
Abstract
For the past three decades, the pharmaceutical industry has undertaken many diverse approaches to discover novel antibiotics, with limited success. We have witnessed and personally experienced many mistakes, hurdles, and dead ends that have derailed projects and discouraged scientists and business leaders. Of the many factors that affect the outcomes of screening campaigns, a lack of understanding of the properties that drive efflux and permeability requirements across species has been a major barrier for advancing hits to leads. Hits that possess bacterial spectrum have seldom also possessed druglike properties required for developability and safety. Persistence in solving these two key barriers is necessary for the reinvestment into discovering antibacterial agents. This perspective narrates our experience in antibacterial discovery—our lessons learned about antibacterial challenges as well as best practices for screening strategies. One of the tenets that guides us is that drug discovery is a hypothesis-driven science. Application of this principle, at all steps in the antibacterial discovery process, should improve decision making and possibly the odds of what has become, in recent decades, an increasingly challenging endeavor with dwindling success rates.
Keywords
Introduction
For nearly a century, antibiotics have enabled modern medicine. Nevertheless, a continuing rise in antibiotic resistance threatens these medical advances.1–3 Concerns around antibiotic resistance and limited treatment options call for increased investment in antibiotic discovery and development and are resonating in academic, biotechnology, government, and pharmaceutical laboratories.4–8 Every major pharmaceutical company has led antibacterial high-throughput screening (HTS) screening efforts; however, there are more reports detailing failed efforts than those pointing to successful outcomes.9–12 In a typical antibacterial screening campaign, the overarching outcome is to find compounds, which (1) modulate a target that would ultimately lead to inhibition of bacterial cell growth and (2) can be optimized via medicinal chemistry to achieve desired features of specificity, selectivity, potency, and druglike properties. The HTS outcomes commonly fall into one or more of these scenarios: (1) hits lacked desired spectrum; (2) hits lacked acceptable druglike properties such as solubility, low plasma protein binding, and low lipophilicity; (3) hits lacked properties to leverage mechanistic tools such as crystallography or phenotypic mechanism of action studies; (4) hits had flat structure-activity relationship (SAR), such that the driver for activity was an undesirable moiety that once removed, activity dropped off; and (5) hits had SAR that lacked bacterial specificity, whereby active compounds were toxic to all cell types. Early steps to address these issues dealt with the target selection criteria and the quality of the assays and screening platforms. One of the target selection criteria was to understand the role target features play into their druggability.13–17 Analysis of screening campaigns led to several lessons learned and best practices regarding assay development, rigor in validation of assays, and quality control over HTS data generation, and analyses of this work have been published.18–20 To augment primary HTS approaches, several orthogonal platforms such as structure-based drug design and virtual screening have been employed in an attempt to leverage existing chemical libraries for SAR optimization.21–24 To further explore chemical space, fragment-based approaches have been attempted.25–28 Despite the vast target and chemical space explored, none of these approaches has delivered the desired lead molecules. The cumulative review of these outcomes speaks to a deeper appreciation of the attributes “lead” compounds need to possess for antibacterial spectrum, developability, and the amount of resources involved in converting an unoptimized structure to possess those features.6,9–11,29–32
This perspective attempts to convey our learnings in antibacterial discovery so that the broader scientific community can strengthen discovery efforts in this notoriously difficult area. In addition to elucidating inherent challenges in antibacterial discovery, one of the key learnings from these numerous campaigns is that the applied science of antibacterial lead discovery must be as hypothesis driven as any basic science. In this perspective, we highlight some of the hypotheses and assumptions that underlie antibacterial screening strategies, including target selection, assay development, hit identification, and hit characterization. For each of these areas, we drill down into some specific examples of testable hypotheses for these assumptions. Table 1 outlines key terms typically encountered in lead discovery efforts. Although there are numerous starting points for the discovery of antibacterial leads, we focus this perspective on identification of novel small-molecule templates based on medium-throughput screening (focused) and HTS of synthetic and natural small-molecule compound collections. The scope of this perspective is limited to the very earliest stages of antibacterial discovery—screening rationale through lead identification—but the emphasis on hypothesis-driven science applies to all aspects of drug discovery.
Common Terms Used in Antibacterial Discovery.

Screening Strategy
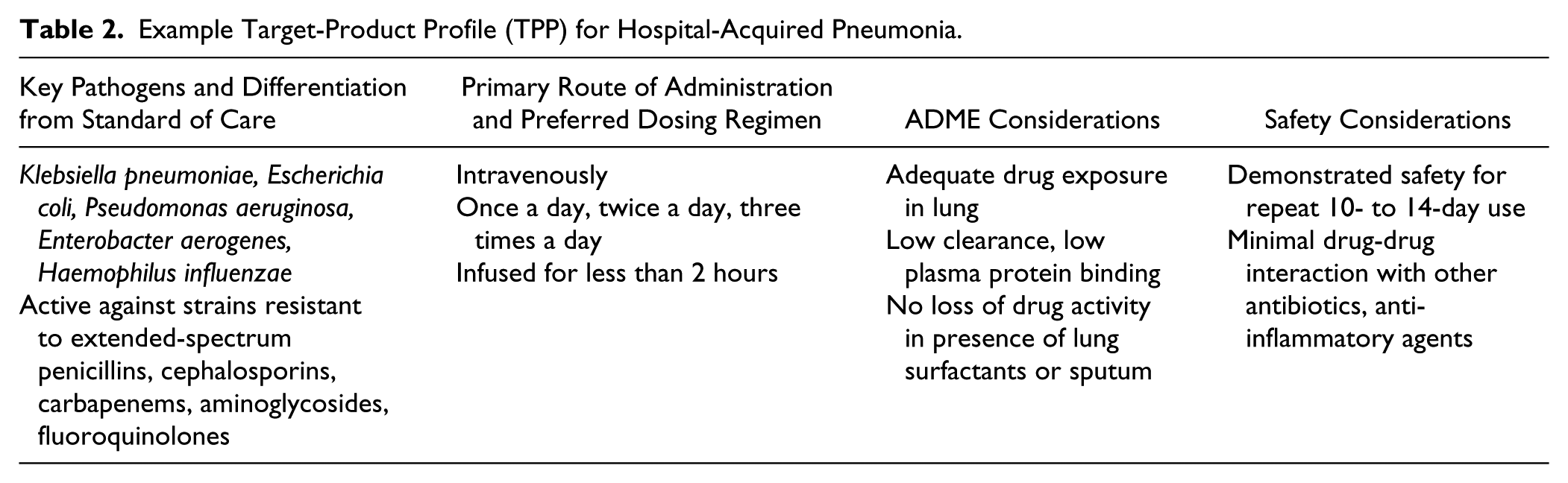
There is an adage: “you get what you screen for.” Most pharmaceutical companies pursued specific therapeutic indications based on frequently encountered hospital or community infections for which there is an unmet medical need. Examples include hospital-acquired pneumonia (HAP), complicated urinary tract infections, or complicated skin and soft-tissue infections. Each of these indications outlines a preferred set of pathogens, dosing regimens, and attributes to enable differentiation from standard of care drugs used to treat these infections. A target product profile for a therapeutic indication is shown in Table 2 using HAP as an example.
Example Target-Product Profile (TPP) for Hospital-Acquired Pneumonia.
Although this is a tall order to fill, antibacterial screening strategies have been relatively simplistic in nature. Two main types of antibacterial compound screening platforms deployed are cellular phenotypic and target-based assays.33,34 The fundamental format for a phenotypic screen is to treat live cells with single compounds or mixtures of compounds (e.g., from a bacterial or fungal extract). The simplest phenotypic cell-based assay measures cell survival; this is historically the most common screening strategy for the discovery of novel antibacterial agents. Target-based approaches are the most direct approach to identify mechanisms associated with a specific biological process. These assays are typically composed of a purified enzyme target or complex in which the functional (enzymatic) or biophysical (binding or displacement) activity of compounds can be detected and quantified.
In early HTS campaigns of the 1990s, the most common screening strategy was based on a single pathogen to be screened in a bacterial killing assay format (typically monitored by optical density) against the largest chemical library available, hopefully 1M+ compounds. For these HTS screens, the typical organisms selected were Staphylococcus aureus or an efflux-deficient Escherichia coli mutant for the following reasons: (1) these species represent gram-negative and gram-positive pathogens, respectively; (2) genetic and mechanism of action tools were available; and (3) good hit rates were generated. Triage would consist of testing hits for antibacterial activity across a diverse spectrum of pathogens, attempting to understand the compounds’ mechanism of action and evaluating the hit’s physical properties. The typical outcome would be thousands of “ugly” hits for which very few had activity across multiple pathogens and even fewer for which one could deduce the mechanism of action. Although whole-cell assays have the advantage of screening all targets essential under the assay growth conditions, the disadvantages include the considerable downstream efforts to determine the mechanism of action and target of the hit. Bacterial killing can be due to optimizable antibacterial mechanisms as well as nonoptimizable, nonspecific mechanisms, such as detergent-like membrane disruption, metal chelation, and so forth. In addition, the identification of the target can be challenging when hits have weak cellular potency, for example, because of the activity of efflux pumps and other intrinsic resistance. The learnings from these single pathogen-screening efforts emphasized the need to incorporate a mechanistic bias into the primary screen as a means of mitigating nonspecific hits.
A target-based approach is the most direct path to identifying mechanisms associated with a specific biological process. Such targeted assays typically comprise a purified enzyme target or complex in which the functional (enzymatic) or biophysical (binding or displacement) activity of compounds can be detected and quantified. To identify conserved and essential genes for HTS campaigns, a significant amount of effort was expended to compare the genetic and protein composition of organisms. Regardless of the target, the outcomes of HTS campaigns were again thousands of “ugly” hits, of which many were artifacts of the assay system. Of those that had target specificity, almost none had whole-cell activity. Structure-based drug design and fragment-based drug design approaches could be used to augment SAR optimization. However, under typical triage timelines and available resources, these hits were in many cases deemed too challenging for optimization efforts.
The learnings from these basic screening strategies emphasized the need to develop assays and tools that would incorporate mechanistic bias into the primary screen or within triage tools and criteria to eliminate false-positives. It also accentuated the need to develop tools and strategies to build in whole-cell activity across a desired spectrum of organisms from target-based hits.33,34
As with any scientific experiment, there is a hypothesis being evaluated using a large chemical screen. The most common hypotheses probed via compound screening are whether a biological target is valid, whether a novel source of chemical matter can provide specific modulators of biological activity, and whether an innovative screening platform can increase the likelihood of identifying high-value chemical starting points. In the case of the biological driver, the goal is to build confidence in the modulation of this biological function in the treatment of infections. The outcome of this screen could be a “tool” molecule or lead candidate supporting the validity of modulating this biological function as a valuable antibacterial approach. With respect to novel chemical matter, the outcome of the screen could determine which type of modality has antibacterial activity with an optimizable mechanism of action. As to the innovative screening method, a screen can evaluate whether this approach is more sensitive, or robust, than previous methods in identifying modulators of biological function.
A screening strategy should encompass a focused, yet comprehensive, set of primary and profiling assays as well as the decision-making criteria that allow one to test the hypothesis for which the screen was designed. The outcomes should speak directly to whether the biological systems, chemical matter, or screening platforms can be successfully employed in drug discovery efforts. It should also encompass the learnings from past attempts using these approaches to increase the likelihood of successful outcomes. The focus in the following sections will be the hypotheses and assumptions gathered over time in an effort to better use these screening strategies.
Robust Screening Funnel
As the name implies, a screening funnel is a set of assays with defined criteria that enrich for hits that can most directly satisfy the screening strategy. A typical screening funnel consists of three main activities: primary screen, hit identification, and hit characterization (
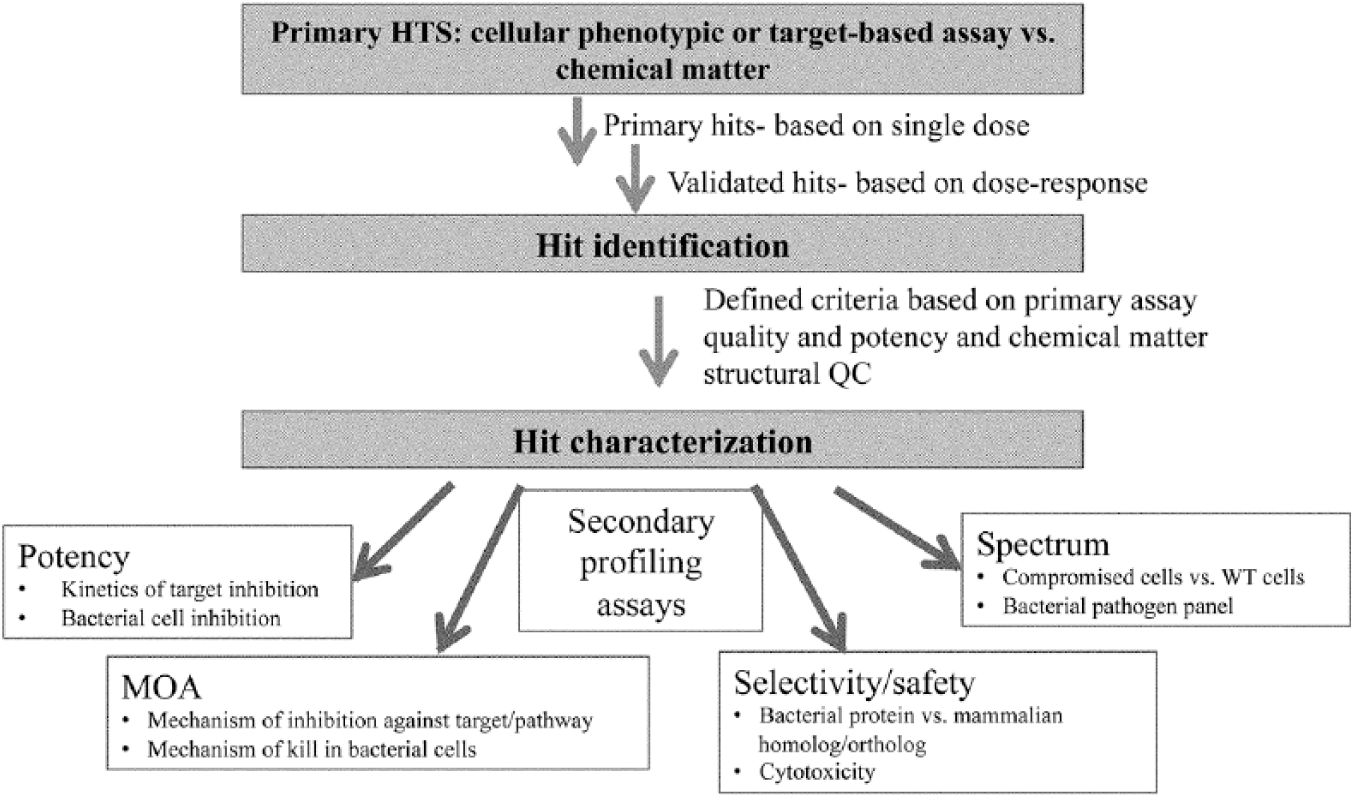
Typical antibacterial screening funnel. The overriding hypothesis is that a specific compound collection contains compounds possessing biological activity that are synthetically tractable in order to optimize toward a drug.
Primary Screen
The primary screening decisions include what to screen (chemical library), how to screen (screening platform and assay parameters), and what to screen against (biological system). Phenotypic bacterial killing assay formats are by far the easiest to deploy. However, lack of mechanistic bias can result in large numbers of nonspecific hits. Variations of the phenotypic screening formats that are biased toward inhibitors of specific antibacterial targets or pathways are typically accomplished with engineered bacterial strains (e.g., through sensitization by target underexpression or through use of a target or pathway-specific reporter genes35,36). Although phenotypic screens may seem to bias toward a specific pathway or target, the complexity of systems biology means hits may be identified that are difficult to relate back to the primary target or pathway.
As the name implies, target-based assay formats are the best choice to identify specific target inhibitors. Typical target assessment criteria include essentiality, novelty, bacterial specificity, and spectrum; however, a “good” target is difficult to define in the absence of clinical success. Below, we address some of the common assumptions associated with selection of the biological screening choices.
Essentiality
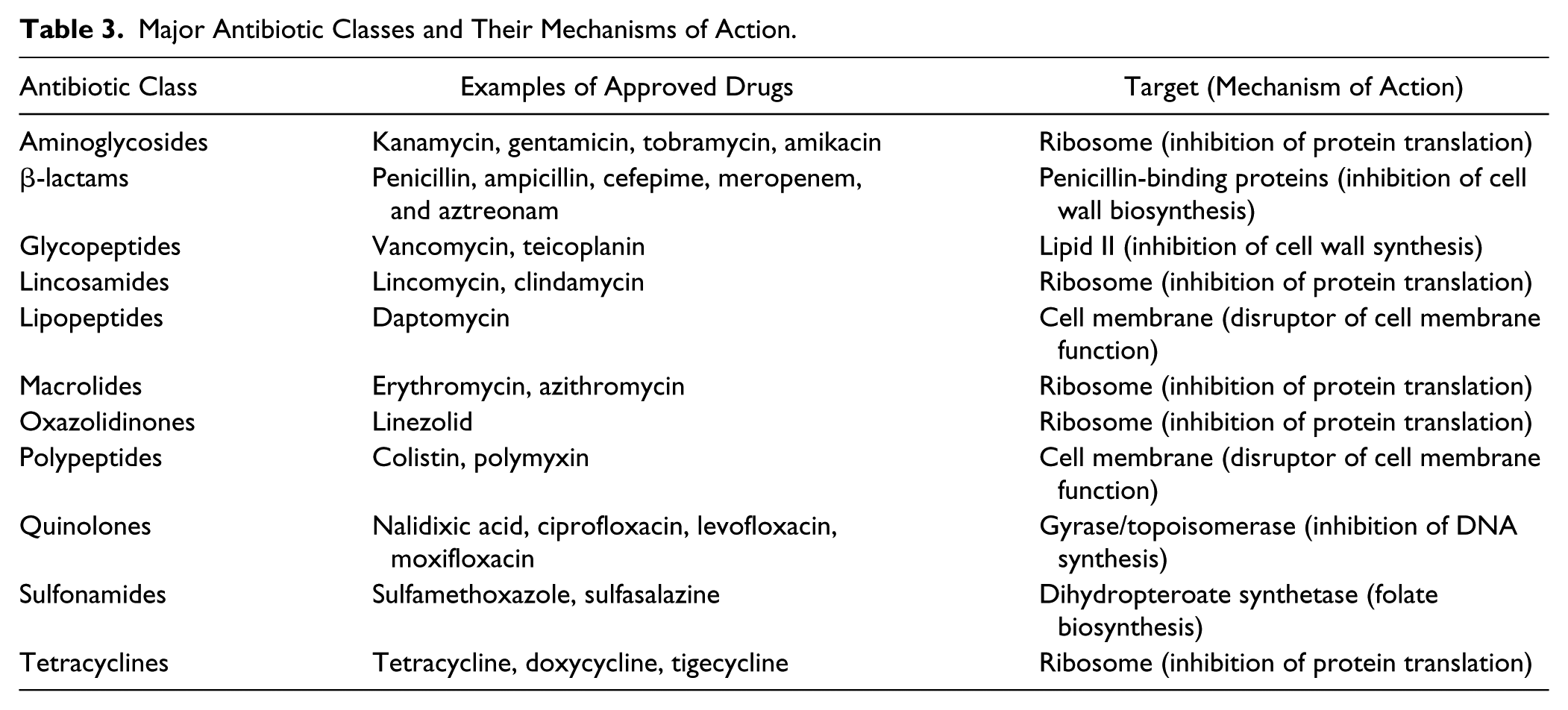
The most salient requirement for an antibacterial target is that it must ultimately have an essential role in bacterial colonization and pathogenesis in the human host. Essentiality of targets can be binned into three general categories based on the conditions and stage of infection: in vitro, in vivo, and virulence. The ideal target is essential under in vitro and in vivo conditions, demonstrating bacterial cell death under standard laboratory testing as well as in animal models and clinical infections. This is a concept that is validated by clinically used antibacterial drugs targeting functions such as RNA synthesis, protein translation, DNA replication, cell wall synthesis, and cell envelope integrity (see Table 3 ). Essentiality under in vivo conditions can also be known as “conditionally essential,” meaning that these genes or pathways are necessary for survival in host settings but not under standard laboratory testing conditions. Virulence factors are functions that are essential at specific stages of infection and can be species and strain specific.
Major Antibiotic Classes and Their Mechanisms of Action.
Essentiality is commonly determined using genetic methods in laboratory strains. If loss-of-function mutations are easily obtained, this suggests that the cell does not require this process under the selected conditions or that it can efficiently compensate for that biological process. Either of these outcomes suggests that there could be a risk for rapid emergence of resistance. In contrast, there are multiple assumptions that accompany the determination of essentiality based on failure to isolate loss-of-function mutants. These assumptions include (1) essentiality in the laboratory translates to essentiality in the diseased host; (2) if a gene is essential in one isolate, it is essential in all isolates of a species; and (3) essentiality in one species is predictive of essentiality in another. Counterexamples exist for all of these,37,38 and testing all of these assumptions may not be possible at the initiation of a project. Beyond testing essentiality via a loss-of-function mutant, one can evaluate the degree of target depletion required for bacterial cell death via regulation of gene expression. This is an imprecise proxy for estimating the outcome of chemical inhibition as the extent of compound potency required to inhibit growth varies considerably from target to target and across all possible mechanisms of inhibition within a given target. 39 By acknowledging these assumptions, assays and approaches can be designed for ongoing validation of target essentiality and optimal mechanism(s) of inhibition.
There is a growing interest in targets important for survival of bacteria in the human host but not required for bacterial survival and reproduction under in vitro conditions.40,41 For these in vivo essential targets, it is important to ascertain whether they are involved in establishing an infection, which may make them more suitable for prophylaxis than therapy,42,43 or in maintenance or dissemination an infection. Validation of this class as therapeutic targets should include a direct test of the hypothesis that inhibition can cure a previously established infection, particularly in deep-seated infections. This may be accomplished through the use of genetically modified strains (i.e., controlled expression of the key gene) or through in vitro replication of the in vivo environment (e.g., elegant work by Fahnoe et al. 44 on the glyoxylate shunt). A target more suitable for prophylaxis is a potential option for addressing many patients at risk, 45 but the development path for this class of drugs will differ from that of conventional antibiotic therapies. Novel clinical susceptibility testing may need to be developed to support appropriate use. One common assumption about in vivo essential targets is that there will be a reduced risk of clinical resistance development. To date, this hypothesis is largely untested and unvalidated; indeed, because survival in the human host can be a selective advantage, it makes sense that compound-resistant mutants will occur. It is critical to test rigorously the resistance hypothesis for target gene and any modulators as early in the discovery process as possible.
Bacterial selectivity of a target
A common criterion for antibacterial target selection is the absence of a human homolog. The underlying hypothesis is that the absence of a human homolog will increase the likelihood of developing a safe compound. The reality is far more complicated. Many successful antibiotics target processes found in both host and pathogen. For example, oxazolidinones, tetracyclines, aminoglycosides, and macrolides all have varying degrees of activity against mammalian protein translation machinery, especially in mitochondria, but the window between efficacy and toxicity justifies appropriate clinical use. At the same time, most antibiotics have side effects caused by off-target activity. Rather than making a blanket pronouncement about target selectivity, it is important to look carefully at each proposed target to formulate testable hypotheses regarding the nature of their selectivity (e.g., setting up counterscreens with known human homologs, performing early safety profiling even if no homologs exist). It is important to have an early understanding of the promiscuity46,47 and other potential safety liabilities of the compound.
Novelty
Documented clinical resistance to marketed drugs places an emphasis on identifying novel targets. One common assumption is that drugs developed against a novel target will be active against bacterial pathogens that are resistant to clinically used drugs. This hypothesis needs to be tested throughout a project as there are many mechanisms of resistance that are target agnostic, including induced or constitutively overexpressed drug efflux pumps and drug-modifying enzymes. It may be more fruitful to focus on the screening strategy, the screening library, and the mechanisms of clinical resistance than to overemphasize target novelty.
Localization of the target
Intracellular targets represent a diverse and wide range of essential functions conserved across bacterial species. These have been attractive targets for many drug discovery efforts, because of the ability to apply biochemical and structural biological approaches for SAR generation. However, these targets face the challenge of cellular accumulation; compounds must be able to traverse the cell wall and the cytoplasmic membrane barriers as well as avoid efflux to stay in the cell. Although it may be easier for compounds to gain access to cell surface and periplasmic targets (in the case of gram-negative species), there are far fewer well-conserved classes of targets.
Whether one starts with a cell-based phenotypic assay or a purified target-based assay, eventually both types of assays will be necessary determine the target inhibition mechanisms and antibacterial action of prioritized leads. Incorporation of assays to assess activity against multiple key pathogens should be placed early in screening funnels to prioritize hits with potential broad-spectrum activity. These are key assay platforms to identify leads with the desired potency, spectrum, and mechanistic properties.
Assay Development and HTS Quality
To support the screening strategy, decisions regarding the bacterial species, demonstrating the quality of primary assay and identifying the screening compound source and concentration, are all highly influential in informing the screening output. As discussed above for the target selection criteria, in addition to essentiality criteria between species, the fundamental physiological differences between bacterial species as well as subtle differences between homologous proteins with a high degree of similarity will bias the hit rate and chemical properties of the hits toward the ortholog that is ultimately chosen for screening. The quality of the assay is driven by the rigor and consistency applied in assay development and assay execution in the HTS campaign. Every screen is biased and has limitations, because of the biological system, the nature of the compounds, or from the screening format.
As a scientific community, we have learned a lot about assay limitations and biases.18–20,46,48 Being vigilant about the sources for artifacts and rigorous testing of the assay system during protocol development for these vulnerabilities will greatly facilitate data interpretation to enhance triage efforts. For phenotypic cellular assays, the main biases in assay development depend on the species, compound concentration, and detection method. As was discussed, the species and assay conditions define which essential processes are interrogated in the screen. The compound concentration selected for the screen has a strong effect on hit rate. Most cellular assays use absorbance to measure changes in cell density. Common confounders for this detection method are solubility, color of the sample, and micelle formation. 49 Solubility artifacts occur where the chemical matter comes out of solution via colloidal aggregation and contributes to a higher optical density (OD) that can look like growth.50–52 One study that evaluated their hit list for aggregators found that 95% of the hits were due to this artifact. 52 Compound color or diffraction can interfere if these are in the overlapping range of the OD wavelength used to monitor the assay. Micelle formation can sequester enzymes and substrates. Higher compound concentrations exacerbate all of these confounding assay artifacts. 50 Although it may be tempting to increase the compound concentration to attempt to include weaker hits, this could result in just a higher false-positive hit rate. Typically, this can be averted by comparing a time-zero read with the final incubation read to determine if the compound has high intrinsic background. If one anticipates encountering large numbers of samples that may produce these artifacts, surrogate readouts for cellular growth such as methods that monitor adenosine triphosphate respiration using luminescence or fluorometric readouts (such as Alamar Blue, resazurin) have been employed.53,54 However, care should be taken when using these systems, as there are known chemical scaffolds that interfere with these reagents as well.55,56
Purified protein assay formats can introduce different artifacts. What started as a collaboration between Eli Lilly and the National Institutes of Health to compile best practices for assay development has expanded to a large repository of HTS assay development guidelines 18 and specialized assay formats. This resource covers many of the parameters encountered with assay development as well as best practices for instrument usage and assay detection methods. A common misconception for enzyme assays is that an assay buffer composition generated for one species’ ortholog will work for all species’ orthologs. Even very similar orthologs will behave differently from each other, such as in the difference in binding affinity of substrates, requirements for cofactors, difference in sensitivity to metals, their stickiness to plastic surfaces, and so forth. In many cases, protein orthologs derived from multiple species should be evaluated to determine which would be the most amenable for screening. It is important to profile each under a range of physiologically relevant buffer conditions, such as pH, ionic strength, chelators, and nonspecific binding additives, to optimize the activity window as much as possible. Given the need to demonstrate spectrum of activity across multiple species, these additional bacterial orthologs will be needed at some point during the life of the screening triage.
With respect to compound-derived artifacts, there is growing literature about many of the nonspecific mechanisms observed in screens, referred to as PAIN (Pan-Assay-INterfering) compounds and aggregators,57–59 that can contribute to false-positives in the assay. Not all enzyme assays are affected to the same extent by these types of compounds, so it is important to test each assay system against these to ensure that there are reasonable means to mitigate their impact. Composite plates containing PAIN reagents can be screened in pilot assays to determine how vulnerable an assay system is to each of these types of interfering compounds. Incorporation of appropriate additives into the assay buffers to address these liabilities or the use relevant counterscreens will greatly reduce the triage time to weed out false-positives.
Once assay parameters have been established, it is important to test the reproducibility of the assay on the automated screening system to be used for the HTS campaign. The assumption that the assay protocol will not need to be modified to adapt to an HTS automation system is typically false. Chai et al. 19 provided a detailed review of best practices for assay adaptation for HTS automation and validation. The main advantages of using automation are the accuracy for the liquid dispensing and consistency of timing for each assay step, which result in tighter assay performance over a higher capacity of plates as compared with what can be achieved by manual processing. However, for target-based approaches, factors such as reagent stability and enzyme kinetics can be affected because of the differences in the length of time for a typical assay run. For example, the length of time to run 10 plates by hand as a pilot screen is much shorter than the time to run 400 plates using a robotic arm. For a typical HTS, it is advantageous for reagents to be stable at room temperature for at least 4 hours. In addition, the plastic reservoirs and tubing used for automated liquid dispensing can cause issues because of differences in protein or substrate-binding affinities. Regardless of the format, the assay reproducibility in the presence of known control drugs or reagents that represent the range of activity (no effect to 100% inhibition) should be assessed over multiple days by testing the anticipated number of plates to be screened each day as plate replicates (at least n = 3), whereby the plate order is changed each day. This method aimed to generate statistically valid numbers of data points for all activity conditions within a plate, across plates, and across days. The analysis of these data will indicate the variability within each activity control, as well as the resulting signal window between 0% and 100% activity. This analysis will identify issues with plate heuristics that need to be addressed, such as edge effects, lack of reproducibility, or drift of signal over time. Alternatively, this type of matrixed replicate system can be performed with representative compounds from the library to be screened. An additional step in assay validation is testing representative compounds from the library at multiple single-dose concentrations followed by dose-response assays of these hits. 20 Results of these two studies should help estimate the primary screen hit rate and the probably false-positive rate by the dose-response assay. For example, a representative library was tested at 10 µM, 50 µM, and 100 µM with the corresponding hit rates of 0.01%, 0.1%, and 1.2%. If the confirmation rate of those hits in the dose-response assays was 80%, 50%, and 20%, it is likely that screening at the higher concentration was merely reflected in a higher percentage of false-positives and not enrichment for quality hits.
Chemical Library Selection
The adage “garbage in, garbage out” is highly relevant to the selection of screening chemical matter. The origins of many modern pharmaceutical companies are as natural product apothecaries or companies that specialized in diverse synthetic chemistries. Most available libraries are composed of compounds archived over decades of drug discovery initiatives, covering a diverse range of therapeutic programs as well as some remnants from these past origins. Bearing in mind the industrial nature of most commercial libraries, the frequency of encountering undesirable lead compounds is high. An experienced medicinal chemist can recognize compounds with identifiable risks, such as nucleophiles, Michael acceptors, redox reagents, alkylators, detergents, or other chemically intractable and non-druglike entities. In addition, online resources, such as www.ChemSpider.com, 60 provide information on more than 28 million chemical structures (often including references to biological activities). Whenever possible, it is best to eliminate these problematic compounds from screening libraries.
In addition to ridding libraries of problematic compounds, efforts to build antibacterial-biased screening libraries run into the complication that the rules for what comprises “antibacterial” activity are poorly understood. Guidelines such as Lipinski’s “rule of 5” for orally available drugs do not apply well to antibiotics.32,61 Selection criteria based on retrospective studies and computational analyses conducted by several groups are not universal across species or diverse targets.11,31,32,62–65 To address these challenges, library selection has been driven toward sources that will exploit as much chemical diversification as possible. These include natural products from diverse ecological niches 66 or fragment derived from natural product biosynthetic pathways, 67 fragment libraries that allow one to build out tailored solutions,68–70 and chemical diversification generation methods such as DNA-encoded libraries, 71 positional scanning libraries, 72 and other combinatorial strategies to exploit chemical diversity. An in-depth perspective on the chemical assessment of compound collections is out of scope for this article; however, given the lack of success to date, researchers should look for ways to expand the diversity of chemotypes with physical properties that lend themselves to better bacterial accumulation and safe human delivery (see refs. 9–11, 62–65, 73 for insights).
In addition to traditional library screens, renewed interest is emerging in repurposing registered drugs that are used to treat other conditions as potential novel antibacterial compounds. The often-stated assumption is that because these agents are already approved drugs, they can be rapidly repurposed without extensive optimization. The best-known example involves statins, the commonly used cholesterol-lowering drugs, which are reported to have antibacterial activity (although that antibacterial activity appears to be unconnected to HMG-CoA reductase inhibition74–76). For this approach to work, the exposure necessary for antibacterial activity would have to result in an acceptable therapeutic index. The validity of this hypothesis for any given compound depends on many factors, including the relative potency for bacterial and nonbacterial targets, route of delivery, duration of therapy, and pharmacokinetic/pharmacodynamic drivers, all of which influence the therapeutic index. There are, as of yet, no known examples of success. Given the relatively low doses of drugs used to treat other human health conditions compared with the doses used for most antibiotics, it may prove extremely difficult for such repurposed drugs to achieve therapeutic value as antibiotics.
Hit Identification
HTS of large-compound collections, whether they are natural products or synthetic compounds, will uncover hundreds of “hits” in many screens. One controversial hypothesis around HTS in general is that a high percentage of positives, or a high hit rate, will increase the chances of identifying a good lead. Unfortunately, a higher hit rate usually translates to a higher percentage of nonspecific inhibitors that will invariably lack selectively or potency, leaving few viable chemical starting points. 77
A “hit” is arbitrarily defined based on the quality of the assay and the desired potency. One common definition of a hit is the 3-SD rule. This rule is derived by determining the standard deviation of the negative control wells (wells that do not contain any inhibitor molecules) and then assumes that any compound with activity greater than three standard deviations from this mean would be statistically different from the noise of the assay. The hypothesis supporting this criterion is that activity observed in these control wells represents the intrinsic variation of the assay due to any assay parameter or automation processes; therefore, activity outside of this range is due solely to the compound’s effects on the biological system. The definition of a positive hit would be any compound showing activity greater than three standard deviations of this range. For a “tight screen,” in which the standard deviation for these control wells could be as low as 6% activity, this would allow one to call a “hit” as anything with greater than 18% activity. This would likely result in a high hit rate and can give a false sense of optimism—it is the “we won’t miss anything” strategy. In contrast, a different strategy for hit calling is the “biological-meaningful” strategy. If one considers the potency required to show activity in downstream profiling assays, many of the hits with this lower percentage of activity may not be potent enough to generate meaningful data. In addition, many mechanistic profiling assays have lower screening capacities, and flooding these assays with low-activity hits will slow timelines. Being aware of the potency thresholds of downstream mechanism-based assays can help define more relevant hit criteria that allow one to enrich for hits that can be profiled and to improve triage efficiency. Another specific consideration for antibacterial hit criteria is the difference between growth inhibition activity and bacterial killing observed in minimal inhibitory concentration (MIC) assays. Spectrum of activity is usually determined by standardized MIC assays that allow for comparison of antibacterial activity across clinical laboratories. 78 Clinical and Laboratory Standard Institute (CLSI) guidelines dictate reference strains, growth protocols, and activity ranges for control antibiotics for each bacterial species. 79 Note that the activity from MIC assays is reported as the concentration (in µg/mL), where no visible growth is observed, whereas most HTS screens report the concentration (µM) where 50% activity is observed. Therefore, one needs to consider hit criteria decisions and typical solubility ranges of most chemical libraries that bias toward hits with higher activity and solubility. For a number of reasons, it may not always be possible to conduct the primary screen under conditions outlined in the CLSI guidelines. However, taking into account these two parameters will minimize the translational discord and select for compounds for which the growth inhibition and MIC values provide a more direct comparison.
Hit identification can control the number of compounds assessed in later-stage assays; however, the main question for hit identification is whether the hit is valid. As mentioned in the “Assay Development and HTS Quality” section, there are many sources of false-positives in any assay. These can be due to the nature of the assay composition but also based on the compound’s properties. To rule out those due to the nature of the assay composition, primary hits are routinely confirmed by either retesting the compound at the same single concentration as the primary screen or in a dose-response format using the primary assay as well as in an appropriate orthogonal assay. Orthogonal assays are devised to address a number of questions about false-positive outcomes. For example, target-based approaches are used to determine if the compound is interfering with the assay detection method or acting through the reagents, and the same assay format is composed of all the same reagents but performed with a nonrelevant enzyme. Another approach would be to employ an alternate assay format with the same primary enzyme to demonstrate modulation of the target biological activity.
As mentioned earlier, a commonly encountered confounder is solubility. Compounds may precipitate in a concentration-dependent manner due to colloidal aggregation, causing interference with both cell-based and target-based assay formats.50–52 Most often, this is detected as steep curves in IC50 assays (hits with a Hill coefficient >2). Although most aggregator activity can be resolved with the addition of detergent to the assay, depending on the screening concentration, these artifacts may be persistent. One method to evaluate whether the compound’s solubility is affecting the assay outcomes is to run the dose-response assay plate in a nephelometer-based plate reader and compare the activity curves with the primary assay detection method. Hits that have overlapping activity patterns between both reader formats are likely due to aggregation and should be deprioritized.
Another frequently encountered confounder is the presence of trace elements, such as heavy metals, used in synthesis reactions or materials leached off purification columns. These entities can be difficult to detect with routine chemical quality control analytics. Another assumption is that the chemical structure registered to the well matches what is in the well. Many HTS libraries contain compounds that have undergone decomposition or are registered with the wrong structural information. In our experience, there have been a number of occasions on which exciting hits have disappointed because the compound analytics do not support the registered molecular composition. Conversely, there have been examples in which confirmation of a hit’s structure has revealed it to be other than the registered structure, providing a novel starting point. To generate a solid SAR, the structural integrity of the compound should be verified and, whenever possible, the compound should be resynthesized. Verify the route of synthesis and structure before investing too much time and resource to pursue these initial hits.
Given the wealth of literature documenting the nature of artifacts and their abundance in chemical libraries, one needs to be vigilant and adhere to strict criteria that will enrich well-validated hits that have the best potential for satisfying the screening strategy. The number of hits does not define a successful screen; it is defined by the quality of the leads or tools that it delivers.
Hit Characterization
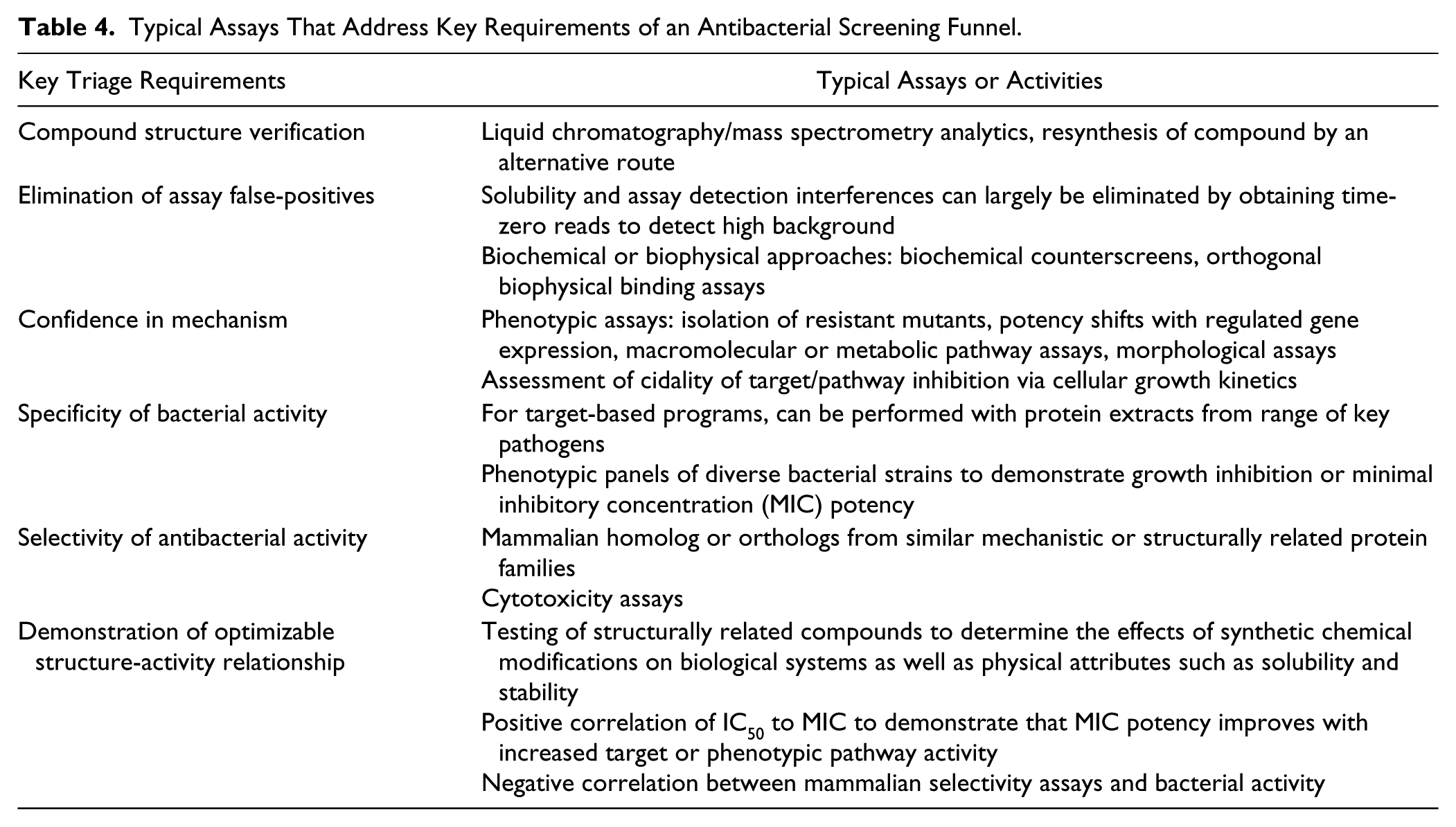
The hit identification phase should have weeded out structurally invalid hits and the false-positives based on the assay format; the characterization phase focuses on determining how these hits are acting on biological systems. Hit characterization consists of multiple, orthogonal tests, which can provide independent assessments of the antibacterial spectrum and mechanism of action in these diverse organisms as well as selectivity against mammalian cells. Table 4 outlines many of the common assays used to profile hits. The vast majority of hits will possess off-target or promiscuous activities. Even if one can demonstrate biochemical activity against a specific target, this does not imply that the compound cannot interact with other components of a biological system. It is just as likely that the antibacterial activity is due to effects of the compound unrelated to specific inhibition of the intended target. Profiling assays should potentially disprove promiscuity (e.g., membrane-damaging activity and cytotoxicity testing) as well as demonstrate specificity for a biological process. Here are a number of common methods used to assess the specificity and selectivity of screening hits and some hypotheses associated with each of them.
Typical Assays That Address Key Requirements of an Antibacterial Screening Funnel.
Generation of resistant mutants
Selection of resistant mutants to an antibacterial compound is one of the most effective tools for identifying a potential antibacterial target. One hypothesis is that a gain-of-function substitution in the target can influence the interaction with the compound, generally by reducing affinity. Target-based activity is confirmed if transferring that mutation into a naive background confers resistance or if the altered mutant target confers a shift in the IC50 for the compound. If no mutants are selected, the underlying hypothesis of many researchers is that the target cannot be modified in a way that prevents the compound from binding, without compromising viability. This is not impossible, but it would be essentially unprecedented for a true single-target inhibitor and should be taken as a sign that the compound has interactions with other entities, necessitating additional studies to validate any interaction with the presumed target. Mutations that reduce bacterial fitness are often said to predict that resistance is unlikely to develop in the clinic. The underlying hypothesis is that bacteria cannot easily compensate for the fitness defect; this should be tested experimentally, because it is known that compensatory mutants can arise in the continued presence of drug.80,81
IC50/MIC correlation
With the availability of a series of related compounds, it may be possible to correlate target potency (typically measured as IC50) with antibacterial activity (typically MIC). While the fundamental assumption is that an increase in target potency should translate into a decrease in MICs, this hypothesis often fails because the SAR for enzyme potency and cellular activity can be divergent or due to an increase in nonspecific activity. Highly potent biochemical inhibitors can have poor whole-cell activity because of insufficient cellular accumulation; thus, IC50 improvement does not always correlate with increased MIC activity. Hits whose primary mechanism of action is nonspecific, detergent-like activity can show increases in both IC50 and MIC activity without truly proving linkage to the target. However, the converse can be true; strongly improved MICs seen in the absence of improved enzyme inhibition suggest there is a possibility that nonspecific activity (or another target) may be contributing. One cannot strongly rely on IC50/MIC correlations for providing confidence in the mechanism of action.
Pathway profiling
Pathway profiling is useful for confirming activity against a specific target in a pathway as a means of deducing mechanism of action from a cellular phenotypic assay. One common approach is to test for inhibition of macromolecular synthesis (e.g., RNA, DNA, protein, cell wall, lipid) in whole cells. Here, the hypothesis is that an inhibitor of a specific component of that pathway (e.g., DNA ligase) should alter the inhibition of a specific pathway (e.g., DNA synthesis). It is important to validate the assay specificity by testing known inhibitors of each macromolecular synthesis pathway. Another powerful approach is to test for specific metabolic changes. For example, inhibition of methionyl-tRNA synthetase leads to accumulation of uncharged Met-tRNA without altering the levels of other transfer RNAs, as well as induction of the stringent response, 33 both of which can be directly measured. Another example is the detection of changes in pathway intermediates upon compound treatment of whole cells by mass spectrometry methods.82,83 As above, it is important to validate this hypothesis by testing suitable controls. It is also important to note that inhibition of a primary target does not rule out the possibility that secondary targets contribute to antibacterial activity. Thus, showing an expected biochemical effect in the cell does not exclude modulation of other systems. Combining the results of target-specific assays with results from cell-based biochemical/metabolic assays (i.e., macromolecular synthesis) provides stronger support for the specificity of the compound.
Physiological or morphological methods
For some targets, genetic studies or previous experience with validated inhibitors may predict a specific morphological or physiological change. Some examples include morphological changes in filamentation or formation of spheroplasts observed with cell division or cell wall inhibitors.84–86 Physiological reversal of the inhibition by metabolic complementation is also helpful (e.g., reversal of dihydrofolate reductase or thymidylate synthetase inhibition by addition of thymine or thymidine) 87 A variety of agents can generate these physiological or morphological changes; therefore, other hit characterization assays should corroborate these readouts.
Under-/overexpression of the target
Specificity of early-stage hits is frequently interrogated using strains engineered for adjustable expression of the proposed target. The underlying hypothesis of these methods is that changing the expression level of the specific target will affect the stoichiometry of target to inhibitor; thus, underexpression is thought to sensitize strains to inhibitors, whereas overexpression is thought to provide some level of protection. These assays should be validated to determine the amount of inducer required for inhibition (via RNA or protein target levels) and frequency for emergence of spontaneous mutations in the induction system. These assays also have the potential for broader effects of target modulation on bacterial physiology that may confound interpretation of these results. Rigorous testing with agents that act via alternate mechanisms (including nonspecific detergents) can be used to build confidence that the modified strain uniquely reports specific target effects. It is also worth noting that if a compound does bind the (presumed) target, then increasing levels of that protein’s expression may reduce the amount of free compound available to interact with other targets, meaning that expression levels could alter the MIC of a compound, even if growth inhibition is not occurring via inhibition of that target.35,36
Genomics
Expression profiling (transcriptome, proteome, metabolome) can help to establish the mechanism of action of an inhibitor. 88 The hypothesis is that treatment of bacteria with an exploratory compound will produce an expression profile similar to that observed upon treatment with other inhibitors of the same pathway or upon mutation or depletion of the target in the same test strain. The challenge in using these approaches is that there will typically be hundreds of genes, proteins, or metabolites with altered expression levels, some of which will match the pattern observed with the control conditions and others that will not. These methods can provide supporting data and help to fine-tune mechanistic hypotheses but rarely provide solid proof of mechanism in isolation.
Cytotoxicity
A very common approach for demonstrating selectivity of hits is testing for cytotoxicity against mammalian cells. There are methodological assumptions that one needs to be cautious about with respect to cytotoxicity testing based on their vastly different experimental conditions. One strong difference is the presence of serum in the mammalian assays, which can reduce free drug concentrations in the assay and create the appearance of no activity. 89 To allow direct comparison, cytotoxicity and antibacterial activity assays should contain the same level of serum protein. Similarly, compounds can have different solubility outcomes between the two media types. Poor solubility may mean that the amount of active drug in the test well is significantly lower than the intended concentration. Although lack of cytotoxicity is a desirable result, appropriate caution is warranted because of confounding factors that can lead to false-negatives. Changes in division, morphology, metabolism, or gene expression are other potential safety issues. A tangential hypothesis regarding lack of cytotoxicity is that the compounds will be safe in vivo; this is not supported by the example of aminoglycosides, which are known to cause nephro- and ototoxicity yet show no in vitro cytotoxicity. Other types of toxicity not detected in common cytotoxicity assays are hERG channel inhibition and allergenicity. Toxicity can come from many directions; obtaining a maximum tolerated dose of selected leads can provide a more relevant insight into toxicity over cellular cytotoxicity assays.
In addition to carefully assessing the biological activities of test compounds, physicochemical properties of the hits should be evaluated with a critical eye to provide support for the hypothesis of selectivity. In the absence of literature references, it is still possible to see if a compound modulates other cellular activities. Central facilities perform most HTS campaigns, whether in industrial or nonindustrial settings, running compound collections against targets from multiple therapeutic areas. The first question to ask is, “In which screens was this compound identified as a hit?” It is worth noting that a compound with nonspecific activity may not necessarily be a bad starting point, but it is critical to be aware of these other activities right from the beginning and to incorporate assays to track such activity. 46
This section described many of the necessary assays to confirm or deduce the mechanism of action of the hit. Hits with an acceptable mechanism of action could be used to further validate a novel target or pathway. However, most hits will lack the attributes necessary to enable spectrum, in vivo efficacy studies, or safety or other requirements that support a drug discovery effort. The hypothesis that an HTS library hit will possess all the necessary druglike properties for antibiotics is usually false.
SAR and Synthetic Optimization
Several key criteria define a suitable chemical starting point for antibacterial drug development. Most antibacterial drugs require significantly higher doses than other human therapeutic drugs to achieve efficacious outcomes. Intravenous injection is the preferred route of administration to treat patients with serious infections requiring hospitalization. These specifications place a greater emphasis on the physical properties that drive solubility, metabolism, and safety of the compound.8,10,11,62 Antibiotics are commonly coadministered with other drugs; therefore, they need to be largely devoid of metabolic hepatic clearance mechanisms to avoid drug-drug interactions or generation of toxic metabolites. Lipophilicity, planarity, and polarity are key parameters that drive solubility, plasma protein binding, and promiscuity of the compound. Planar and lipophilic compounds with LogD >2.0 are more prone to lower solubility and higher plasma protein binding. Compounds that tightly bind protein require significantly higher doses to achieve efficacious exposures.
In selecting hits for SAR optimization, physical properties and structural characteristics are crucial components for evaluation. Alterations to reduce lipophilicity and planarity while maintaining biological activity is not trivial. In many cases, lipophilicity is required for target binding, and any changes to improve solubility or protein binding could result in a loss of biological activity, resulting in “flat SAR” and no means to optimize the lead.62–65
The hardest criterion to meet is the optimization for bacterial spectrum across diverse pathogens. In most cases, because of efflux and permeability limitations, the compound cannot reach adequate intracellular concentrations to induce antibacterial killing. Thousands of hits have been identified that inhibit valid targets yet failed to kill targeted bacterial cells. This may have been the greatest obstacle for target-based screens. In addition, most screening campaigns focus on a single species for their primary assay and characterization studies, yet the target product profiles for most antibacterial development programs require the potential to treat infections caused by multiple bacterial pathogens. As discussed previously, most clinical indications used to register antibiotics, such as HAP or complicated intra-abdominal infection, are caused by a number of different bacterial species. Therefore, a desirable antibiotic to treat these infections would need to cover multiple species or be compatible with other antibiotics that would need to be coadministered to achieve the desired spectrum. By far, one of the greatest obstacles for antibacterial lead optimization is the ability to achieve adequate intracellular accumulation of the compound.
The difficulty in optimizing potent and selective antibacterial leads is largely a result of evolution. Bacteria have evolved elaborate permeability barriers to survive the wide variety of environmental insults that they encounter in their ecosystems.90–93 The gram-negative dual-cell membranes present an even greater challenge. Three main routes exist to bypass the outer membrane: passive diffusion via a lipophilic charged membrane or via hydrophilic water-filled porin or active transport. Once across the outer membrane, compounds need to traverse the hydrophobic cytoplasmic membrane. These routes dictate very different physical properties. For example, positively charged polar molecules may be able to gain access to the periplasmic space via porins, but if the target is in the cytoplasm, the phospholipid inner membrane presents a challenge. In concert with the permeability barriers, gram-negative bacteria in particular have efficient efflux systems that minimize the intracellular accumulation of molecules that do not belong there. Several orthogonal approaches have been undertaken to understand how different species maintain cell wall integrity, substrate specificity, and kinetics of efflux pump mechanisms.94–98 Chemical probes and analytical platforms to measure bacterial accumulation along with application of artificial intelligence and machine learning are being used to gain insights into the chemical properties that govern intracellular accumulation.99–107 Efflux systems have a broad spectrum of substrates and therefore very promiscuous chemical properties. One proposed hypotheses for evading efflux includes the rate of influx and affinity to the target. In this hypothesis, if the influx rate across membranes is fast enough and the compound target affinity has a fast on rate and extremely slow off rate, it may be able to overwhelm the efflux system. Now consider that each gram-negative species, and even within a species, has porins of various dimensions, membranes composed of different lipid and phospholipid species, and redundant efflux systems, and you can start to imagine the complexity of designing compounds that can navigate all of these biological requirements. The rules for bacterial accumulation will be complex, consisting of multiple parameters beyond simple logD and charge; the shape, flexibility, and positional relationship of these attributes will complete the “lock and key” concept for each target as well as the permeability characteristics of organisms.62–65 At this time, there are only limited examples in which groups have monitored the permeability/efflux characteristics of their leads along with more typical activity-based approaches, including the efforts by Trius Pharmaceuticals in discovering dual inhibitors of bacterial DNA gyrase and topoisomerase IV with good activity against gram-negative bacteria108,109 and Novartis’ work pairing metabolite accumulation assays for their CoaD inhibitor programs. 82 Even with the success of these approaches in improving whole-cell biological activity, these programs were not able to bring all the requisite drug activities into one molecule. Greater investment into gathering and analyzing data across diverse species and strains, leveraging these innovative analytical platforms, is necessary to begin to unravel the rules for bacterial compound disposition that afford spectrum, adequate exposure, and safety to treat infections.
SAR optimization is a very resource-intensive endeavor; each potential series must be evaluated for whether it will provide value based on a net equation of assets and liabilities. One should have a focused SAR strategy to address specific liabilities. SAR optimization is a very iterative process; the requisite criteria for biological activity need to be evaluated with each modification to the scaffold. It is a fundamental hypothesis that toxicity can be positively correlated with bacterial potency; one needs to firmly disprove that hypothesis. Not all hits possess features that would be amenable to SAR optimization, hence the “flat SAR” outcome. Chemical matter considered for these efforts requires critical evaluation by an experienced medicinal chemist to determine whether the scaffold affords synthetic handles necessary to support this use of resources.
Like other experimental science, antibacterial discovery requires a strong scientific rationale. A weak scientific rationale or lack of rigor in testing these hypotheses is likely to lead to failed outcomes. Scientists launch antibacterial discovery efforts with the hypothesis that they can find good leads. Finding a good lead requires disproving the null hypotheses for the strategy undertaken. Here are some of our overarching perspectives to aide antibacterial discovery efforts.
Antibacterial discovery requires a committed strategic focus to test a hypothesis.
Chance favors a prepared mind. There is a strong element of serendipity in drug discovery, but you have to be prepared to exploit it. Know the desired outcomes and design a screening funnel that will enrich for those hits.
Utilize the learnings from fellow scientists; appreciate the hurdles encountered by others. How will your approach overcome those obstacles?
A high hit rate is not necessarily the hallmark of a successful primary screen. Set the criteria at a level to quickly to enrich for compounds with desirable attributes; sometimes, less is more.
When contemplating the use of a “cool, new” technology, think critically of whether this approach will solve any of the critical issues that lead to the failure of so many previous HTS campaigns to identify useful antibacterial leads.
Discovery of a new drug is only one of many possible research outcomes. Other valuable outcomes could involve discovery of tool molecules or biomarkers to help understand new mechanisms of action or to provide useful diagnostic tools.
Antibacterial activity is the first of many hurdles necessary to be overcome in the drug development process; in vivo efficacy, optimal pharmacokinetics and safety, and compound developability are crucial for antibiotic success.
Our goal with this perspective is to enable antibacterial discovery by helping to emphasize the importance of hypothesis-driven science as the most essential component of rigorous screening campaigns. Most screening strategies aim to identify lead molecules for a desired therapeutic indication. In actuality, because of the lack of whole-cell activity or inability to generate divergent toxicity from bacterial activity, most antibacterial efforts concluded with the discovery of molecular tools for novel targets or pathways or validated innovative screening platform hypotheses. We realize the incremental nature of scientific questioning and learnings to build on this foundational knowledge. These screens educate the community at large about target properties and screening platforms. The lack of success in finding leads could be due to a dearth of leadlike molecules in the screening deck to start with as well as the complexity of defining the rules for bacterial intracellular accumulation. Analytical and computational tools are needed to understand the rules for bacterial intracellular accumulation across species, not just for E. coli. If the rules are limited to a single species or are scaffold specific, resulting in single-species inhibitors, rethinking clinical development strategies for these molecules needs to be further explored to satisfy regulatory requirements. If this could be achieved, there is a treasure trove of hits that could be further evaluated for lead optimization. This work will not be easy, but it is necessary to the advancement of novel agents needed to address the antibacterial resistance issues emerging in the patient populations.
Footnotes
Acknowledgements
We gratefully and humbly acknowledge all of the marvelously talented and tenacious colleagues that we have had the pleasure of learning from and working with over the years of our tenure in antibacterial lead discovery. Through our many joint endeavors we have tested many hypotheses and learned much together, thank you!
Declaration of Conflicting Interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: Laura McDowell and Jennifer Leeds are shareholders and employees of Novartis Institute for Biomedical Research.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.