
Abstract
G-protein–coupled receptors (GPCRs) still offer enormous scope for new therapeutic targets. Currently marketed agents are dominated by those with activity at aminergic receptors and yet they account for only ~10% of the family. Progress up until now with other subfamilies, notably orphans, Family A/peptide, Family A/lipid, Family B, Family C, and Family F, has been, at best, patchy. This may be attributable to the heterogeneous nature of GPCRs, their endogenous ligands, and consequently their binding sites. Our appreciation of receptor similarity has arguably been too simplistic, and screening collections have not necessarily been well suited to identifying leads in new areas. Despite the relative shortage of high-quality tool molecules in a number of cases, there is an emerging, and increasingly substantial, body of evidence associating many as yet “undrugged” receptors with a very wide range of diseases. Significant advances in our understanding of receptor pharmacology and technical advances in screening, protein X-ray crystallography, and ligand design methods are paving the way for new successes in the area. Exploitation of allosteric mechanisms; alternative signaling pathways such as G12/13, Gβγ, and β-arrestin; the discovery of “biased” ligands; and the emergence of GPCR-protein complexes as potential drug targets offer scope for new and much improved drugs.
Introduction
Historically, G-protein–coupled receptors (GPCRs) have been the most successful protein target class for drug discovery. While the estimates vary somewhat, analyses of approved drugs typically quote 30% to 50% of all medications as exerting their effect via members of this family.1–4 In light of this long track record of success, it might be tempting to speculate that many of the opportunities in this area have been exhausted. However, a review of the GPCR drug discovery landscape still reveals enormous, as yet largely untapped, opportunities. Given the exceptional precedence of successful drug discovery at the GPCR target family—in stark contrast to emergent, “new” areas—it seems essential to be aware of those opportunities and how best to exploit them.
Marketed Drugs
A fresh analysis of marketed drugs as listed in DrugBank (http://www.drugbank.ca)5–7 initially yields very similar data to those published elsewhere. Of 1448 approved small molecules, 131 approved biologics (termed biotech in the database), and 84 nutraceuticals, 437 (26%) have their activity linked to GPCRs. There are 365 nonolfactory GPCRs that are typically thought of as being potential drug targets, of which 284 are categorized as Family A/rhodopsin, 15 Family B/secretin, 33 Family/adhesion, 22 Family C/glutamate, and 11 Family F/frizzled receptors. 8 Allowing for two modalities, agonists, and antagonists, there are perhaps crudely 365 × 2 = 730 potential drug targets in the family. We might not expect every modality/target combination to have a therapeutically useful end point, so a total of 437 drugs for a group of 730 potential targets sounds, on the face of it, quite well served. However, this logic fails for two key reasons: first, the pharmacology of GPCRs is far more subtle than a single target agonist or antagonist, so the number of potential targets is far higher; second, the currently available therapeutics do not exert their effect(s) via an obligingly even distribution of different receptors.
To reveal the latter point, the DrugBank data were analyzed further. From a total of 1479 underlying targets for the action of 1663 drugs, 109 (7%) were GPCRs or GPCR related (e.g., receptor-activity modifying proteins or RAMPs). This immediately reveals an issue: 26% of drugs target GPCRs, but they account for only 7% of the underlying targets. The results are heavily skewed by certain receptors that have far more than their “fair share” of drugs. The most commonly targeted receptors are as follows: histamine H1 (77 occurrences), α1A adrenergic (73), muscarinic M1 (72), dopamine D2 (62), muscarinic M2 (60), 5HT2a (59), α2A adrenergic (56), and muscarinic M3 (55)—notably, these are all aminergic GPCRs. Even the calculation that the available drugs exert their effects via 109 GPCR or GPCR-related targets is almost certainly an overestimate since it includes a fair proportion where there are only a very small number of active agents, and they all have a pharmacological action that is “unknown”; in truth, we have probably yet to discover an agent with a compelling activity at the target in question, let alone one with exactly the right pharmacology and appropriately tuned pharmacokinetics (PK), pharmacodynamics (PD), and selectivity to give clinical efficacy for our disease of choice. A prime example of this would be the eight metabotropic (mGluR) receptors, many of which have only been “drugged” according to this analysis due to the availability of the endogenous ligand (L-glutamic acid) as an approved nutraceutical. There are also a considerable number of targets for which the only known agents are peptides, rather than small molecules. There may, of course, be some drugs that exert their effects via GPCRs, but where that activity has yet to be determined, although this is becoming increasingly less likely as known agents are broadly profiled through drug repositioning programs9–11 and given the widespread availability of panels of GPCR assays. Hence, it seems that >400 drugs exert their effect via no more than about 100 receptors, being less than 30% of those expressed in the human genome and a considerably smaller proportion of the possible target/pharmacology combinations.
Recent Food and Drug Administration Approvals
GPCRs are still a mainstay for new drugs, as seen by examination of recent regulatory approvals in the United States. There were 21, 30, and 39 new molecular entities (NMEs) approved by the Food and Drug Administration (FDA) in 2010, 2011, and 2012, respectively. Of these, 5 of 21 (2010), 5 of 30 (2011), and 7 of 39 (2012) target GPCRs, giving a total of 17 of 90 (19%) over the 3 years. Perhaps more informative than the raw numbers, though, are the activities of those NMEs and an assessment of whether they represent a new activity/target combination as opposed to an improved agent acting via a known mechanism. This is summarized in Table 1 . Also highly pertinent is whether the agent is a small molecule or peptide/biological agent.
NMEs Targeting GPCRs Approved by the FDA in 2010–2012, Their Quoted Key Receptor Activity, Compound Type, Disease Indication, and Assessment of Whether the Activity Represents a New Receptor Targeting Relative to Existing Agents Listed in DrugBank.
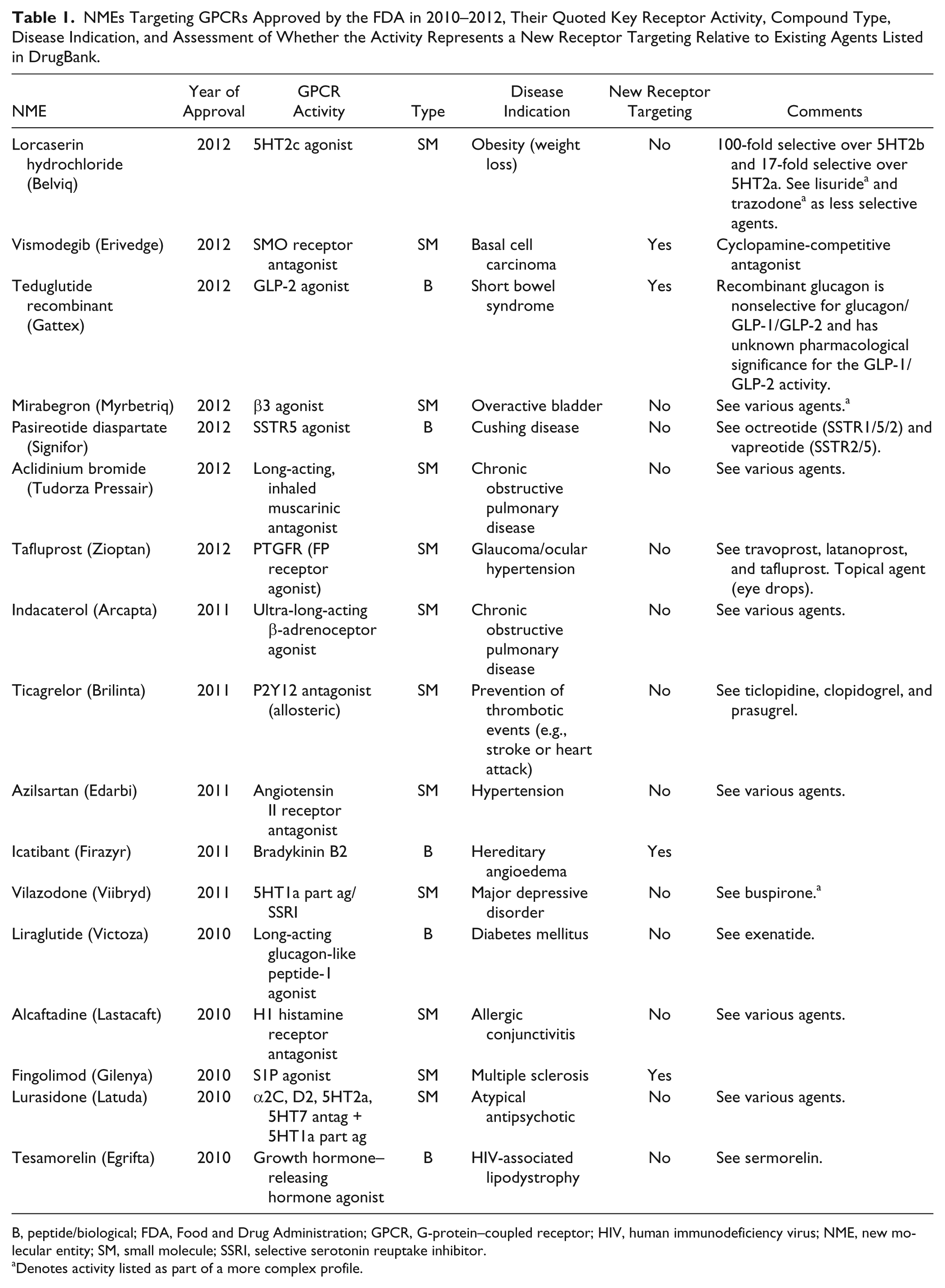
B, peptide/biological; FDA, Food and Drug Administration; GPCR, G-protein–coupled receptor; HIV, human immunodeficiency virus; NME, new molecular entity; SM, small molecule; SSRI, selective serotonin reuptake inhibitor.
Denotes activity listed as part of a more complex profile.
Within the 17 approvals, two small molecules stand out: (1) vismodegib, as the first example of a SMO-receptor antagonist used for the treatment of basal cell carcinoma (BCC) and is in clinical trials for a number of other indications, and (2) fingolimod, as the first example of an S1P receptor agonist/functional antagonist for the treatment of multiple sclerosis (MS) (see Fig. 1 ). Also noteworthy are the regulatory approvals for two peptide/biological agents: teduglutide recombinant as the first glucagon-like peptide 2 (GLP-2)–selective agonist for the treatment of short bowel syndrome and icatibant as the first selective bradykinin B2 receptor agonist for the treatment of hereditary angioedema (HAE).
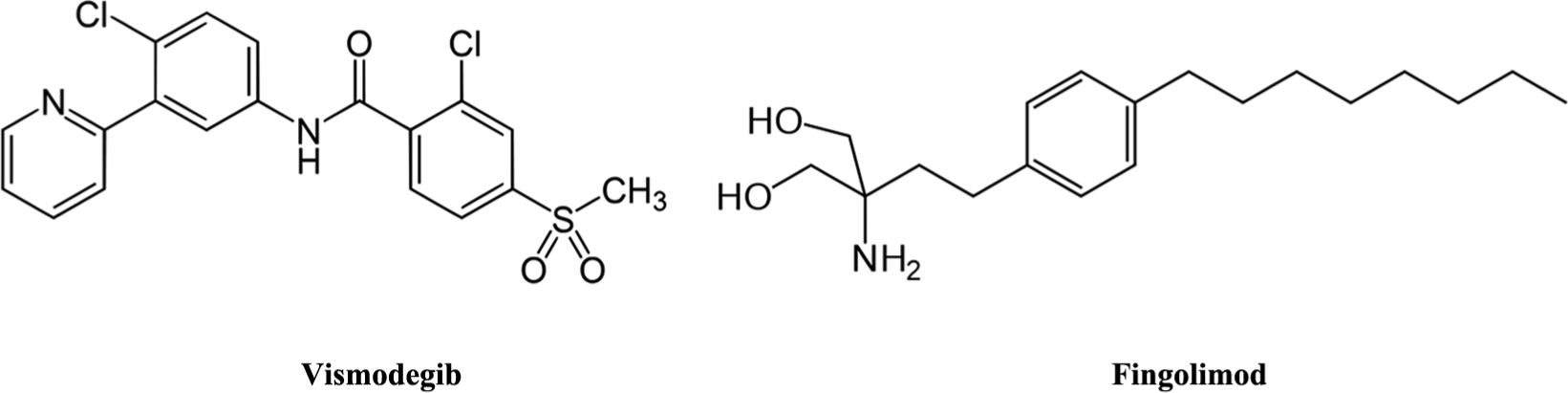
Key small molecules from recent regulatory approvals: (left) Vismodegib (Erivedge; Genentech, San Francisco, California, USA), a SMO-receptor antagonist for the treatment of basal cell carcinoma (BCC), is also in clinical trials for metastatic colorectal cancer, small-cell lung cancer, advanced stomach cancer, pancreatic cancer, medulloblastoma, and chondrosarcoma; (right) Fingolimod (Gilenya; Novartis, Basel, Switzerland), an S1P receptor agonist/functional antagonist, is an immunosuppressant used for the treatment of multiple sclerosis (MS).
In addition, there are a number of agents where improvements in the selectivity profile appear to provide benefit, including new disease indications: lorcaserin hydrochloride is a small-molecule 5HT2c agonist approved for weight loss with 100-fold and 17-fold selectivity, respectively, over the closely related 5HT2b and 5HT2a receptors; mirabegron is a selective small-molecule β3 adrenergic receptor agonist approved for the treatment of overactive bladder; and, while there are a couple of other, less selective, peptidal SSTR5 agonists available, pasireotide diaspartate has recently been approved for the treatment of Cushing disease (an orphan disease).
The presence of a number of peptide/biological agents in the approvals list seems noticeable. GPCRs are more amenable to this approach than many target classes given that they are cell surface expressed. A considerable proportion of receptors also are activated by endogenous peptide ligands, which, in turn, tend to be receptors that have proved relatively intractable to small-molecule drug discovery. Hence, there can be a considerable body of biological evidence supporting the target for a particular disease developed around peptidal tool molecules; if the issues around route of administration/enzymatic stability can be overcome, peptidal agents could be an attractive route forward. Once these moieties achieve proof of concept in the clinic, it provides a very strong rationale for the subsequent development of small-molecule (oral) medications with the same mechanism of action. Of the 437 drugs found to target GPCRs, 21 are classified as “biotech” (i.e., biopharmaceuticals) with the rest as “small molecules.” However, that definition seems rather generous given that the molecular weight (MW) of the “small molecules” extends as high as 1623. Using a fairly modest threshold of MW <600 suggests that ~387 are more truly small molecules and ~50 are non–small molecules, being roughly an 80:20 split. Pursuing the 20%, while not being novel targets/mechanisms, could still provide important new oral/small-molecule medications with the comfort of excellent existing clinical validation. For this reason, targets such as GLP-1 agonists for diabetes mellitus are perennial “hot” targets in GPCR research, although they have consistently faltered up to this point due to the difficulties in finding good lead compounds. However, a number of recent developments around improved protein structure determination, improved screening paradigms, and improved compound design approaches offer scope to crack these areas open.
Heterogeneity of the GPCR System
It is well known that the endogenous ligands of GPCRs are highly diverse, ranging from ions through amino acids, small-molecule neurotransmitters and lipophilic signaling molecules to peptides and proteins. 12 Perhaps unsurprisingly, this translates itself into a diversity of recognition mechanisms across the different receptor families and in ligand binding sites within the families. 8 Most currently known ligands are thought to act via the “orthosteric” transmembrane (TM) bundle binding site in Family A receptors. The similarities of these receptors have been assessed based on a subset of 44 residues that are thought to form the majority of the binding site. 13 The exact similarity threshold to be used in partitioning the receptors into subfamilies is subjective, but arguably the data suggest that, in addition to the very widely exploited aminergic subfamily, there are perhaps ~11 peptide subfamilies and a similar number that bind lipid moieties, together with single purine, retinal, adenosine, and melatonin subfamilies. There are also the more structurally diverse Family B/secretin, Family B/adhesion, Family C/glutamate, and Family F/frizzled receptor families.
This has important implications for drug discovery and relates directly to the success observed to date in screening: success within one receptor subfamily may not translate into improved chances of success when cross-screening compounds against other subfamilies in an effort to identify hits/leads. The tractability issue seen with certain GPCRs is arguably due to a lack of the right compounds in screening decks, despite the long history of success with the target family. That long history is largely dominated by the aminergic area and will not necessarily translate into success for other targets. Indeed, success within “the” peptide or lipid family will not necessarily translate to success at other peptide or lipid receptors; it depends rather strongly on which of the subfamilies the ligands are active at, given differences in the binding sites. However, there are emerging techniques for exploiting the tractability in one subfamily and using it for successful lead design at other, more distant receptors: so-called chemogenomics methods. 14 While the precise details vary in different versions, the methods each exploit similarity/identity in subpockets of the binding sites to direct the design of hybrid or modified structures for new targets. Successful examples to date include MCHR1 (peptide) antagonists derived from dopamine (amine) compounds, 15 DP2 (GPR44) (lipid) antagonists from AGTR2 (peptide)/GHSR (peptide)/LTB4R (lipid) compounds, 16 SSTR5 (peptide) antagonists from histamine H1 (amine) compounds,17,18 and C3AR (peptide) antagonists from AGTR2 (peptide)/GHSR (peptide)/LTB4R (lipid) compounds. 19
With this in mind, it is important to consider the receptor subfamilies that have been successfully targeted to date. From the 109 receptors in the marketed drugs list, 36 are aminergic, 12 are Peptide #3 (CCKAR, CCKBR, EDNRA, EDNRB, GnRHR, NPSR1, NPY2R, OXTR, TACR1, V1a, V1b, and V2), 10 are Family B, 10 are Family C, 8 are Lipid #3 (Prostanoid), 6 are Peptide #5 (opioid/somatostatin), and 4 are adenosine receptors, with no more than 2 for receptors in each of the other subfamilies. The dominance of the aminergic receptor subfamily is even more pronounced considering the number of marketed drugs in each case and that many of the marketed agents in other groups are far from ideal. For example, the 10 agents targeting the 10 Family B receptors are all peptides, and of the 3 ligands targeting the 10 Family C receptors, only cinacalcet is really what we might call drug-like. Even within the Peptide #3 subfamily, only 7 agents are really drug-like (conivaptan, danazol, aprepitant, ketamine, fosaprepitant, bosentan, and sitaxentan) and are active at only 6 of the 12 receptors that have supposedly been “drugged.”
There are, therefore, substantial gaps in our success to date. This is most notable for the various peptide and lipid subfamilies. Reviewing the entire peptide receptor space is a significant task since it covers ~100 receptors plus a dozen or so orphans. We can get a feel for the issues by examining the lipid group, which is more manageable in size and highly pertinent given recent regulatory approval of fingolimod. There are 30 receptors formally classified as lipid, 1 as dual lipid/peptide and potentially as many as 20 orphan receptors, which are most likely lipid receptors based on their phylogenetic grouping. Indeed, 3 of these have fairly recently been classified as lipid receptors: GPR23 (LPA4), 20 GPR92 (LPA5), 21 and P2Y6 (LPAR6). 22 Exact partitioning will depend on the level of binding site similarity used, but broadly the lipid receptors could be placed into 11 different groups:
EDG receptors (S1P1-5, LPA1-3, plus the orphans GPR3, 6, and 12) Prostanoid receptors (TP, EP1-4, FP, DP1, IP) Cannabinoid receptors (CB1, CB2) G-protein–coupled bile acid receptor 1 (GPBAR1) Leukotriene receptors (LTB4R, LTB4R2, DP2/GPR44, GPER, GPR1) Chemokine receptor-like 1 (CMKLR1) Free fatty acid receptors (FFAR1-3, GPR42, GPR55) Orphan group (GPR4, GPR65, GPR68, GPR132) Cysteinyl leukotriene receptors (CysLT1-2) PAF group (PTAFR, EBI2, GPR23, GPR92, GPR174, P2Y5, P2Y10) OXER1 group (OXER1, GPR81, GPR109A, GPR109B, possibly also GPR31)
This diversity in receptor binding sites is perhaps to be expected given the diversity of the endogenous ligands (see Fig. 2 ), but it is important to beware of “false friends” within the receptor subclassification. For example, DP1 does not cluster with DP2 (GPR44); the binding site of the latter is more similar to the leukotriene B4 receptors (LTB4R and LTB4R2) than the prostanoids. Similarly, LPA1-3 do not cluster with newly paired LPA4-6, and GPR55, a putative cannabinoid receptor, clusters considerably more closely to the free fatty acid receptors than CB1/2, implying different recognition modes/binding sites for the same ligand at different receptors.
There is considerable structural diversity in endogenous ligands for G-protein–coupled receptors—even within a single subclass such as “lipid” receptors: (
Key approved drugs in the lipid family include “leukotriene” receptor antagonists such as montelukast, zafirlukast, and pranlukast, which are actually cysteinyl leukotriene receptor antagonists and hence a different subgroup to the leukotriene receptors (note, especially, that the drugs are used for the treatment of asthma and that the LTB4R and LTB4R2 receptors do not appear to be causally linked to that disease 23 ), and various prostanoid receptor ligands such as dinoprostone (EP1-4), epoprostenol (IP), alprostadil (EP1/2), travoprost (FP), misoprostol (EP2-4), and bimatoprost (FP, EP1/3), which are mostly naturally occurring compounds or fairly close analogues of the endogenous ligands. Similarly, there are also a couple of approved tetrahydrocannabinol (THC) derivatives that are active at CB1/CB2 receptors, dronabinol and nabilone, as well as rimonabant, a CB1 inverse agonist for the treatment of obesity, although the latter has been withdrawn due to adverse effects.
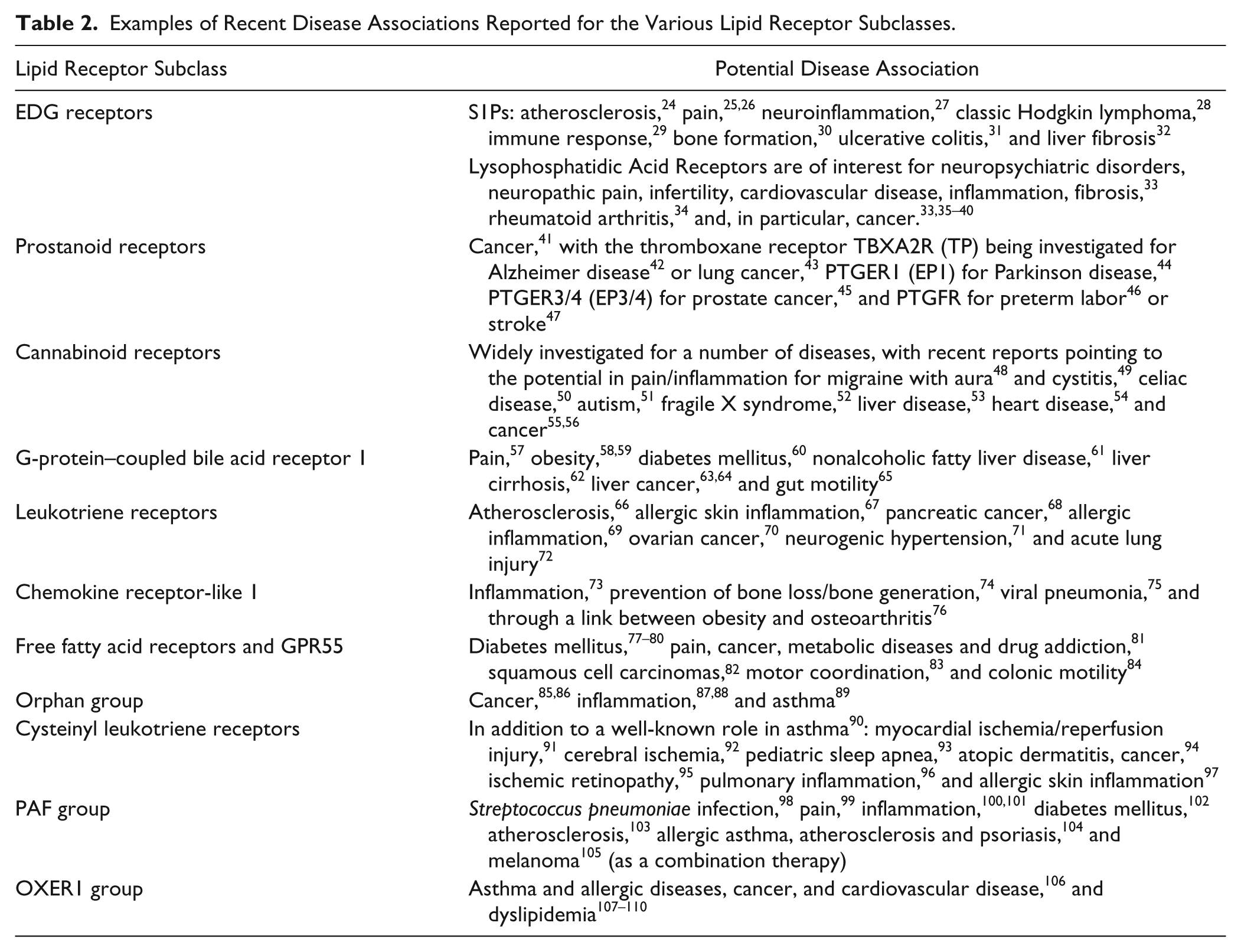
The area is ripe for substantial further discovery. While the degree of the target validation will vary enormously, a sense of the potential can be obtained from the recent literature summarized in Table 2 .24–110 The studies have been selected based on simple searching and review, with the only requirement being a clear proposed link to a specific disease as opposed to more general interest in the target per se. This list of potential disease application for compounds targeting the lipid receptors is by no means exhaustive but shows the enormous scope across a wide range of therapeutic areas. The range of opportunity for GPCRs as a whole is obviously far wider given the additional, larger peptide receptor subfamilies, as well as Family A/purine, Family B, Family C, Family F, and orphan receptors, which have been largely unaddressed up to this point. Within the list, there seems, perhaps, to have been a general increase in interest for GPCRs in cancer (e.g., S1Ps, 28 LPAs,33,35–40 prostanoids, 41 cannabinoids,55,56 GPBAR1/TGR5,63–65 leukotrienes,68,70,111 FFARs,81,82 orphan group,85,86 CysLTs, 94 PAF-R, 105 and OXER1 group 106 ). It also seems notable that progress within the different subfamilies is linked to the availability of tool molecules with which to probe the underlying biology. This is most pronounced for orphan receptors, where the lack of tool molecules extends to a lack of knowledge around the endogenous ligand itself and potentially also around receptor signaling pathway(s), severely limiting disease association work.
Examples of Recent Disease Associations Reported for the Various Lipid Receptor Subclasses.
Availability of Tool Molecules
There are a number of means by which a target can be associated with a particular disease, some of which rely on genetic and other biological data, but it is usually highly beneficial to have good-quality tool molecules available with which to probe the pharmacology in vitro and preferably achieve some initial target validation in vivo. 2 The compounds in question need to be reasonably potent and selective for their target to avoid confounding effects but do not need to be novel or necessarily have a particularly good ADME profile—these issues can be addressed in subsequent hit identification and lead optimization campaigns. There are specific initiatives—most notably the National Institutes of Health (NIH) Molecular Libraries Program—aimed at the identification of novel tool molecules which are then put into the public domain to stimulate further investigation around the targets. Also, given the long track record of research around GPCRs, there is a substantial body of literature data, so the question arises as to how much these efforts provide compounds with which to explore new targets and associate them with disease.
The output from the NIH Molecular Libraries Program is summarized in the probe reports for years 1 to 4 (2009–2012) available online. 112 Targets are nominated by academic research groups for high-throughput screening (HTS) against a 350,000 set of diverse compounds, which is then performed by one of the screening centers. Validated hits undergo initial optimization by affiliated medicinal chemistry teams to yield tools useful for in vitro experimentation, with the results placed in the public domain. Reviewing the reports shows that, of 123 target/modality combinations screened, 23 (19%) are GPCRs. These are listed in Table 3 . This reveals a broad interest across a wide range of disease areas, as well as a mix of different modalities (agonist, antagonist, inverse agonist, positive allosteric modulator, negative allosteric modulator, allosteric agonist) and receptor subclasses, including aminergic, peptidergic, lipid, amino acid, and orphan.
National Institutes of Health Probe Reports (Years 1–4, 2009–2012).
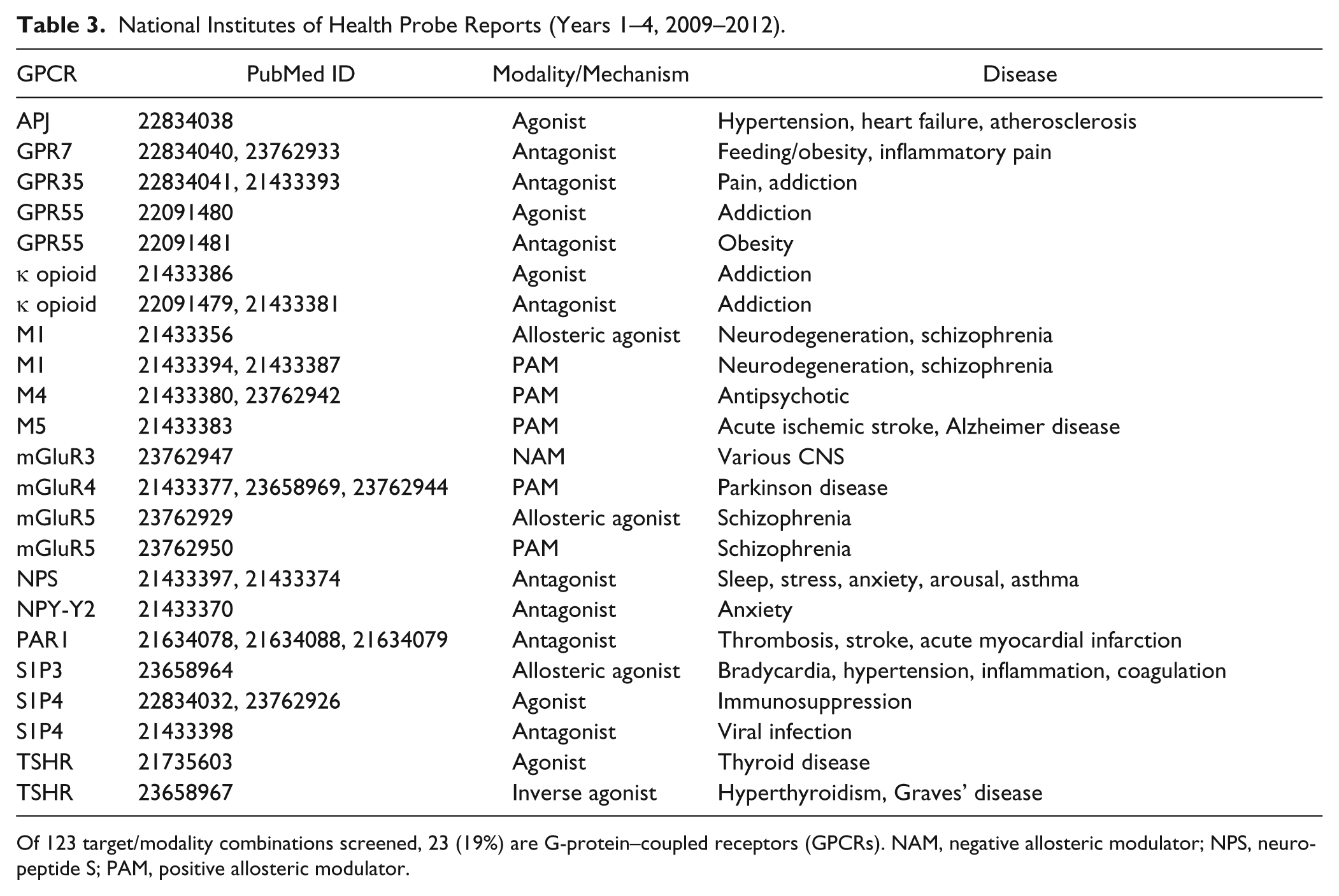
Of 123 target/modality combinations screened, 23 (19%) are G-protein–coupled receptors (GPCRs). NAM, negative allosteric modulator; NPS, neuropeptide S; PAM, positive allosteric modulator.
A broader view of the availability of active compounds for GPCRs can be obtained by analysis of the literature data collated within the ChEMBL database. 113 Data were found for 747 membrane-bound receptors, of which 649 are GPCRs. Looking at human sequences only and single targets (not necessarily single-protein [e.g., CGRP and GABAb] receptors, which are heterodimers) shows there are published activity data for 189 targets, of which 160 are Family A (3 are listed but have no data), 17 are Family B, and 12 are Family C (1 listed but with no data). Hence, if existing drugs target at most 109 receptors, there are reported biological activity data for an additional ~80 targets and around 50% of all GPCRs in total. However, these data include screening as selectivity targets where the compounds can be inactive, as well as single-shot percentage inhibition or activation data without full curve results; this becomes important where there are relatively few data points since it can mean that there are not any useful actives reported—found to be true for at least a dozen targets upon review.
The emergence of new tool compounds within this set was examined by ranking the ChEMBL activity data by the number of reported bioactivities and then working down the list, focusing on those with no or very few, suboptimal, marketed agents and those for which the screening data are all relatively recent (within the past 5–6 years):
TSHR agonists have nearly 30,000 data points, of which a considerable proportion are from the associated NIH probe report (from 2009). 114 The target is strongly implicated in thyroid cancer, where recombinant TSHR is currently used in patients receiving thyroid hormone suppression therapy, but there are issues with production, and the agent must be administered intramuscularly. An orally available small molecule with selectivity over other glycoprotein hormone receptors such as LHCGR and FSHR would be highly beneficial. There is an earlier publication for a series of TSHR agonists, but they lack selectivity over LHCGR 115 ; the probes described by the NIH project are selective.
GPR109A agonists have >700 compounds reported with ~1400 bioactivities all from 2007 onwards. The target has particular potential for dyslipidemia. While the endogenous ligand, niacin (vitamin B3), is an approved nutraceutical, its use suffers from modest potency at the receptor and a subcutaneous flushing side effect. Developments in our understanding of receptor pharmacology have led to proposed mechanisms that should elicit the desired response without the side effect (see later). There are a number of recent examples from the literature,116–126 including at least one example of a positive allosteric modulator (PAM) rather than a conventional agonist (see later). 116
Aside from these, G-protein–coupled bile acid receptor 1 (GPBAR1, TGR5) agonists have >200 compounds with >400 bioactivities reported, all from 2007 onwards; the target has been explored for its potential in diabetes mellitus, with a number of medicinal chemistry studies published.127–133 Galanin 2 (GALR2) and galanin 1 (GALR1) receptor antagonists have ~100 to 200 compounds reported with associated biological data all from 2007–2008 onwards; compounds targeting these receptors appear potentially useful for the treatment of seizures and epilepsy.134–137 Kiss-1 receptor (Metastin GPR54) antagonists have ~150 compounds with reported biological data all from 2007 onwards; compounds targeting this receptor appear potentially useful in cancer metastasis and sex hormone–dependent diseases such as prostate cancer or endometriosis.138–140 Furthermore, while the number of published compounds/bioactivities is fairly modest, there are also potentially interesting reports of FFAR2 agonists providing PAM tools for exploring function of the receptor, 141 TAAR1 antagonists for exploring amphetamine-related addictions,142,143 P2Y14 agonists with a putative immunological role,144,145 NPFFR1 and NPFFR2 with peptide tool molecules, 146 PAR-4 with potential for antiplatelet drugs, 147 and LTB4R with potential for various inflammatory diseases. 148
Also within the list are some “perennial” favorite targets, such as MC4R agonists. This target has considerable potential for the treatment of obesity, but hit identification has proven quite difficult, possibly because melanocortin receptors, although peptide receptors, cluster separately from all the other peptide receptors and hence have quite different binding sites. However, there are signs of recent progress here with modified peptides, 149 a novel methodology for compound design (in this case, based on turn mimetics), 150 the development of pharmacophore models, 151 the discovery of potent partial agonists with reduced side effect liability, 152 and the discovery of novel chemotypes.153,154
Another area that is receiving widespread attention, as well as yielding substantial progress, is in the rediscovery of aminergic receptors through allosteric mechanisms (see later). This provides a means by which to develop therapeutics with a substantially improved selectivity profile where the orthosteric binding sites are highly similar. This problem is particularly acute within the muscarinic receptor group (M1–M5) and is reflected in a number of the NIH probe reports, among other literature.155–161
Increased Availability of Structural Data
One of the most notable areas of progress in recent years has been the determination of X-ray crystal structures of GPCR targets. It took 7 years to go from the first GPCR crystal structure, that of rhodopsin, solved in 2000, to those of the β2 adrenergic receptor, which provided the first views of small-molecule drug targets with drug-like moieties bound. Since then, substantial progress has been made with the determination of further structures for aminergic (β1 and β2 adrenergic, dopamine D3, histamine H1, muscarinic M2 and M3, 5HT1b and 2b serotonin receptors), nucleoside (adenosine A2a), chemokine (CXCR1, CXCR4), peptide (κ, µ, δ and ORL-1 opioid, neurotensin NTSR1, protease-activated PAR1), and lipid (S1P1) receptors. 162 Most recently, the structure of the smoothened receptor (SMO) has been determined, providing the first structure of a Family F receptor (and, indeed, a non–Family A receptor). Many of the structures have been co-crystalized with small-molecule ligands, providing detail on how those compounds bind at their targets. While a number of the structures relate to an antagonist/inactive form of the receptor, there are an increasing number with agonist ligands bound or that are otherwise biased toward an agonist state and are starting to provide insight into the activation mechanism; this includes a structure of the β2 adrenergic receptor in complex with its G-protein.
The structures are revealing details of the conventional “orthosteric” binding site within the TM bundle and, in particular, how the extracellular loops can fold differently to form the “top” of the binding site. The availability of structural data can facilitate compound optimization, particularly in relation to achieving enhanced potency/selectivity, and they pave the way for the application of fragment-based drug design (FBDD), which is difficult to apply in the absence of structure. 163 In addition to details of the receptor activation mechanism, the structures are also revealing other insights, such as an apparent allosteric site for cholesterol on the external (lipid) face of the receptor, between TMs 1, 2, 3, and 4. 164 The presence of such a site has been predicted for ~44% of Family A receptors based on sequence similarity and could potentially be exploited therapeutically.
It is worth noting, however, that none of the crystal structures determined to date is of the native form of the protein. All have been modified somewhat to enhance stability and/or make them amenable to crystallization. It is usually demonstrated that the modified receptor retains appropriate binding affinity for the ligand(s) and is therefore thought to be a good surrogate, at least in the binding site. There are, however, alternative techniques such as photo-affinity labeling studies 165 or solid-state nuclear magnetic resonance (NMR) that can provide supporting data or additional insight. 166 There is also a substantial amount of expertise in the use of homology models for drug design. 167 This is still highly relevant given the relatively small number of receptors for which there is currently structural information. There remain major gaps in our structural knowledge as compared with the GPCR classification scheme outlined above, although the availability of crystal structures for an increasing range of aminergic receptors, certain peptide receptors, and a lipid receptor is a major step forward.
There have been recent reports that an X-ray crystal structure for a Family B/secretin receptor—namely, CRHR1 (CRF1)—has been solved by researchers at Heptares Therapeutics (see http://cenblog.org/the-haystack?s=GPCR). This is highly significant given differences in receptor topology and ligand binding relative to Family A, plus the strong disease association for many members of this family, in many cases proven clinically through the use of peptidal agents. Of the 15 members of the secretin family, glucagon/GCGR, GIPR, and GLP-1R are implicated in diabetes mellitus; GLP-2R in short bowel syndrome and inflammatory bowel disease; PTHR1, PTHR2, CALCR, and CRLR in osteoporosis; PAC1, VPAC1, and VPAC2 in inflammation and neurodegeneration; CRHR1 and CRHR2 in stress; GHRHR in dwarfism; and SECR in gastrinoma. 168 CRLR is also strongly implicated in migraine through formation of the CGRP receptor (a CRLR/RAMP1 heterodimer).169–171 The availability of structural information for this family will be hugely informative, aid substantially in compound optimization, and potentially also lead identification through, for example, FBDD approaches or the design of orally available, small-molecule peptidomimetics.
Greater Complexity of Receptor Signaling and Improved Screening Paradigms
Partial/Inverse Agonism and Allosteric Modulation
It is plainly too simple to regard the pharmacology of GPCRs as being restricted to agonists and antagonists. Constitutive receptor activity gives rise to the potential for antagonists to be neutral antagonists or inverse agonists, and there is scope for partial agonism. Compounds do not have to bind at the same site as the endogenous ligand to function with any of these modalities. This scope for “allosteric” receptor regulation has been appreciated for 50 years or more but has required advances in assay technology to progress, not least making the transition from binding to functional assays. Now, a substantial number of allosteric GPCR ligands are known, 161 and there are examples such as cinacalcet and maraviroc on the market, as well as numerous others in clinical development. There are most likely at least four different allosteric binding sites in Family A GPCRs: the cholesterol site, 164 an intracellular site, 172 a secondary site within the TM bundle 173 (beside but not overlapping with the orthosteric site), and in the extracellular loops. 174 When binding allosterically, further pharmacological texture is possible: compounds can potentiate the activity of the endogenous ligand but not signal in their own right (PAMs), appear to be antagonists (negative allosteric modulators, NAMs), block the allosteric site but have no effect on the potency/efficacy of the endogenous ligand (silent allosteric modulators, SAMs), or potentiate the endogenous agonist while also activating the receptor in their own right (ago-allosteric modulators or ago-PAMs). It is worth noting, though, that many “allosteric” agonists may actually be “bitopic” ligands, binding to both orthosteric and allosteric sites of the receptor.
Deliberate targeting of allosteric sites can have a number of advantages: there is no evolutionary pressure for the sites to remain conserved, so greater selectivity is possible, particularly between receptor subtypes; the sites are quite different, so they may provide greater chemical tractability where the orthosteric site has proven challenging; the upregulation of receptor activity by PAMs is saturable, providing a “ceiling effect” and protecting against potential overdose, as well as toxicity seen through overstimulation, desensitization, internalization, or downregulation of receptors; and there is spatial and temporal control with PAMs (less so with ago-PAMs) borrowed from the endogenous ligand because the receptor activation only occurs in the presence of the native moiety. 175
On the down side, the lack of evolutionary pressure within allosteric sites can lead to greater species differences, with attendant difficulties in progression of compounds, and the structure-activity relationship (SAR) can tend to be relatively “flat,” making optimization difficult. It is important to screen PAMs in the presence of the true endogenous ligand rather than a surrogate agonist due to the potential for “probe dependence”; there is a cooperativity between agonist and PAM in activating the receptor, which can change quite markedly if the ligand changes. It is also very hard to know the basal tone of the endogenous ligand at the site of action and hence the degree of potentiation that is required for clinical efficacy, and it is possible to modulate agonist affinity, efficacy, or both (including obtaining >100% efficacy). Allosteric binding can give rise to distinct receptor conformations, with potential for biased signaling (see below). Despite these challenges, many molecular tools are being identified with exquisite selectivity and rich pharmacology with which to probe the role of receptors in disease, with a number of examples progressing into the clinic.
Alternative G-Protein Signaling: G12/13 and Gβγ
GPCRs are conventionally viewed as signaling via G-proteins (hence the name), although in reality various additional mechanisms have therapeutic significance (see, e.g., β-arrestin below). Even within the space of G-protein signaling, however, there is significant scope for further discoveries. G-proteins are heterotrimeric, comprising α, β, and γ subunits, of which there are 21, 6, and 12, respectively, encoded by the human genome. In principle, this gives a very large number of potential combinations, but in practice, only a smaller subset form active complexes. The G-proteins are typically classified into four families based on the sequence similarity of the α subunit: (1) Gαs, which stimulates cAMP production; (2) Gαi, which inhibits cAMP production; (3) Gαq/11, which leads to intracellular calcium mobilization; and (4) Gα12/13, which actives the monomeric GTPase RhoA.
Receptor signaling is complex. Some receptors couple to only one type of G-protein, but others couple to a combination. Conventional assays tend to measure only a single signaling pathway, so the profile of GPCRs is not always fully characterized, and the G12/13 pathway has, in general, not been well explored.176,177 Even so, there are a number of reports suggesting potential significance: G12/G13 is thought to be important in embryonic development, oncogenesis, and cancer metastasis 178 ; 5HT7/G12 signaling is involved in the formation of initial neuronal networks during early postnatal development 179 ; GPR55/G13 signaling stimulates neurite retraction 180 ; G13 interacts with the β subunit of integrins 181 ; G13 signaling has a role in cardiac remodeling leading to heart failure 182 ; and there is a suggestion that selective inhibition of CXCR4/G13 signaling could be useful in preventing the metastatic spread of basal-like breast cancer cells. 183
Even within the other, broad classes of G-protein, there are potential opportunities for further refinement. There is a suggestion that G15, while nominally a member of the Gq class, is somewhat diverse. 184 It is structurally divergent from other members of the family with some different properties, and its expression is only detected in highly specific cell types (hematopoietic and epithelial cells). Moreover, signaling of the G-protein βγ subunit arguably mediates as many responses as α but has received substantially less attention. 185
β-Arrestin Signaling and Biased Agonism/Functional Selectivity
Agonist binding to a receptor promotes not only G-protein activation but also desensitization. Rapid phosphorylation at the C-terminus and intracellular loops (ICLs), in large part by the G-protein receptor kinases (GRKs), together with the change in receptor conformation, leads to a dramatic increase in affinity for the GPCR adapter proteins, β-arrestin1 and/or β-arrestin2. This process blocks subsequent activation and leads to removal of the receptors from the cell surface.
Originally, β-arrestins were thought of simply as negative regulators of G-protein signaling, but it has recently become appreciated that they can signal in their own right, through a diverse range of pathways, including kinase activation (e.g., MAP and Src families), transcriptional regulation, and receptor transactivation. The signals are often spatially and temporally distinct from those of G-proteins and can result in unique cellular and physiological/pathophysiological consequences. This area has been reviewed in detail,186–188 including examples linking β-arrestin signaling to various diseases. 189
The presence of multiple signaling pathways originating from GPCR activation has led to the concept of “biased agonism” or “functional selectivity”—that is, the selective activation of one pathway over another. Balanced ligands bind to a receptor in a way that stabilizes the conformation(s) capable of signaling through each of the various different downstream pathways, whereas biased ligands stabilize only those conformation(s) capable of signaling via a subset of the pathways. This provides considerable scope for the identification of compounds that selectively target clinically useful signaling pathways and are more neutral or even block alternative pathways, which give rise to undesirable side effects.
Repeated use of a drug can induce a decrease in responsiveness and a need for higher doses to achieve the same effect (“tolerance”), a process thought to be mediated by β-arrestin–mediated desensitization. G-protein–biased ligands therefore have scope for improved agonist therapies with more sustained efficacy. Examples include β1-adrenergic receptor agonists in heart failure, β2-adrenergic receptor agonists in asthma, and µ-opioid receptor agonists for the treatment of pain. 189 There is also scope to avoid side effects with such biased ligands; β-arrestin2 knockout mice are resistant to morphine-induced respiratory suppression compared with wild type, and there is evidence that GPR109A (niacin) receptor agonists could be developed for the treatment of dyslipidemia in the absence of the subcutaneous flushing side effect, which is thought to be β-arrestin mediated.
Equally, there are examples where β-arrestin biased signaling could be advantageous. There is evidence to suggest that biased β1-adrenergic receptor agonists could provide improved treatments for heart failure, leading to transactivation of epidermal growth factor receptor (EGFR), which is cardioprotective, while avoiding cardiotoxic effects thought to be associated with chronic receptor activation, in large part driven by G-protein signaling. With the angiotensin II type 1A receptor, the β-arrestin–biased ligand Sar 1 ,D-Ala 8 angiotensin II (TRV120027) has been shown to increase cardiac performance in anesthetized rats, whereas unbiased ligands reduce cardiac performance. 190 There is also potential for improved PTH receptor agonists used for the treatment of osteoporosis. The β-arrestin–biased ligand (D-Trp 12 ,Tyr 34 )-PTH(7-34) stimulates β-arrestin while blocking G-protein signaling and promotes anabolic bone formation in the absence of bone resorption, an effect that is abolished in β-arrestin2 knockout mice. More generally, β-arrestin is involved in the activation of a number of signaling pathways associated with cancer, be they proliferative or suppressing tumor growth/metastasis, and as such may offer a number of potential future targets.
There is at least one biotechnology company, Trevena, dedicated to the discovery of improved GPCR-based drugs exploiting biased signaling (http://www.trevenainc.com). It currently reports TRV027, a β-arrestin–biased ligand of the angiotensin receptor (AT1R), in phase II development for the treatment of acute heart failure, as well as four different opioid receptor–biased ligand programs for pain. The most advanced compound from those, TRV130, is a G-protein–biased µ-opioid receptor agonist in phase I development for the treatment of acute postoperative pain.
GPCRs as Multiple Targets and System Dependence
GPCRs are not just more complex than might be expected in terms of their pharmacology and signaling profiles. They also offer important opportunities for new drugs given scope to target multiple receptors simultaneously or from the fact that they are most likely homo/heterodimers or higher order oligomers in the first place and bind to a considerable range of additional proteins.
The development of drugs with multiple receptor activities is well precedented, particularly within the aminergic receptor field. Certain disease indications, notably antipsychotics, most likely require a complex mixture of receptor activities for clinical efficacy. However, given the high similarity in binding sites between, for example, the various dopamine and serotonin receptor subtypes, it is quite likely that further, undesirable activities are also present. This provides scope for the design of compounds that retain complex polypharmacology profiles but with enhanced selectivity over those targets that give rise to the side effects. 191 There are related opportunities in the chemokine receptor field: receptor activation is controlled by some 50 ligands, which often act in a redundant and overlapping manner, providing a complex regulatory system 192 with significant implications for the design of clinically effective agents; the system may simply compensate if challenged with an agent that is too selective. There are numerous other cases of targeted design of compounds with multiple activities, including multiple GPCR activities and also multiple activities across target classes (see, e.g., Lin et al. 193 and Gattrell et al. 194 ).
It is well known that certain GPCRs are heterodimers. Two key examples of this would be the Family C receptor GABAb and the Family B receptor CGRP. GABAb is an obligate dimer formed between GABAbR1 and GABAbR2. GABAbR1 contains the ligand binding site but requires heterodimerization with R2 for proper transport to the cell surface 195 as well as G-protein coupling. 196 The CGRP receptor is a heterodimer formed between the CRLR GPCR and receptor activity modifying protein 1 (RAMP1), a protein with a single transmembrane helix and N-terminal domain (NTD), the latter of which contributes to the binding site of known CGRP receptor antagonists along with the NTD of CRLR. 197 The interaction of the three RAMPs with CALCR and CRLR yields seven distinct receptor phenotypes for the four different calcitonin peptides. There is a potentially far wider role for RAMPs, including trafficking receptors to the cell surface, notably with Family C receptors. 198
Many—but perhaps not all 199 —GPCRs interact with each other to form dimers or potentially higher-order complexes. 200 It is generally difficult to distinguish dimers from higher-order oligomers, and the latter term is usually used to encompass both. There are several examples of the functionality of this oligomization within Family A. 200 It can have effects on receptor expression, signaling, and ligand binding, including positive or negative cooperativity via allosteric mechanisms. 201 Oligomer formation is also implicated in the pathophysiology of disease. For example, δ- and κ-opioid receptors expressed individually have low affinity for either δ- or κ-selective ligands; however, high affinity is restored when the two are coexpressed, suggesting positive cooperativity between the two. This implies that there is scope for the development of compounds that selectively interact with a specific heteromer without affecting the individual receptors. 202 The µ-δ heteromer could also be a therapeutic target in the treatment of chronic pain and addiction due to its distinct signaling properties. 203 More broadly, there is interest in developing biased TPα-TPα homodimer antagonists that avoid IP-TPα heterodimer formation, with potential superiority in cardiovascular disease, 204 oligomer-targeted therapeutics with improved profile for thrombosis, 205 with chemokines,192,206,207 5HT4 and 5HT7 serotonin receptors, 208 and others.202,209 There are also data that point to a ligand-induced formation of the GLP-1/GIP heterodimer in response to GLP-1 peptide, which is reversed by GIP peptide. A functional role for this heterodimer is indicated with reduced β-arrestin signaling via GLP-1R. 210 There is, therefore, an apparently intriguing interplay between two receptors for integrin hormones that together regulate postprandial blood glucose levels and have a strong rationale for treating diabetes mellitus 211 with an associated scope for the discovery of highly targeted new treatments.
GPCRs interact with a myriad of other proteins, not only to attenuate their signaling but also to couple the receptors to G-protein–independent signaling pathways. Intracellular and transmembrane proteins can regulate processing in the endoplasmic reticulum (ER), trafficking to the cell surface, compartmentalization to plasma membrane microdomains, endocytosis, and trafficking between intracellular membrane compartments. Examples include β-arrestin, receptor activity–modifying proteins (RAMPS), regulators of G-protein signaling (RGS), GPCR-associated sorting proteins (GASPs), Homer, small GTPases, PSD95/Disc Large/Zona Occludens (PDZ), spinophilin, protein phosphatases, calmodulin, optineurin, and Src homology 3 (SH3)–containing protein interactions with GPCRs. 212
GPCR-interacting proteins fine-tune GPCR pharmacology and signal transduction, and they potentially provide the opportunity for novel, exquisitely targeted therapeutics. With the RGS proteins, for example, the amino-termini represent potential sites for interaction with both GPCRs and other signal transduction proteins, suggesting the formation of protein complexes that could contribute to the regulation and generation of GPCR-mediated signals. The interaction of GPCRs with RGS proteins is a key factor in resetting the pathway following stimulation. The precise GPCR/G-protein/RGS combination determines the nature and duration of the response. It is difficult to examine in cells containing multiple variants of each component, but studies have been performed in which these elements have been dissected, 213 indicating scope for precisely targeted agents. 214 An RGS inhibitor would be expected to enhance GPCR signaling and could do so in a tissue- or pathway-specific manner.
Label-Free Screening
The complexities of GPCR signaling provide a need and an opportunity for new assay systems. Label-free whole-cell assays have emerged within the past few years and have the potential to substantially change some aspects of GPCR screening.215,216 Traditional assays tend to the reductionist view; they are very robust, provide a good measure of activity via a specific pathway, but do not provide the bigger picture. It is possible to measure integrated, cumulative responses rather than individual events, typically by detecting changes in cellular features such as adhesion, proliferation, migration, and cell death via either optical or electrical biosensors. The sensitivity, precision, and high-throughput nature of some label-free instruments enable their use in HTS, although the cost of consumables might be limiting. These systems have also been used for selectivity studies and endogenous receptor profiling in commonly used cell lines, among other aspects of GPCR research.
Scope for Biological Agents: Antibodies
In a sense, GPCRs are already the target of a number of biological—as opposed to small-molecule—agents. Several marketed peptide agents, typically the endogenous ligands of peptide receptors, are used to treat a variety of diseases. Recent commentary on FDA approvals has focused on the emergence of biologicals, and a number of companies have committed to strengthening their pipeline with such moieties given scope with otherwise “intractable” targets.
While the tractability issues for small-molecule drug discovery remain with certain GPCR subfamilies, there is scope for biotherapeutics such as monoclonal antibodies (MAbs). 217 There have been issues generating antigen up to this point due to the low levels of receptor expression and the lack of protein stability when purified, but advances from the X-ray crystallographic area provide a potential solution (see, e.g., http://www.morphosys.com/pressrelease/morphosys-and-heptares-sign-alliance-develop-antibody-therapeutics-targeting-gpcrs). The antigen would also be conformation specific, offering potential scope to develop agonist and antagonist antibodies. In addition to being potential therapeutics, these could be useful as research tools with which to probe the role of the receptors, particularly in the Family B/adhesion and Family F/frizzled families, where relatively little is currently known. Activating antibodies could be of particular utility for orphan receptors, where it is otherwise very difficult to explore their function.
An analysis of the scope for biotherapeutics has suggested that around 100 receptors are strongly implicated in disease, of which around 80 are amenable to this approach and ~25% would require activating molecules. 217 Most opportunities seen are in the cancer, inflammation, or metabolic disease areas. There are believed to be no marketed antibodies for GPCRs as of yet, although there is growing interest, with MAbs reported to be in development for C5AR (rheumatoid arthritis, systemic lupus erythematosis), CXCR4 (cancer), CCR5 (human immunodeficiency virus), CCR2 (inflammation/rheumatoid arthritis), CCR4 (cancer), and GCG-R (type 2 diabetes mellitus).
Conclusions
GPCRs still offer enormous scope for the discovery of new therapeutic targets. The long history of success has been largely dominated by compounds targeting the aminergic receptor subfamily. These receptors account for only about 10% of GPCRs; the vast majority of the receptors have yet to be successfully tackled. This is particularly true for the orphan receptors, which account for about 25% of the targets, and where progress is hampered by a lack of knowledge around the endogenous ligand. In other areas, notably the peptide, lipid, and purine Family A receptors plus the more unusual Family B/secretin, Family B/adhesion, Family C/glutamate, and Family F/frizzled receptors, the issues have been more around the identification of high-quality small-molecule lead compounds for optimization.
Progress is clearly being made, however, as the recent regulatory approvals for vismodegib and fingolimod testify. The former targets the SMO receptor (Family F/frizzled) and the latter S1P receptors (Family A/lipid). Despite the wealth of existing drugs, there is still potential for further advances in the aminergic field, most notably through the identification of more highly selective compounds, perhaps acting through allosteric mechanisms. There is also a potentially significant role to be played by biotherapeutic agents, and existing peptidal agents could represent a platform for further discovery given clinical validation for the target(s) and improvements in our ability to identify/design small molecules.
Technical advances in assay technology, X-ray crystal structure determination, and ligand design methods offer substantial promise that the key issues can be addressed and further progress substantially accelerated. Screening advances are opening up previously neglected signaling pathways such as G12/13, new pathways such as β-arrestin, and more holistic investigation of receptor function through label-free methods. GPCR X-ray crystallography has made enormous strides in recent years, affording structures for an ever increasing range of receptors, providing detail of ligand binding sites and binding modes, identifying detail of receptor activation as well as novel binding sites, and facilitating hit identification through FBDD and the lead optimization process. Computational methods have contributed to our understanding of the lack of hit identification success in certain areas due to the heterogeneous nature of GPCR ligands/binding sites coupled to unbalanced screening decks, with novel design methods demonstrating success at bridging the gap and maximizing the substantial body of existing data. It is also worth noting the key role that open innovation initiatives are playing in this respect: known drugs are stored and annotated in the drug bank database; the NIH Molecular Libraries Program is identifying tool molecules for nominated targets and putting them in the public domain to facilitate target validation studies; those compounds, plus many thousands of other reported biologically active compounds from the literature, can be searched effectively via the ChEMBL or PubChem websites; and the protein X-ray crystal structures, once published, are put into the public domain via the RCSB database (http://www.rcsb.org/).
The potential scope of the unaddressed targets is almost bewildering. The Family A/lipid receptors alone represent a group of ~50 receptors that have been largely unaddressed up to this point; as such, this group is about the same size as the whole of the nuclear receptor protein family. The Family A/peptide group is about twice the size, with limited, patchy progress up to this point, in a number of instances being achieved via peptide rather than small-molecule agents. The target validation studies are often facilitated by the availability of high-quality molecules, which are somewhat lacking; even so, there is already a very substantial body of literature suggesting wide-ranging potential disease association, and progress will likely be stimulated as further tool molecules are described.
Improvements in assay technology coupled to enhanced understanding of receptor pharmacology are presenting substantial additional opportunities. Allosteric mechanisms can be targeted to improve chemical tractability, selectivity, and safety; alternative signaling pathways such as G12/13, Gβγ, or β-arrestin are providing new ligand pairings for orphan receptors and links to disease; and “biased” ligands can be used to alleviate problems associated with drug tolerance or to avoid unwanted side effects. The appreciation that GPCRs are most likely oligomers, associate with a wide range of additional proteins, and can operate through subtle interplay with one another provides yet further opportunities for new, improved, and exquisitely targeted therapeutics through the selective interaction with homo/heteromers, other protein partners such as RGS, and tuned polypharmacology profiles.
Footnotes
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.