favorable predicted drug-likeness (molecular weight: 328, TPSA: 72, LogP: 3.8, HB donors 2, HB acceptors 4, no PAINs). Further, its screening history suggests a lack of pan-assay activity; it had emerged as a hit in only 3 of 68 bioassays run previously at Scripps Research.
Concentration response curves from the first 100k activators screened generated a total of 86 hits. The most potent hit from the activator screen was mitoxantrone ( Fig. 5 ; Suppl. Fig. S2 ), a marketed type II topoisomerase inhibitor used as an anticancer agent. Many other hits were seen with EC50 values less than 1 µM and maximum activation >40%, including several nitroaromatic compounds and quinone or quinone-derived compounds. Also active were indirin oxime and a related analog. Slightly less potent activators were benzoisoquinoline diones ( Fig. 5 , rows 1–2). However, many of these compounds also had known pan-assay inhibition, and many of the compounds were also hits in more than five assays performed at Scripps Research, suggesting they may be nonspecific hits. Potentially tractable compounds with EC50 values in the 2 to 10 µM range are shown in the bottom row of Figure 5 .
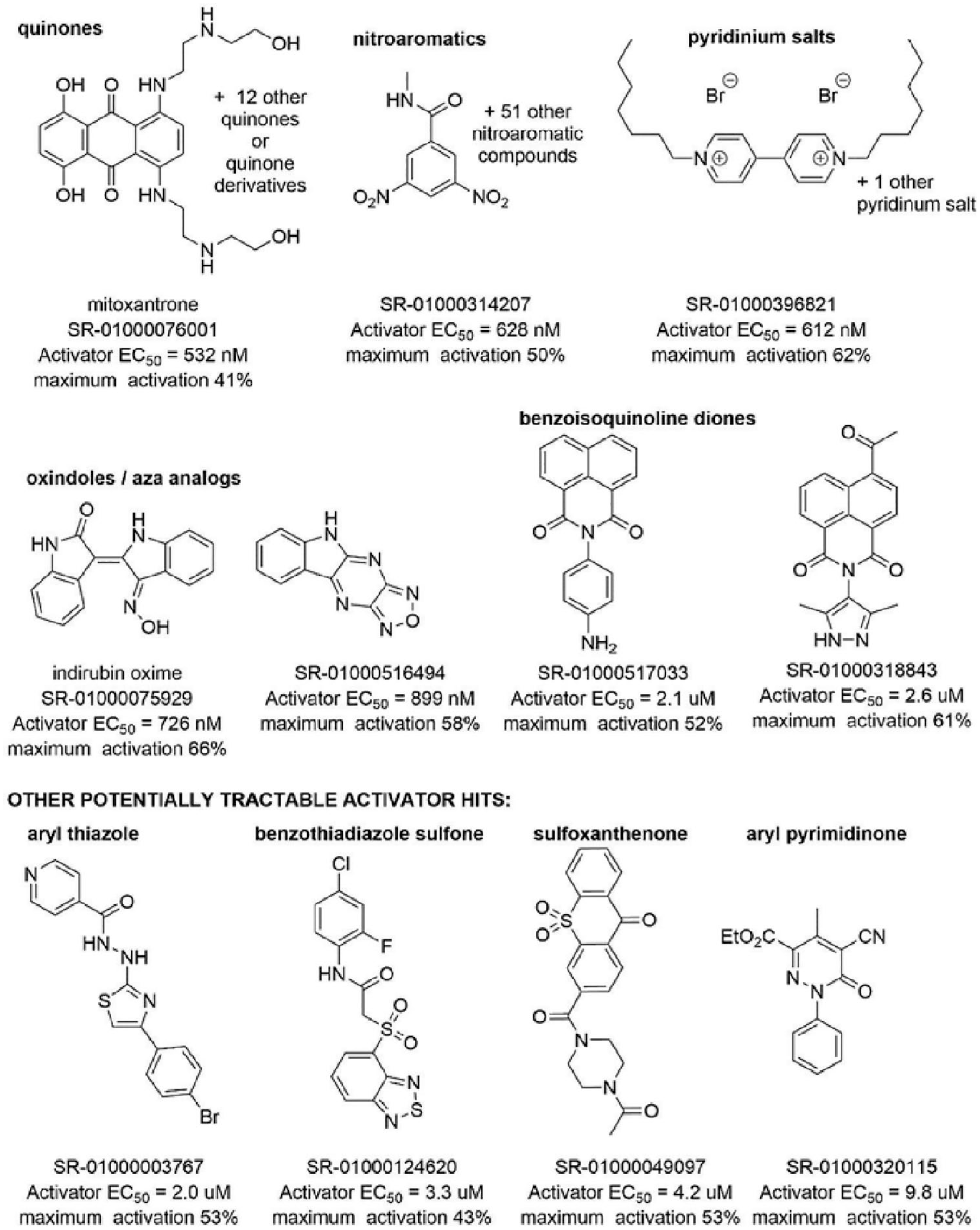
Structures of top hits from the 100k screen. The top and middle rows represent the most potent compounds identified in the first 100k screen. The bottom compounds, although less potent, represent the most tractable hit compounds for further development.
Properties of Identified Inhibitory Hit Compounds
Because many of the activating hits demonstrated broad promiscuity in assays performed at Scripps Research and contained known PAINs compounds, we focused our compound analysis on the inhibitory hit compounds. 22 Figure 4 shows the chemical structures of 20 screening hits of high interest from the 2 HTS rounds plus the most active compound mentioned above. Compounds shown in the top row were from screen 1 and had IC50 values ranging from 2.0 to 9.3 µM. Criteria for selection of these five compounds were as follows: >50% inhibition in the primary and confirmation screens, dose-response IC50 <10 µM, no PAINS alerts, no structure alerts, and no overt stability or assay promiscuity concerns. Compounds shown in the next three rows were from screen 2, for which dose-response data were not run. Criteria for selection of these 15 were as follows: >65% inhibition in the primary and confirmation screens, no PAINS alerts, no structure alerts, and no overt instability or assay promiscuity concerns. At the bottom of the figure is shown the most active compound, the bisthioamide compound SR-01000562568-1, which (as mentioned above) has assay selectivity concerns. A total of 17 of the 20 hits shown have molecular weight less than 350 and have other favorable drug-likeness attributes. Certain hits are structurally related, increasing confidence in the chemotype for further study (e.g., ureas SR-01000272704-1, SR-01000202074-1, and SR-01000200314-1; see also quinolines SR-01000093160-1 and SR-01000093158-1, which may relate to top druggable hit, the azaquinazoline SR-01000003727-1). Many of these compounds could prove suitable for further development as RGS7 complex inhibitors ( Fig. 6 ).
Overview of hits from primary, secondary, and tertiary screening. (
Discussion
Several RGS proteins have been shown to regulate antinociception in mice. RGS4, RGS9-2, and RGS7, for example, have profound effects on morphine-mediated antinociception. 18 Some of these proteins have additionally displayed specific expression patterns in the central nervous system or have been shown to function as GAPs for specific Gα proteins. Given the current opioid crisis in the United States, there is a pressing need to target the opioid-signaling axis effectively and creatively with the hopes of increasing the therapeutic window for analgesia while diminishing undesirable reward and tolerance side effects.18,30,31 By decreasing or altering its interaction with R7BP, the function of RGS7-Gβ5 would similarly be blunted, as has been shown. Modulation of the protein-protein interaction between R7BP and RGS7-Gβ5 has been shown to be sufficient to alter antinociception mediated by the MOR signaling axis.15,16
The fact that R7BP knockout mice do not develop drug-induced tolerance holds promise to selectively reduce these opioid side effects. 20 The aim of the current study was to identify compounds that interfered with the interaction between RGS7/Gβ5 and R7BP to alleviate the negative signaling pressure on the MOR to permit more sensitivity to endogenous opiate peptides and/or to exogenously administered opioids. In addition, specifically targeting this protein-protein interaction could theoretically lessen or abolish tolerance generated through repeated opioid exposure, as happens upon genetic loss of R7BP. 20 Thus, we screened nearly 200,000 compounds to identify such potential small-molecule modulators. This identified several promising compounds with no known PAINs, apparent selectivity, and good druglike properties that may be bona fide inhibitors and warrant further investigation.
The screen relies on changes in the FRET-based signal between RGS7/Gβ5 and R7BP. It is possible that small molecules will bind the complex and not prevent dissociation but could alter the GAP activity of RGS7-Gβ5 on Gαo. In the same vein, the small-molecule modulators may bind in such a way to disrupt the FRET interaction of the complex without actually altering the complex per se. The premise of this screening strategy is based on the ability of a small molecule to disrupt the protein-protein interaction between fluorescently labeled RGS7/Gβ5 and R7BP. It is possible, however, that compounds may not disrupt the protein-protein interaction per se, and instead, they may alter efficient energy transfer between the FRET pair. The compounds would then serve as allosteric moieties to enhance or diminish protein activity and not necessarily fully dissociate or stably associate the complex. Similarly, small molecules could bind the complex and not inhibit the complex formation while still acting to inhibit the GAP activity of RGS7-Gβ5 on Gαo.
The use of the small molecule 6-hydroxydopamine as an inhibitory control, although not behaving as robustly as the R7BP control in the first 100k screen, enabled us to explore more physiological modulators of the R7BP/RGS7/Gβ5 complex. The large binding interface between R7BP and RGS7 is unlikely to be completely disrupted by a small chemical compound. Because compounds are more likely to adjust the orientation and activity of the complex, the cutoff from the second screen allowed us to find molecules suitable for physiological interference while also still capturing the high-performance scaffolds, comparable to the use of untagged R7BP instead.
Finally, it is worth mentioning the potential pitfalls related to the identification of compounds based on TR-FRET assay. For one, compound autofluorescence could enhance the signal perceived as interaction. This is a common problem for homogenous in vitro assay platforms. 32 Counterscreening to identify these auto emitters is one way to identify these false positives. Conversely, compounds that physically or chemically quench the employed fluorophores may appear to be complex inhibitors. The assay we employed cannot distinguish between these scenarios and must be resolved through filtering compounds with known pan-assay inhibition or activation and testing cellular behavior. To address some of these difficulties, we took steps to increase confidence in our hits through analysis of common pan-interacting chemical moieties and through comparison with assays performed at Scripps Research. This strategy enables ignoring compounds that nonspecifically enhance or decrease FRET signals.
Future compound characterization and development efforts would include testing hits for activity in the cellular environment. Such testing could rely on cell surface localization in the presence and absence of R7BP and, ideally, on assaying the catalytic activity of RGS7-Gβ5/R7BP complex in living cells. Finally, the activity of the promising leads will need to be validated in the endogenous setting. The role of RGS7-Gβ5/R7BP has been well established in controlling the efficacy of opioid-mediated antinociception and in the development of tolerance. Active and specific inhibitors of the complex are expected to mimic the opioid-related behaviors observed in knockout mice. Despite the need for further validation, the screening campaign described herein has yielded a rich crop of compounds that could be used to enhance, disrupt, or allosterically tune the interaction of the RGS7-Gβ5/R7BP protein complex. Ultimately, this strategy offers the possibility to creatively increase opioid efficacy and mitigate unwanted side effects without directly pharmacologically targeting MOR.
Supplemental Material
sj-pdf-1-jbx-10.1177_24725552211020679 – Supplemental material for Identification of Potential Modulators of the RGS7/Gβ5/R7BP Complex
Supplemental material, sj-pdf-1-jbx-10.1177_24725552211020679 for Identification of Potential Modulators of the RGS7/Gβ5/R7BP Complex by Hannah M. Stoveken, Virneliz Fernandez-Vega, Brian S. Muntean, Dipak N. Patil, Justin Shumate, Thomas D. Bannister, Louis Scampavia, Timothy P. Spicer and Kirill A. Martemyanov in SLAS Discovery
Footnotes
Acknowledgements
The authors thank members of Martemyanov laboratory for helpful discussions throughout the project.
Supplemental material is available online with this article.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Funding for this project was provided by the NIH National Institute on Drug Abuse (NIDA) DA042746 to K.A.M. and L.A.S. and F32 DA047771-03 to H.M.S. ChemAxon (![]() ) academic licensing was provided for the use of Instant JChem (ver. 15.10.12.0) to perform compound mining of the SDDL deck for CNS/BBB compliance. This work was also supported in part by the NIH S10 instrument award (1S10OD025279-01) that provided Scripps Research with the acoustic transfer system integrated into HTS operations for compound management.
) academic licensing was provided for the use of Instant JChem (ver. 15.10.12.0) to perform compound mining of the SDDL deck for CNS/BBB compliance. This work was also supported in part by the NIH S10 instrument award (1S10OD025279-01) that provided Scripps Research with the acoustic transfer system integrated into HTS operations for compound management.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.