
Abstract
Innovation and new molecular entity production by the pharmaceutical industry has been below expectations. Surprisingly, more first-in-class small-molecule drugs approved by the U.S. Food and Drug Administration (FDA) between 1999 and 2008 were identified by functional phenotypic lead generation strategies reminiscent of pre-genomics pharmacology than contemporary molecular targeted strategies that encompass the vast majority of lead generation efforts. This observation, in conjunction with the difficulty in validating molecular targets for drug discovery, has diminished the impact of the “genomics revolution” and has led to a growing grassroots movement and now broader trend in pharma to reconsider the use of modern physiology-based or phenotypic drug discovery (PDD) strategies. This “From the Guest Editors” column provides an introduction and overview of the two-part special issues of Journal of Biomolecular Screening on PDD. Terminology and the business case for use of PDD are defined. Key issues such as assay performance, chemical optimization, target identification, and challenges to the organization and implementation of PDD are discussed. Possible solutions for these challenges and a new neoclassic vision for PDD that combines phenotypic and functional approaches with technology innovations resulting from the genomics-driven era of target-based drug discovery (TDD) are also described. Finally, an overview of the manuscripts in this special edition is provided.
Keywords
Introduction
Welcome to the first of a two-part special issue of the Journal of Biomolecular Screening (JBS) on phenotypic drug discovery (PDD).
Recently, there has been growing uncertainty about the commercial impact of the genomics revolution and the monolithic use of molecular targeted drug discovery (TDD) strategies1–5 by the pharmaceutical industry since new molecular entities (NMEs) output in the post-genomic era has been significantly below expectations.6–9 In light of the increased realization that the topology and regulation of biological processes are complex in their molecular components and integrative wiring,9 –11 investigators are starting to reconsider the use of complex biological systems to screen compounds in a target agnostic fashion.1,2,7 This phenotypic drug discovery strategy may be considered “neoclassic” since it is similar to physiology-based approaches used prior to the wide use of recombinant DNA technology but is enhanced by technology advances, such as those presented in both parts of this special issue.
This impetus for this special issue arose from a growing grassroots movement. Following publication of an article by Swinney and Anthony 4 titled “How Were New Medicines Discovered?” PDD became the subject of multiple discussions at SLAS2012 (the Society for Laboratory Automation and Screening Annual Conference and Exhibition, which was held February 4–8, 2012, in San Diego, CA) and led to the formation of the SLAS Phenotypic Drug Discovery Special Interest Group (PDD SIG). The SLAS PDD SIG quickly grew to become the largest SLAS SIG with the most active online forum. The JBS Editorial Board recognized the emerging importance of PDD and commissioned this special issue. A call for papers resulted in 95 manuscript proposals from 18 different countries. From this unprecedented response, JBS Editor-in-Chief Bob Campbell and Guest Editors Ellen Berg and Jonathan Lee decided that two special issues on PDD were warranted. This issue (JBS 18.10 December 2013) is part one. Part two will be published in JBS 19.1 January 2014.
This “From the Guest Editors” column serves multiple purposes. Initially, it defines terminology and provides the background and business case for the use of PDD. It then discusses key issues to PDD researchers, including technical performance, chemical optimization, target identification, and challenges to the organization and implementation of PDD. Finally, it provides an overview of the articles showcased in this two-part special issue.
Definitions
Figure 1 illustrates differences between drug discovery strategies based on target mechanism versus biological function. Following the integration of recombinant DNA technology into pharmaceutical research more than 20 years ago, most drug discovery efforts have used a molecular target-centric approach. In TDD, the molecular target important to the disease biology is typically identified from in vivo human/animal and/or cellular studies. The complementary DNA (cDNA) encoding the molecular target is used to express recombinant protein for purification or to generate cell lines that overexpress the target. In either case, assays are enabled that measure the activity of the recombinant molecular target of interest. The foundation of the TDD strategy rests on the experimental studies that link the specific molecular target to the in vivo model or disease condition, a process called target validation (TV). TV is discussed in more depth below.
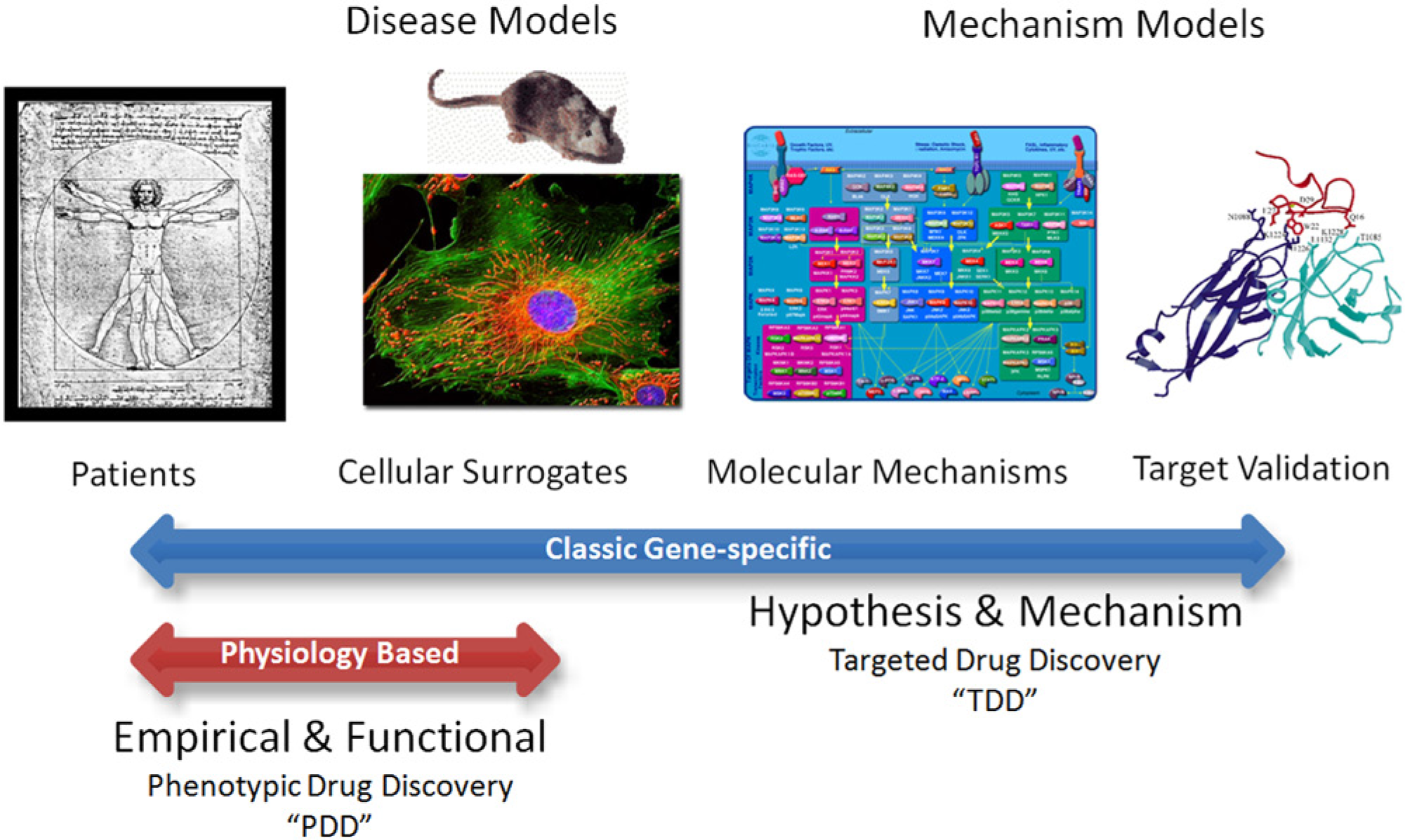
Drug discovery strategies are based on target mechanism or biological function. Most current drug discovery strategies are directed toward specific molecular targets (targeted drug discovery [TDD]) and use a hypothesis-driven, mechanism-based approach where linkage of the molecular target to in vivo biology (target validation) is foundational. In contrast, drug discovery strategies based on biological function and/or phenotype (phenotypic drug discovery [PDD]) enable interrogation of relevant signaling pathways and molecular targets in an agnostic and empirical manner. Such physiology-based PDD screens mitigate target validation risk and have contributed more first-in-class small-molecule therapeutics than TDD. 4
Prior to the widespread use of molecular biology in drug discovery, TDD approaches were less numerous because they relied on protein purification from natural sources. As an alternative, physiology/pharmacology-based readouts such as tissue-based muscle contractility or in vivo–based behavioral studies guided drug discovery. In contrast to TDD, functional assay end points do not usually measure the activity of a specific molecular target but instead assess the effect of compounds on an integrated physiological response at a systems level with minimal assumptions concerning the molecular mechanism of the overall biology.
The term phenotypic drug discovery or PDD has been recently used to describe drug discovery based on functional biology for primary screening. PDD and TDD are complementary approaches to drug discovery, each with their inherent strengths and weaknesses. The major feature that differentiates TDD from PDD is the requirement for unambiguous identification of a relevant molecular target for the former while the latter uses a target agnostic approach. While phenotypic assays are frequently used as follow-up assays in TDD flow schemes, the term PDD as used here is restricted for drug discovery programs that are target agnostic.
The Business Case for Phenotypic Drug Discovery
In recent years, the readership of this journal has been directly or indirectly affected by the turmoil and instability within the pharma and biotechnology sectors. The reasons for these issues are many and, with the exception of research productivity, are beyond the scope of this discussion. Figure 2 is a summary of the gross productivity of the pharmaceutical industry. The total number of NMEs (biologics and small molecules) approved by the Food and Drug Administration (FDA) has shown a slow, consistent growth since 1970, with the exception of the period around 1997 6 and possibly 2012–2013 ( Fig. 2 ). The apparent spike in NMEs in the mid-1990s has been attributed to U.S. passage of the Prescription Drug User Fee Act (PDUFA), resulting in increased numbers of FDA examiners to facilitate the registration of backlogged FDA submissions. 6 Although the use of molecular biology has facilitated the launch of bioproducts (recombinant protein factors and therapeutic antibodies), 6 the overall production of total NMEs has not been increased by recombinant DNA technology introduced in the mid to late 1980s and the availability of new molecular targets identified by deep sequencing and mining of Expressed Sequence Tagged (cDNA) libraries 12 or the draft human genome13,14 ( Fig. 2 ). Additional metrics, such as the number of novel molecular targets modulated by launched drugs from the early 1980s to mid-2000s (roughly five per year), are consistent with the notion that genomics has had a minimal impact on overall pharma innovation.8,9
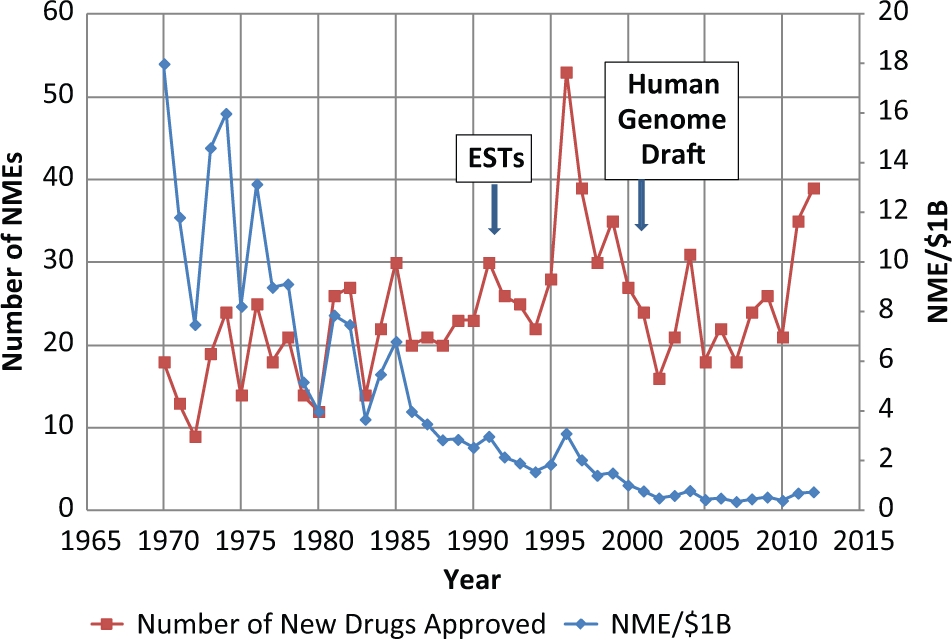
Productivity of the pharmaceutical industry. The total number of new molecular entities (NMEs; biologics and small molecules) approved by the Food and Drug Administration (FDA) is shown (red). With the exception of an apparent spike in NMEs in the mid-1990s, attributed to the registration of backlogged FDA submissions, 6 NME production has remained relatively flat and not enhanced by integration of recombinant DNA technologies in the mid-1980s and the availability of new molecular targets identified by deep sequencing and mining of Expressed Sequence Tagged (EST) (cDNA) libraries 12 or the draft human genome.13,14 The number of NMEs launched per billion dollar of investment in the pharmaceutical sector (blue). Data from FDA and PHRma, courtesy of Ellen Berg and Bernard Munos.
The clinical success of TDD rests on successful TV, the linkage of a specific drug target molecule to in vivo biology. Although molecular biologists in the early 1990s considered TV an actionable hurdle, contemporary analysis indicates that most published TV studies suffer from experimental reproducibility issues to such a degree that the majority of projects require termination.15,16 In addition, analysis of clinical studies indicates that lack of drug efficacy, the successful modulation of the disease by the therapeutic agent, is the most frequent cause of phase 3 clinical failures. 17 Although the increased availability of novel molecular targets by the genomics revolution has not resulted in increased numbers of new molecular targets for launched drugs8,9 or the overall production rate of NMEs 6 ( Fig. 2 ), the precise reasons for this disconnect are unknown. The drug discovery and development process is long and complex, with many factors that may potentially contribute to this disconnect. However, among the various potential factors, lack of drug efficacy as the principal failure mode of clinical trials 17 suggests, but does not prove, that TV issues are a significant contributing factor. Taken together, these studies highlight the difficulty in validating a molecular target for drug discovery.
These difficulties related to TV may contribute to the proliferation of “Me Too” drugs and redundant drug discovery efforts, as illustrated by a recent Forbes magazine article titled “Cancer Drug Targets: The March of the Lemmings” (June 7, 2012). This study analyzed approximately 1000 oncology drug discovery efforts from across the industry and found that >20% of research programs focused on eight molecular targets, each of which had >24 ongoing clinical stage efforts. 18 Analysis of the Thomson Pharma database for molecular targets associated with metabolic disorder and cardiovascular, neurological, and endocrine indications reveals a similar scenario: multiple discovery programs focusing on a few targets where at least an equal number of clinical efforts are ongoing and frequently marketed drugs already exist (JAL, personal observations). This “Me Too” drug discovery mentality may reflect the experimental difficulties associated with TV. As a result, resources are focused on a small but highly validated set of molecular targets where short-term biologic and chemical risk is lower but long-term risk of product differentiation skyrockets. 18 The difficulty of TV is widely recognized and has led to proposals such as conducting TV studies as a precompetitive pharma consortium. 19
In contrast to TDD, where identification and validation of specific molecular targets are foundational, drug discovery strategies that use functional, physiologically relevant end points enable direct chemical interrogation of biological systems in a target agnostic manner. One would expect functional biology approaches such as PDD to mitigate TV risk, be more likely to identify novel molecular targets, and enhance innovation in drug discovery.
In a landmark article (which is updated in this issue 3 ), Swinney and Anthony 4 examined how “first-in-class” and “follower” drugs were discovered from 1999 to 2008. For first-in-class small-molecule drugs, PDD and TDD contributed 28 and 17 NMEs, respectively, whereas for small-molecule follower drugs, PDD and TDD contributed 30 and 83 NMEs, respectively. 4 Perhaps more significant was the rate of first-in-class small-molecule drug launches; PDD exhibited a higher overall rate than TDD, which was roughly linear from 1999 to 2008. 4 These results are particularly significant considering that <10% of pharma lead generation efforts during this period used PDD approaches, suggesting that the overall launch rate for first-in-class drugs underestimates the intrinsic probability of technical success (pTS) of PDD. 4
Taken together, the business case for using a modern PDD strategy comes into focus. Functional drug discovery approaches such as PDD directly interrogate physiologically relevant biology in a molecular target and signaling pathway agnostic manner. These features of PDD are expected to mitigate TV risk, enhance drug discovery innovation, and decrease redundant “Me Too” drug efforts. These expectations are consistent with PDD contributing 34 first-in-class small-molecule NMEs between 1999 and 2012 3 and advancing several molecules to clinical phase trials for indications related to inflammation, oncology, neuroprotection, and antivirals.20–23
It is important to note that these attributes of functional drug discovery were not apparent to most scientists in the mid-1980s. During this period, a convergence of genomics and high-throughput technologies promised to revolutionize the manner in which drug discovery was practiced. Integration of molecular biology into pharma processes provided the ability to purify any recombinant protein, advances in laboratory automation and high-throughput screening provided the capability to test unprecedented number of compounds, and the development of parallel synthesis chemistry promised access to new chemical space. These breakthrough technologies, in conjunction with the identification of novel molecular targets from exon and genome sequencing,12–14 foreshadowed a revolution in drug discovery. 24 The exponential increase in the cost of drug launches before the mid-1980s ( Fig. 2 ) drove the business case, advances in cell biology and molecular biology provided the foundation for a molecular-centric view of biology, and broad scientific successes in molecular approaches reinforced reductionist viewpoints. The molecular paradigm for drug discovery made a lot of sense; we embraced it, excluded other approaches, and evolved a molecular-centric strategy. 24 Clearly, molecular approaches have enabled advances in the identification, modification, and production of protein therapeutics and contributed to small-molecule TDD successes such as kinase inhibitors Sutent, Nexavar, Gleevec, and Iressa for oncology. However, few drug hunters anticipated the difficulties associated with TV studies, ranging from the lack of reproducibility of published discovery phase experiments15,16 to late-stage clinical failures attributed to poor target validation and the lack of predictive biomarkers. 17 The phrase “hindsight is always twenty-twenty,” attributed to film director Billy Wilder, is appropriate. From the perspective of the 21st century, where the emergent properties of signal transduction networks,25–27 poly-pharmacology,9,11 multitargeted drug discovery,28,29 and systems biology 30 are contemporary discussion topics and issues with TV studies are recognized,15–17 molecular-centric approaches such as TDD and the “one drug, one target” paradigm should be reevaluated. The pendulum of drug discovery that swung from physiology-based systems of the pre-molecular era to the promise of a genomics revolution in the early 1990s is now positioned for a return. Perhaps it is time to establish a middle ground that fully leverages the complementary natures of PDD and TDD and provides a more integrative approach.
Phenotypic Drug Discovery: A New Pharma Movement
The dominant role of PDD in the development of first-in-class small-molecule drugs3,4 is somewhat surprising considering that most lead generation projects conducted in pharmaceutical research have been TDD based since the early 1990s. In contrast, academic researchers have successfully used PDD to identify tool compounds and elucidate novel molecular details involved in cell cycle, cell migration, cytokinesis, metastasis, tissue regeneration and stem cell induction, renewal, and differentiation.31–39 These academic studies underline the advantages of PDD, simultaneous interrogation of multiple targets/signaling pathways in a physiologically relevant context, the potential for identifying compounds acting through poly-pharmacology mechanisms, and the use of tool compounds to identify new biology and molecular targets. However, these academic studies did not address perceived issues of PDD related to drug discovery such as poor assay performance, difficulty in determining compound structure-activity relationships (SARs), uncertain applicability of chemoinformatics, and the difficulty/requirement for elucidating a molecular target.
Some of these tactical issues were addressed in a recent study where a medium-throughput primary screen and hit expansion follow-up were conducted using a co-culture angiogenesis assay with primary human cells. 1 This screen identified structurally diverse compounds that inhibit endothelial cord formation through a variety of target classes, including kinases (highly validated in angiogenesis) and molecular targets not typically associated with angiogenesis such as G protein–coupled receptors (GPCRs), phosphodiesterases (PDEs), and nuclear hormone receptors (NHRs). Interestingly, most confirmed actives and their close structural analogues were not known to modulate activity in any biochemical assay tested. Following a successful structure-based hit expansion, compounds were identified that were structurally and mechanistically distinct from standards of care (SOC), potently inhibited endothelial cord formation in vitro, and inhibited angiogenesis in vivo. 1 These results indicate that modern phenotypic assays can interrogate complex biology in a target agnostic manner, be statistically robust, support compound SAR, and readily identify active compounds that are structurally/biologically diverse. Therefore, at the tactical level, modern phenotypic approaches are neoclassic drug discovery strategies compatible with current pharmaceutical discovery processes. 1
The implementation of PDD in biopharma brings with it additional specific issues and concerns, and there has been much discussion over the pros and cons of this approach. Many of these issues have been the topic of discussions, and indeed the article by Swinney and Anthony 4 initiated multiple conversations at SLAS2012. This interest led to the establishment of the SLAS PDD SIG and the associated SLAS PDD LinkedIn subgroup in May 2012. 40
SLAS Phenotypic Drug Discovery Special Interest Group
The SLAS PDD SIG currently (August 2013) has more than 780 members and serves as a discussion forum for the global research community to share, discuss, and debate topics related to PDD research. Participants have shared their experiences, perceptions, and thoughts on the advantages and disadvantages of PDD and whether/how PDD complements TDD strategies. Related topics such as target identification/validation, chemical diversity, chemical genomics, chemoinformatics, poly-pharmacology, translational pharmacology, biological model systems, and patient tailoring are also in scope. The SLAS PDD SIG held its first in-person meeting at the SLAS2013 (the Society for Laboratory Automation and Screening Annual Conference and Exhibition, which was held January 12–16, 2013, in Orlando, FL). Seventy individuals from pharma, biotech, and academia participated in the energetic discussions.
Among the advantages discussed by the group were the use of biologically relevant systems to monitor physiologically relevant readouts that are closer to in vivo and clinical end points. Indeed, implied in the definition of PDD is the use of a biological system that mimics in vivo/disease-relevant biology as close as practical. This includes model organisms, tissues and tissue mimetics, cell culture in 2D, 3D, and co-culture using primary cells, stem cells and derived cell lineages, progenitor cells, and cell lines. Use of recombinant cell lines overexpressing specific molecular targets such that the signal is principally related to the expressed protein was not considered phenotypic, although cell lines expressing reporter gene constructs and/or a factor that alters the differentiation or functional state of the transfected cells is a gray area leaning toward phenotypic because these systems may interface with multiple signaling pathways composed of known and unknown molecular components. Other advantages of PDD include the ability to interrogate molecular targets in a native context, direct chemical testing of functional biology in a target agnostic manner, consideration of the cellular microenvironment and multiple cell processes signal transduction pathways, and the opportunity to select compounds with poly-pharmacology. However, simulating complex biological and physiological processes in vitro comes with a price. Not infrequently, appropriate cellular models do not exist or existing in vitro models have not been correlated with in vivo models. In addition, complex cellular assays used for PDD are more difficult to enable, are typically more expensive to run, have longer operational cycle times, and have lower capacity than their biochemical counterparts. Therefore, when strong TV exists in an area where differentiation from competitor compounds or SOC is not an issue, TDD is the more efficient path forward. In contrast, in situations where TDD has previously failed, TV is poorly understood, complex biology is suspected, or differentiation from competitor compounds or SOC is imperative, the target agnostic PDD approach may be more advantageous. Moreover, use of a complex biological system does not guarantee better translation to animal or clinical models; it is therefore imperative that phenotypic actives be tested in vivo to establish a qualitative in vitro–in vivo correspondence.
Discussions on the broader use of PDD in pharmaceutical research by the SLAS PDD SIG members and other researchers have included topics related to the targets of phenotypic actives such as the necessity of molecular target identification, methods for target deconvolution, operational considerations of running PDD programs, and institutional and organizational hurdles related to the broader adoption of PDD.
Targets of Phenotypic Drugs
There has been some misunderstanding by many researchers as to the requirement by the FDA for knowledge of the molecular target of a new therapeutic. Guidance from the Center for Drug Evaluation and Research and the Center for Biologics Evaluation and Research at the U.S. FDA indicates that identification of the molecular target for a drug is not required for initiation of clinical trials or approval. 41 In accordance with this guidance, the molecular targets for cyclosporine, FK 506, rapamycin, fibrates, thiazolidinedione, and various classes of Ca2+ channel blockers were identified only following drug launch (see reviews23,42), and indeed, 8% of the FDA small-molecule drugs approved between 2000 and 2012 had unknown mechanism of action (MOA). 7 A recent Nature manuscript by Miller et al. 43 regarding the MOA of biguanides such as metformin, the first-line medication for diabetes introduced in the United Kingdom in 1958, highlights how a safe and efficacious drug may be used for decades with few details concerning the identity of the molecular drug target(s).
Despite this, researchers are in agreement that additional knowledge concerning the molecular targets, signaling pathways, and cellular mechanisms modulated by a phenotypic active is important to discovery programs. It has been suggested that phenotypic actives are best optimized using biochemical assays,44–46 implying that target identification is required for compound lead optimization. Target identification is certainly preferred by many medicinal chemists as it enables structure-guided SAR determinations. In addition, there is bias that likely originates from the notion that phenotypic/functional assays have inherent quality issues and are not statistically robust. This limitation was discussed at the SLAS PDD SIG meeting at SLAS2013, but approximately 12 participants reported that they have successfully supported compound SAR with phenotypic assays. Our experience with enablement and operations of a variety of phenotypic assays is consistent with the experience of the SLAS PDD SIG members. Although phenotypic assays are initially more difficult to enable and validate, they can be operationally robust and pass statistical validation metrics defined for biochemical assays, 47 be as responsive to compound structural changes as biochemical or cellular substrate modification assays, 48 and be sensitive to stereochemical changes at compound chiral centers. 1 This is not to say that activity cliffs cannot occur and, as discussed by the SLAS PDD SIG members, may be more frequently observed in phenotypic than in biochemical assays since additional potential factors such as cell permeability, metabolism, and cellular efflux may obscure a PDD SAR.
Target identification is also frequently considered crucial to support medicinal chemistry efforts aimed at mitigating compound toxicity, a view potentially related to the perceived limitations of phenotypic assays to support general compound SAR (above). Compound toxicity effects can be mediated by the molecular target of interest, other molecular targets, or less specific mechanisms. In PDD, without a known target, it is unclear which if any of these three mechanisms dominate. Among the online SLAS PDD SIG members who participated in online discussions about this, there was agreement that if the “on target” or “on phenotype” SAR could not be differentiated from the SAR observed for toxicity, it was very unlikely that the compound series would be continued. Similar to compounds derived from TDD, compound series showing differences between “on” phenotype/target activity and toxicity SAR provides a potential path forward for lead optimization, provided that the functional assay is statistically validated and robust. Thus, target identification is not a prerequisite for advancing a compound into surrogate or early stage in vivo toxicity studies, with the caveat that results from animal model studies do not necessarily translate to clinical studies in general.
In contrast, target identification is needed to overexpress, crystallize, and obtain 3D structural information by X-ray crystallography. Many contemporary medicinal chemists frequently rely on structured guided SAR (see molecular mind-set below), but it should be noted that compound SAR has been conducted and drugs developed for GPCRs long before the 3D structure determination49 –51 of rhodopsin or the β2-adrenergic receptor.
The topic of target identification (TI) remains an important issue for PDD researchers. TI has been the subject of multiple articles,52–56 and there is continuing interest by SLAS PDD SIG members in TI methods used, best practices, timing of TI efforts, and pTS of TI. Furthermore, in contrast to the pharmacology-dominated approaches in drug discovery prior to the genomics era, we now have the tools and methods for more rapid assessments of MOA.
In discussions at the SLAS PDD SIG meeting at SLAS2013, Edward Ainscow (Genomics Institute of the Novartis Research Foundation) estimated that in his experience, 65% of phenotypic actives could be associated with a signal transduction pathway/cellular process, an important step toward identifying a molecular target. Regarding the use of tool compounds to identify potential molecular targets of PDD actives, Fabian Vincent (Pfizer) described the assembly of a “chemical genomics cassette” composed of 6000 compounds representing various chemotypes with optimal target selectivity and cell permeability that modulates approximately 1200 mechanisms. Scientists from GlaxoSmithKline, Pasteur Institute, and Eli Lilly & Company reported the assembly of similar compound collections within their organizations. The utility of such collections for characterization of phenotypic systems, formulation of project flow scheme, development of SOC differentiation strategies, and serendipitous lead generation and target identification was discussed. Examples of the serendipitous identification of molecular targets previously not associated with a biological function are numerous (Tina Garayantes, Chromocell). Published examples include findings that inhibitors of acetyl CoA carboxylase, functional β-secretase, and HMG-CoA reductase inhibit endothelial cord formation.1,57
Regarding the use of tool compounds, results should be interpreted with caution (John Moffat, Genentech; ELB and JAL, personal observations). Target selectivity and compound efficacy need to be considered, and biologists should acquire an increased appreciation of the poor selectivity of many “tool” compounds. If care is taken to collect a panel of selective tool compounds and test these at appropriate concentrations, these tools can be very helpful for characterizing phenotypic assays, such that known mechanisms for hit compounds can be quickly identified. Demonstrated SAR correlations between phenotypic and target activities within a compound series and ideally with additional, structurally unrelated compound series 1 increase the confidence of phenotype-target correlations.
Another approach for target deconvolution was illustrated in an SLAS2013 podium presentation by Veronica Solvenka (Institute Pasteur, Korea) that described combining genome-wide siRNA target knockdown with high-throughput compound dose-response data to identify molecular targets that sensitize the functional response to phenotypic actives, a method used successfully by Richard Wobbe (personal communication 2012), among others. 58 Label-free proteomics methods based on the elution pattern of a compound following target engagement within cellular lysates following chromatography 59 or capillary electrophoresis 60 were highlighted by Alfred Ajami (DCAM Pharma) and Wobbe. Combined use of metabolome and primary human cell profiling was also useful for identifying undesirable cellular processes associated with a phenotypic active identified in an angiogenesis assay (JAL, unpublished data).
Affinity capture proteomics appears to have the widest potential applicability for TI of phenotypic actives.52–56 In general terms, this method relies on the capture and enrichment of the molecular target by a derivative of the phenotypic active suitable for affinity capture, enrichment of the proteins specifically bound to a matrix, and identification of bound proteins by mass spectroscopy–based methodologies. Although straightforward in principle, the experimental devil is in the details. Challenges with these methods include identification of a phenotypic active with compatible SAR, synthesis of an affinity derivative that maintains high potency in the functional assay, performance of numerous control studies addressing capture of nonspecific proteins, development of non-denaturing isolation conditions without a binding assay to monitor target engagement, protein enrichment under nonequilibrium conditions, and consideration of a target-affinity ligand dissociation rate. 54 Successful studies using the technology have been published (see reviews52,53,55,56), but it is less clear how frequently this methodology fails and does not lead to publication due to the otherwise “phenomenological” nature of the observations. Moreover, a gray zone for success is apparent; SLAS PDD SIG participants have relayed examples where the TI exercise worked but the molecular targets identified were not consistent with known literature, totally unexpected and so were not pursued (more on our molecular mind-set later), or could not be validated. In the online SLAS PDD SIG forum, a study 61 that estimated the success of various methods to detect protein-protein interactions was discussed. These include affinity-based methodologies that typically use signal amplification and involve fewer steps following complex formation than ligand-based affinity purification approaches. This study estimates that various protein-protein interaction methods capture between 22% and 35% of the total protein interactions, 61 arguably an upper limit estimate for affinity purification methodology. An informed estimate of the overall pTS of affinity capture proteomics is not clear (at least to SLAS PDD SIG members), although participants have articulated gut feelings indicating a low likelihood of success. Researchers running the proteomics platform of the Broad Institute have written “the problem of target identification will not generally be solved by a single method but rather by analytical integration of multiple, complementary approaches.” 55 Obviously, this is a crucial topic for further discussion and research.
There is also the question of whether, under contemporary market pressures, could/would a company launch a drug without knowing an MOA or molecular target. In part, this question is addressed by the recent analysis by Munos, 7 who found that 8% of the FDA small-molecule drugs approved between 2000 and 2012 have unknown MOAs. There was broad agreement in the SLAS PDD SIG online discussion forums that more MOA/molecular target information was better than less, and it was acknowledged that therapeutic area-specific considerations such as possible limitations of existing standard of care, impact of personalized medicine, target validation status, strength of genotype/phenotype linkage, and research portfolio/full-time equivalent (FTE) balance are contributing factors. Marcie Glicksman (Harvard NeuroDiscovery Center) reported less interest from external in licensing groups for PDD leads that do not have an identified target relative to TDD leads given similar in vivo biology efficacy studies. In contrast, metformin has been the diabetes SOC for more than five decades, with few details concerning the identity of the molecular drug target(s). 43
There are also a variety of opinions as to when to start looking for the molecular target or mechanism of a phenotypic active. Some researchers advocate for early engagement following identification of a promising cellular active, and others delay this effort until in vivo efficacy has been established. In part, this may be a reflection on how different scientists define identification of MOA and molecular targets. MOA can be associated with a signal transduction pathway/cellular process by broad and systematic profiling of compound libraries in multiple cellular pathway assays (Edward Ainscow, Genomics Institute of the Novartis Research Foundation and ELB). In contrast, use of genome-wide profiling and affinity chemoproteomics approaches to identify molecular targets of phenotypic actives probably has a higher initial investment and more uncertain outcome. These projects will result in a later “go” decision, perhaps not until a correlation between in vitro and in vivo activity has been established. Most researchers support overall parallel activities for MOA/target identification and therapeutic agent development, and indeed, this has been enabled by new multiparametric or so-called omics profiling technologies. There did not seem to be an example where PDD was used for TI and the resulting gene-specific assay provided the groundwork for a therapeutic entity. This notion is consistent with the historical superiority of PDD for identifying first-in-class therapeutics, 4 the narrow time window (<2 years) for commercially viable fast follower drugs, 62 and the average and median time between first- and second-in-class drugs (2.8 and 2.0 years, respectively) launched between 2000 and 2012. 7
There is also the question as to whether drugs with a single, molecular drug target exist at all. Mestres et al. 11 mined public databases to estimate the selectivity of drugs approved by the FDA and found on average that known drugs interact with an average of six molecular targets. Notably, this is probably a lower estimate because drugs and tool compounds are not systematically tested in all assays, and the estimated druggable genome is less than 10% of the proteins encoded by the human genome.8,9,13,14 It is currently not clear whether all the molecular targets identified by this analysis are pharmacologically relevant. However, given the known complexity of drug target9,11 and signaling30,63 networks and the error tolerance of complex networks,25,26,64 drug targets may be less likely to be a single gene product than a collection of molecular targets that modulates an integrated signaling network or biological system. This concept is consistent with the increased interest and historical effectiveness of multitargeted drug discovery (MTDD), where activity on multiple molecular targets is desired.28,29,65 In principle, the target agnostic approach used by PDD may more efficiently identify compounds working through MTDD mechanisms, but operationally, chemists on PDD teams should realize the difficulties in physiochemical properties and SAR that are associated with MTDD approaches with known targets.28,66 Perhaps we incorrectly attribute the pharmacological activity of many drugs to a specific molecular target when poly-pharmacology may be more realistic.
How Is PDD Being Used?
Although we would like to think that there has been an optimal derivation of best practices or best processes concerning PDD, the reality seems less clear-cut. PDD approaches have been used by scientists in academia, biotech, and the pharmaceutical industry and broadly include discovery of new biological mechanisms, identification of novel molecular targets, and identification of novel therapeutic agents. Ann Carpenter (Broad Institute) pointed out that academic PDD screens using small collections of bioactive molecules often identify targets of interest. Analysis of the small-molecule, nonnatural product NMEs approved between 1999 and 2008 indicates that 37% and 62% of the total and first-in-class small-molecule NMEs were derived from PDD, respectively. 4 Marcie Glicksman (Harvard NeuroDiscovery Center) has used PDD approaches in academia and biotech and has noted that “the climate for phenotypic assays has been more acceptable than, say, 20 years ago.” Informal conversations with scientists from various academic and biotech organizations indicate that 50% to 100% of projects are phenotypic, whereas pharmaceutical companies, formerly the domain of TDD, are trending toward an increased number of PDD projects, from <10% to some as high as ~20% of lead generation activities. Tina Garayantes (Chromocell) shared her experience in running phenotypic and biochemical screens in parallel. She indicated that double actives from a dual PDD-TDD screen can identify active compounds distinct from the biochemical assay in isolation. As a result, the SAR trajectory is changed, and compounds active in primary cell tertiary assay and animal studies are identified earlier. Despite these examples, institutional acceptance of PDD in pharma has been challenging.
Why is PDD accepted more in academia than in the pharmaceutical industry? Dhara Patel (Washington University) indicated that her research group realized that they did not understand enough of the underlying biology to make critical mechanistic assumptions and chose a phenotypic strategy before being aware of the buzzwords PDD and TDD. This scenario illustrates key differences between academic and industrial research environments; academia tends to be more risk tolerant, with direct or localized accountability, and uses fewer research processes/timelines; generally, academic teams tend to be more directly empowered to make science-driven decisions. Academic research is also better suited for biology-first approaches, where tenure of experience in a given area of biology can be much longer (10+ years) than in pharmaceutical research, where companies shift therapeutic areas of focus every 3 to 5 years. There are also multiple rationales for conducting PDD research, and objectives are expected to vary between research environments with dissimilar missions. In academia, a small screen may be sufficient to identify new compound activities or interesting new target biology or to formulate a novel hypothesis worthy of publication or providing data for grant funding. PDD in the biotech and pharmaceutical sectors has a longer term mission—making novel, safe, and effective medicines. These distinct missions change the entire complexion of risk, governance, and process within these distinct research environments. As discussed in the SLAS PDD SIG, one also needs to consider differences in the principal goals and reward systems in operation within the very diverse environments encompassing an academic, nonprofit research institute and for-profit drug discovery research. Robert Ames (GlaxoSmithKline) pointed out that PDD flow schemes tend to be more complicated and nonlinear than TDD projects. This creates concern in industry because of increased direct/indirect costs and resulting difficulties in strict adherence to project timelines and milestones, concerns that are probably less important to academic and nonprofit research efforts.
One of the largest hurdles limiting broader implementation of PDD in the pharmaceutical industry is related to mind-set and risk perception rather than technical and scientific issues. For the most part, contemporary biological sciences are viewed from a molecular mind-set. Nearly all practicing biologists and chemists have been trained in an era where many molecular targets have been identified and are readily accessible on a genome-wide scale, the structure of protein targets is frequently available to influence compound SAR, cell biology and pharmacology are frequently viewed as elegant mathematical models, and successful discovery of novel molecular mechanisms is rewarded by funded grants, publication in high-profile journals, academic praise, and the shear satisfaction of “understanding” biology at such a level. In addition, molecular-targeted approaches to drug discovery are more aligned with the hypothesis-driven, deductive methodology that is widely but incorrectly considered to be the primary means in which scientific progress is thought to occur. 67 It is difficult to break away from our training and significantly change the intellectual framework or molecular mind-set in which we conceptualize science.
In addition to this intellectual inertia, the availability of breakthrough capabilities from technology advances in laboratory automation, biology, and chemistry in the late 1980s and the availability of novel targets from exon/genome mining promised to transform drug discovery, 24 a concept that readily gained acceptance given the exponential cost increase in developing drugs during the mid-1980s ( Fig. 2 ). From a process perspective, Frank Sams-Dodd has suggested that the rapid uptake and popularity of gene-specific strategies in the pharmaceutical industry may also be related to the apparent linearity and simplicity of TDD, which portrays drug discovery as a systematic, rationale, and scalable endeavor. Such a narrative is compatible with organizational structures seeking to incorporate defined processes and quantified metrics and is also appealing to investors, shareholders, and managers because of the promise of a fast return on investment. 2
Thus, molecular-targeted drug discovery approaches are appealing from an intellectual, science/technology, process, and business perspective. With such a confluence of rationales and the promise of a genomic revolution, a molecular mind-set with regard to life science research was inevitable. However, given the unexpected success of phenotypic approaches for the identification of first-in-class drugs,3,4 it may be time to challenge our reliance on this mind-set and reestablish a biology-first, neoclassic drug discovery paradigm in which the molecular mind-set is integrated with but positioned in service to phenotype.
The JBS Phenotypic Drug Discovery Special Issue
Thirty-five manuscripts emerged from the 95 abstracts submitted in response to the call for papers. In addition to this perspective, three other articles provide perspectives on relevant issues. Swinney 3 summarizes the role of mechanistic information on the discovery of first-in-class NMEs by PDD. Young 23 provides a retrospective on the identification and subsequent pharma impact of identifying p38 MAP kinase as the molecular target of cell-active inhibitors of cytokine secretion. Patel 68 discusses the relevance of target identity and mechanism of drug action from the perspective of pharma customers, clinicians, and patients. Two articles describe the use of PDD to investigate neurodegenerative disorders; Zhang 69 provides an overview, whereas Figuera-Losada 70 reviews the role of activated microglia. Harpreet et al. 71 used a phenotypic approach to identify an antimalarial drug (DHA) that attenuates BCR-ABL activity under kinase inhibitor (dasatinib) resistant conditions; a potential for drug repurposing is indicated by reduced primary leukemia burden and improved long-term survival in a murine model following cotreatment of DHA and dasatinib.
Of course, the day job of most SLAS members focuses on technologies/methodologies related to the enablement and operations of in vitro assays. Therapeutic areas covered by this JBS special issue include infectious disease,72,73 cardiovascular and metabolic disease,74–77 CNS,69,70,78 –80 oncology and angiogenesis,81–86 allergy, 87 and renal disease. 88 In addition, this two-part special issue covers a wide range of topics of particular interest to phenotypic drug discovery scientists. These include descriptions of novel physiologically relevant screening systems, approaches for MOA deconvolution, and novel analysis methods for phenotypic assays.
Many of the phenotypic screens described in this issue incorporate challenging but more physiologically relevant cell types and systems. Several articles describe assays using human stem cell types such as erythroid progenitors, 75 induced pluripotent stem cell–derived cardiomyocytes, 74 and neural cell types. 79 Primary human cells and co-cultures, including endothelial cells, fibroblasts, mast cells, peripheral blood mononuclear cells, mast cells, keratinocytes, smooth muscle cells, bronchial epithelial cells,82,84,87,89 and patient cells, 69 were also used in a number of the studies. Several articles describe screening in 3D systems and the comparison of 2D to 3D systems,81,83,86 as well as screening in zebrafish. 88
Articles using multiparametric analysis of data from high-content screening (HCS)82,90 or phenotypic assay panels 89 to generate predictive activity profiles are presented. Reisen 91 compares 16 methods for multiparametric feature reduction and similarity measures using a large-scale HCS data set to evaluate their performances in HCS-fingerprint analysis. Advances in analytical capabilities include real-time, single-cell Ca2+ imaging 78 and label-free quantitative interferometric microscopy. 92 The development and validation of a 1536-well compatible muscle cell triglyceride accumulation assay and the quantitative comparison using a confocal, high-content reader and conventional plate reader 76 is also noteworthy.
Steps Forward
The availability of novel molecular targets from the genomics revolution has not translated to increased number of new drugs6,18 ( Fig. 1 ) or increased innovation in the pharmaceutical industry.8,9,18 We have observed that drug discovery based on biological function and empiricism rather than molecular target approaches based on a therapeutic hypothesis has contributed to a disproportionate number of drugs in an era dominated by molecular research.3,4 The obvious, although not exclusive, factor contributing to lower pharma productivity may be related to the overdependence on a molecular target-driven drug discovery strategy and the prevalence of a molecular mind-set within contemporary business and scientific leadership.2–7 Discussions among SLAS researchers and within the SLAS PDD SIG indicate that this molecular mind-set is probably the single most influential factor that limits the use of PDD in the pharmaceutical industry. To be more successful, we need to reconsider this mind-set and better understand how PDD might be used to enhance innovation and productivity in the pharmaceutical industry. Clarity and philosophical equilibrium can only be achieved through discussion, debate, and education. The publication of this two-part JBS special issue provides a meaningful step toward this goal. SLAS webinars, scientific sessions, and special interest group activities provide additional opportunities for discussion and education. Taken together, these venues provide an important way forward to rethink the molecular mind-set that has dominated our collective thinking in pharma and to reimplement a neoclassic drug discovery paradigm based on empirical, functional biology rather than hypotheses derived from molecular targets.
Footnotes
Acknowledgements
We thank Steve Hamilton, Amy McGorry, and Lynn Valastyn for their encouragement and contributions in establishing the SLAS Phenotypic Drug Discovery Special Interest Group (which is free to join—for more information, visit ![]() ). We also are grateful to Alfred Ajami, Dhara Patel, Ray Perkins, Ken Seidenman, Richard Wobbe, Edward Ainscow, Tina Garyantes, Marcie Glicksman, Viktor Lakics, John Moffat, Anand Patel, Fabien Vincent, John Westwick, and other SLAS Phenotypic Drug Discovery Special Interest Group participants who keep the online discussions lively and moving forward with such vigor. Finally, we are indebted to JBS Editor-in-Chief Bob Campbell and SLAS Director of Publishing Nan Hallock for their expertise and guidance through the editing and production of these special issues of JBS.
). We also are grateful to Alfred Ajami, Dhara Patel, Ray Perkins, Ken Seidenman, Richard Wobbe, Edward Ainscow, Tina Garyantes, Marcie Glicksman, Viktor Lakics, John Moffat, Anand Patel, Fabien Vincent, John Westwick, and other SLAS Phenotypic Drug Discovery Special Interest Group participants who keep the online discussions lively and moving forward with such vigor. Finally, we are indebted to JBS Editor-in-Chief Bob Campbell and SLAS Director of Publishing Nan Hallock for their expertise and guidance through the editing and production of these special issues of JBS.
Viewpoints expressed by the authors do not necessarily reflect those of Eli Lilly or DiscoveRx.
Declaration of Conflicting Interests
ELB and JAL are employees of DiscoveRx and Eli Lilly, respectively.
Ellen Berg, PhD Scientific Director Bioseek, a division of DiscoveRx South San Francisco, CA (USA)
Jonathan A. Lee, PhD Senior Research Advisor Eli Lilly Research Labs Indianapolis, IN (USA)