
Abstract
Fragment-based lead discovery has emerged over the past two decades as a successful approach to generate novel lead candidates in drug discovery programs. The two main advantages over conventional high-throughput screening (HTS) are more efficient sampling of chemical space and tighter control over the physicochemical properties of the lead candidates. Antibiotics are a class of drugs with particularly strict property requirements for efficacy and safety. The development of novel antibiotics has slowed down so much that resistance has now evolved against every available antibiotic drug. Here we give an overview of fragment-based approaches in screening and lead discovery projects for new antibiotics. We discuss several successful hit-to-lead development examples. Finally, we highlight the current challenges and opportunities for fragment-based lead discovery toward new antibiotics.
Introduction
Antibiotic resistance (AR) has grown into a major global health problem over the past three to four decades. Over the same period, many warnings about the dangers of emerging AR have been issued, but these have not yet resulted in improvement of the current treatment options for bacterial infections. Only in the last decade or two have national and international agencies (like the European Centre for Disease Prevention and Control [ECDC] and World Health Organization [WHO])1,2 proposed and implemented actual policy changes to address AR, such as strengthening prudent use of antibiotics (especially in veterinary medicine), improving surveillance and diagnostics, and increasing awareness of the need for the development of new antibiotic drugs.
The incidence of AR is increasing according to recent reports.3,4 Of particular concern is resistance to last-resort drugs like carbapenems, cephalosporins, and polymyxins in hospitals. There are also hints of transferability of multiple resistance genes outside the clinic.5,6 Lastly, the incidence of multi-drug-resistant Mycobacterium tuberculosis infections outside the clinic is also increasing. 7 Tuberculosis (TB) is often discussed separately because of its differing disease and treatment characteristics.
One of the biggest current problems is that the clinical pipelines do not contain innovative new compounds to address this rising AR.8,9 This is often attributed to low incentives for development (any new antibiotic would be used as little as possible to avoid the generation of resistance) 10 and technical hurdles compared with other diseases (discussed later in this review). 11 The most recent new class of marketed broad-spectrum antibiotics (oxazolidinones) was discovered in the early 1980s, 12 and the most recent first-in-class narrow-spectrum antibiotic to be marketed (anti-TB synthetic diarylquinoline) was discovered around the turn of the millennium. 13
This somewhat bleak picture of the current state of antibiotics development reveals the need for increased basic research efforts and antibiotic discovery programs that can feed into the clinical pipeline. Current research does offer many promising new concepts and ideas to work with. Some examples are systematic screening for inhibitors of apparently nonessential genes as antibiotic adjuvants (β-lactamase inhibitors are a classical example) (reviewed in Wright 14 ), increased understanding of mechanisms of bacterial membrane influx and efflux,15,16 and renewed interest in natural products using modern synthetic17–19 and bioinformatic tools.20–22
The last two decades have also seen technical advances in structure-based drug discovery, 23 in particular a variety of developments in biophysical techniques. 24 These advances have supported the evolution of the methods of fragment-based lead discovery (FBLD), for which there have been many reviews.25–27 There is a simple rationale behind the testing of fragment-sized compounds, typically with molecular weights of 120–250 Da: fragments cover a large part of theoretically possible atom configurations per molecule because they contain fewer atoms, and because of their small size, they are more likely to bind to a target. Thus, a well-designed, diverse fragment library contains about a thousand-fold fewer compounds than the average high-throughput screening (HTS) library and covers a considerably larger chemical space. However, fragments bind with a much weaker affinity (useful fragments have a dissociation constant, KD, of up to 2–3 mM) than usual lead compounds, and screening relies on the high sensitivity, robustness, and throughput of known and new techniques to both identify and characterize fragment binding. Once the binding of a fragment is confirmed, some initial structure–activity relationships (SARs) can be explored by the purchase of compounds that are similar to the fragment or contain substructures of the fragment (sometimes called SAR by catalog). This can increase confidence in the validity of the fragment hit, but in general, the process of growing the fragment to a lead compound is challenging without structural information generated by either crystallography or nuclear magnetic resonance (NMR) experiments. The structure of the fragment bound provides ideas about how to evolve the structure of the fragment, initially by systematic exploration of SARs, making small changes to the core scaffold. For the development of the fragment, there are three general strategies, all greatly benefiting from structural information and usually supported by biophysical methods to confirm and characterize binding: (1) fragment growing, which increases potency by optimally engaging the binding pocket, based on structural and/or SAR information; (2) fragment linking; and (3) fragment merging, in cases where there are two molecules binding close to each other.
While the main goal of hit-to-lead optimization is to increase the affinity or activity of a series, many researchers now try to optimize every single atom addition, guided by a metric called ligand efficiency (LE), 28 which is the average binding energy of the molecule per ligand nonhydrogen atom (i.e., [–RT ln(KD)]/[heavy atom count]). All in all, the two core concepts of structure-guided and LE-optimized lead design leave relatively little room for serendipity in the FBLD process, but instead try to rationally optimize the chances and opportunities of finding lead compounds with novel scaffolds, favorable physicochemical properties, and target selectivity.
In this mini-review, we first summarize the key features of the techniques that are used in fragment-based discovery. The techniques are primarily considered for their use in screening to identify fragments, but the same methods can be used throughout the optimization process. We then summarize some of the examples published where the methods have been used in antibiotic discovery projects.
Screening Methods in FBLD
A wide range of biophysical, structural, and biochemical assays have been used to identify fragments that bind to a protein target. Although some targets can have highly enclosed binding sites where fragment-sized molecules bind with a KD below 1 µM (such as G-protein-coupled receptors), most fragment screening campaigns need to identify compounds that bind with affinities in the hundreds of micromolar to low micromolar range. This places particular constraints on the screening approach—requiring high solubility for the ligands, high sensitivity of the detection method, and for the assay not to be liable to interference from the high concentrations of fragment that are used. Most screening campaigns use biophysical methods, usually with an orthogonal method used for the validation and characterization of binding. There have been extensive reviews of the different fragment screening methods.26,29,30 The following is a brief summary of the main characteristics and considerations for each of the screening approaches. Key points for consideration of each method are also represented in Table 1 .
Summary of Main Features of Most Widely Used Fragment Screening Methods.
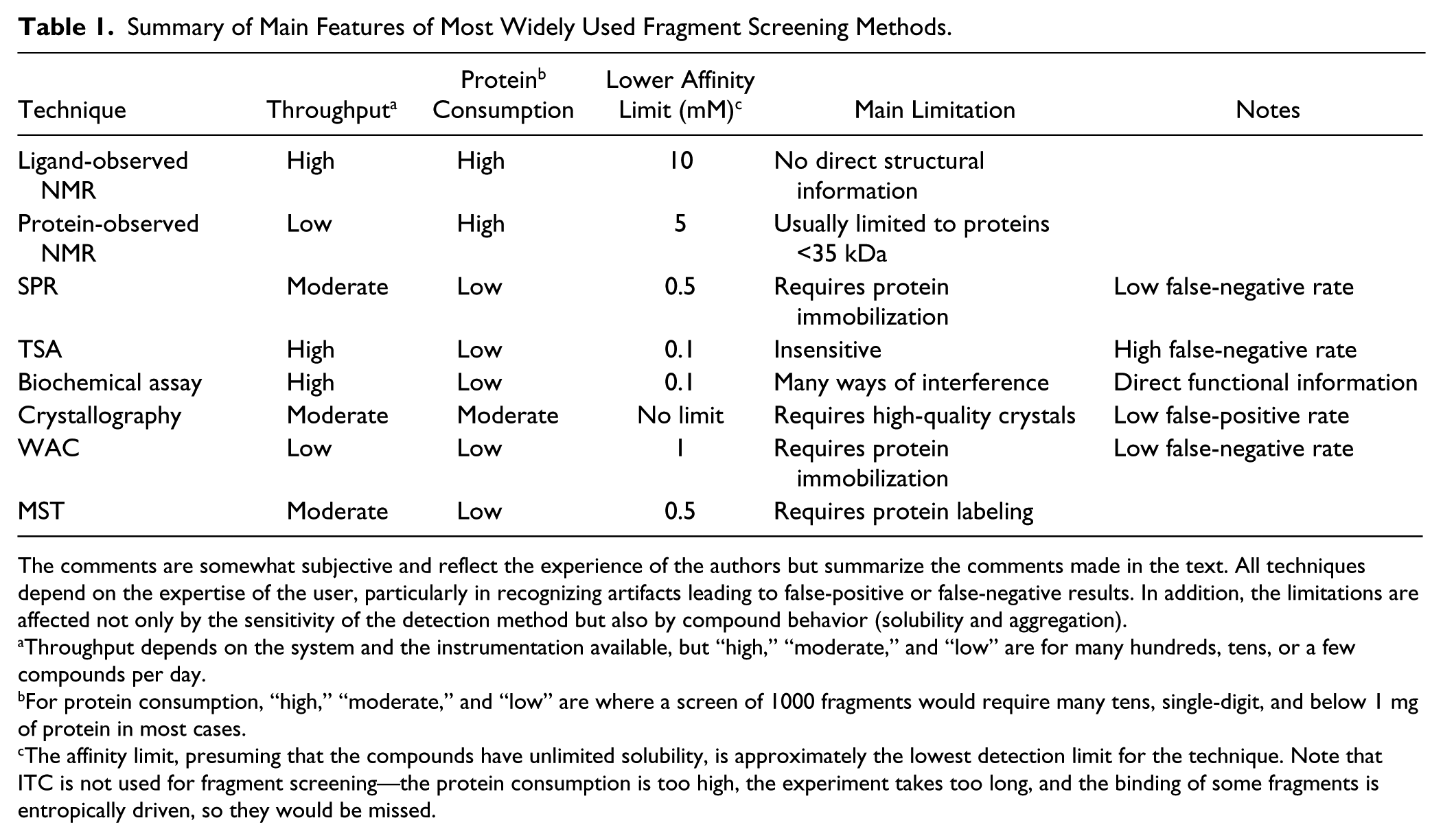
The comments are somewhat subjective and reflect the experience of the authors but summarize the comments made in the text. All techniques depend on the expertise of the user, particularly in recognizing artifacts leading to false-positive or false-negative results. In addition, the limitations are affected not only by the sensitivity of the detection method but also by compound behavior (solubility and aggregation).
Throughput depends on the system and the instrumentation available, but “high,” “moderate,” and “low” are for many hundreds, tens, or a few compounds per day.
For protein consumption, “high,” “moderate,” and “low” are where a screen of 1000 fragments would require many tens, single-digit, and below 1 mg of protein in most cases.
The affinity limit, presuming that the compounds have unlimited solubility, is approximately the lowest detection limit for the technique. Note that ITC is not used for fragment screening—the protein consumption is too high, the experiment takes too long, and the binding of some fragments is entropically driven, so they would be missed.
Nuclear Magnetic Resonance
There are a wide variety of NMR experiments where the spectra obtained are sensitive to the binding of a ligand to a protein. There are two main classes of experiments—protein observed, which detect changes in the spectrum of the protein, and ligand observed, which detect changes in the spectrum of the ligand, both of which are briefly introduced below and described elsewhere in more detail.31–33
Ligand-observed spectra for fragment screening are usually acquired with a large molar excess of ligand over the protein—typically with the protein at 10 µM and the ligand at 500 µM. The three most widely used experiments are saturation transfer difference (STD),34 water ligand observed via gradient spectroscopy (water-LOGSY),35 and Carr–Purcell–Meiboom–Gill (CPMG) experiments,36 in each of which binding is detected through a change in the spectrum of the ligand. For STD, a series of pulses are applied at the chemical shift of a core hydrophobic nucleus in the protein—this energy is transferred through the protein, to the ligand, and persists when the ligand dissociates into solution. This results in a difference in the spectra measured for the ligand with and without protein saturation. In water-LOGSY, the energy is transmitted between water molecules and ligand molecules. The efficiency of the transfer depends on the tumbling speed of the ligand molecules, giving differential signals for free and bound ligands. The CPMG experiment more directly measures the tumbling time of the ligand, which will be different when bound or free. These ligand-observed experiments give no indication of the site of binding, and because of the high concentrations used, it can be possible to obtain false-positive results from very weak nonspecific (and often superstoichiometric) binding. For this reason, a competitive step is usually included to check for changes to the fragment binding signal when a known ligand binds to the site of interest. In addition, the ligand-observed methods require exchange rates (approaching the diffusion limit) that allow the excess ligand in a sample to bind at least once to the protein within the timescale of the experiment, and so can miss high-affinity compounds. Also, the different physical bases of the NMR experiments can give rise to artifacts, and for this reason, it can be prudent to require positive signs of competitive binding in all three NMR experiments.37 The advantages of these ligand-observed methods are that the protein does not need isotopic labeling, there is no limit on the size of the protein, and the spectra that are obtained confirm that both the protein and the fragment are intact and in solution. The main disadvantage is the large amount (typically tens of milligrams) of protein required.
The most widely used protein-observed NMR experiment is heteronuclear single-quantum coherence (HSQC), which was used in the first published fragment-based discovery project from Abbott. 38 Transfer of signal between 1H and 15N or 13C in the isotopically labeled protein results in a spectrum where each amide or methyl group gives rise to a single peak, where the position of each peak depends on the local chemical environment, which can be affected by ligand binding. The main limitations are the size of protein that can be studied (typically 35 kDa), the need for isotopic labeling (which is difficult for proteins produced through nonbacterial expression), and the need for higher protein solubility to give sufficient signal (typically 20–100 µM). This requirement also increases the amount of protein required for screening. However, the dynamic range of HSQC measurements is quite broad, mainly limited by compound solubility, although there can be issues at low micromolar affinity, where the exchange rates between free and bound populations lead to peak broadening. HSQC can also give additional information: the pattern of peaks that shift can confirm that the fragments are binding to the same binding site and, if the spectrum of the protein is assigned, then where this binding site is. Also, as long as the ligand is soluble, it is possible to titrate and obtain a KD from the size of chemical shift on the protein.
A more advanced use of NMR is to determine the binding mode of the ligand bound to the protein. This requires more extensive NMR experiments that give an assignment of which peak corresponds to which nucleus, and then collection of sets of distances between the atoms from nuclear Overhauser effect (NOE) experiments. A full collection of NOEs can give a complete structure for the protein with ligand bound. A variant is to collect limited NOE data (using an isotope-filtered NOE experiment) to identify particular ligand–protein NOEs that can be used to generate an NMR-guided model of how the ligand binds. 39 Such protein-observed NMR experiments underpinned the fragment discovery work at Abbott.38,40,41
Surface Plasmon Resonance
Surface plasmon resonance (SPR) is a method to measure the change in molecular mass when a ligand binds to a protein. The most widely used equipment uses a surface to which either the ligand or the protein is attached—the other component is then flowed over the surface. The refractive index of light shone onto the surface is sensitive to the molecular mass of what is attached. If a protein is immobilized, the increase in mass as the ligand is flowed over gives information about the association rate; if the ligand is replaced with just buffer, then the dissociation rate can be measured. The ratio of the dissociation and association rates is the equilibrium constant. The current generation of instruments is sensitive enough to detect the binding of low-molecular-mass compounds, such as fragments. The main issues are in finding the conditions and strategy to immobilize the protein to provide a homogenous surface where the protein remains folded and having the reagents (and experience) to design suitable control experiments to validate the system, as summarized in the excellent review from Giannetti. 42
Thermal Shift Analysis
The principle behind thermal shift analysis (TSA) is that the temperature at which a protein unfolds will be changed by binding of a ligand. In practice, a protein solution (± the ligand) is heated in the presence of a fluorescent dye—the dye binds to the hydrophobic surface as the protein unfolds. The advantage of the technique is that it uses relatively cheap equipment (a qPCR machine is sufficient), uses small amounts of protein, and is quite rapid. The method works well to identify ligands that stabilize a protein for crystallization 43 and for screening suitable buffers to stabilize the protein, 44 and has been used for screening libraries of larger compounds. 45 TSA has been used for fragments, but there are issues with many false negatives; 46 a weakly binding fragment does not necessarily stabilize the protein to a detectable level. However, it is a fast and economical way of screening, attractive to academic groups.
Biochemical Assay
A biochemical assay (such as a functional enzyme assay or a binding assay, such as displacement of a fluorescently labeled probe) can be used to screen for fragments. The main requirement is that the assay is not compromised by the high concentration of ligand (and sometimes associated solvent) of ligand being used. The advantage of a functional assay over biophysical techniques is that it only reports binders that modulate function (functional relevance of hits is often unclear, especially from crystallography 47 ) and can distinguish between different types of modulators if the assay is balanced properly. 48 The review in 49 includes comparison of biochemical and biophysical assays.
Crystallography
There was a strong focus on high-throughput x-ray crystallography for the screening of fragments from some of the early adopters of the approach, 50 and there have recently been significant improvements in streamlining data collection and structure determination. 51 There is an immediate advantage in providing a model for the binding of the fragment, and the high concentrations required for soaking experiments means quite weak binding fragments can be identified. However, it does require the protein to crystallize with an accessible active site and a crystal packing resistant to moderate solvent and ligand concentrations. It often takes a number of attempts to obtain a crystal structure of a fragment binding to a protein even when it has been confirmed to bind by other techniques. 29
Isothermal Titration Calorimetry
An isothermal titration calorimetry (ITC) experiment measures the heat (enthalpy or ΔH) that is released or taken up when a ligand binds to a protein, and the titration gives the equilibrium binding constant from which the entropy component (ΔS) can also be determined. As long as there is some heat change on binding, the technique is extremely robust and can be used to detect and characterize binding for proteins that are difficult to assay in other ways. Importantly, it can confirm the stoichiometry of binding. However, it is quite expensive in protein and so is not used for screening (see the survey on screening methods 25 ). It also has high requirements for the solubility of the ligand (if titrated).
Other Ideas and Approaches
Many other approaches and technologies have been suggested and used for detecting low-affinity interactions. In general, no particular technique can be regarded as a “best-in-class” solution, but it is useful to be aware of and consider all available options. Some of these other methods are proprietary, such as the capillary electrophoresis method used by Selcia, 52 the target-immobilized NMR screening (TINS) method of ZoBio, 53 and weak affinity chromatography (WAC). 54 Mass spectrometry is limited to situations where the ligand binds with affinities better than approximately 100 µM, but has been applied successfully with covalently bound fragments, 55 with the “tethered” fragments approach of Sunesis, 56 and more generally by NovAliX. 57 Another recently developed biophysical technique that has been successfully applied by some to FBLD is microscale thermophoresis (MST) (Nanotemper GmbH 58 ). In this approach, the mobility of molecules (observed from a fluorescent label) along thermal gradients (thermophoretic mobility) is used to identify changes in molecular hydration and thus molecular interactions.
General Comments on Fragment Screening
There is much debate about the “best” technique to use for fragment screening (e.g., see the Practical Fragments blog 25 ). All the techniques have their pros and cons (see Table 1 )—protein-observed NMR requires labeling and has a size limit, ligand-observed NMR requires large amounts of protein, x-ray crystallography requires a suitable crystal system, and SPR (and MST) requires effective labeling that does not affect function. Each of the techniques also has particular requirements in terms of the solubility of the protein and the solubility of the ligands screened and can robustly detect different dynamic ranges of binding affinity. There are occasionally studies that discuss the different hits obtained from fragment screening with different techniques (e.g., Schiebel et al. 59 ). However, if the limitations of each technique are taken into account (sensitivity, buffers, solubility, etc.), then the same hits should be obtained.49,54 One should take caution if only taking the intersection of hits—this will identify the most robust binder but means the diversity of hits is vulnerable to the least reliable method. As mentioned before, hit validation by orthogonal techniques will filter out some false positives when those emerge from limitations of the techniques (e.g., compound intrinsic fluorescence in some types of biochemical assay or direct saturation in STD NMR). There are still many possible ways in which false positives survive orthogonal validation, as drug discovery practitioners became aware of relatively recently,60,61 but there seem to be no general rules applicable to all projects.62,63 In the end, a balance must be found between false positives and false negatives, depending on the goals of the project (taking into account scaffold diversity, chemical tractability, etc.).
Examples of Fragment-Based Discovery of Antibiotics
Biotin Carboxylase
Bacterial proteins without human homologs have often been prioritized as antibacterial targets. However, unbiased whole-cell screening has yielded good targets, even when homologous to human targets, as in the case of the Escherichia coli enzyme biotin carboxylase (BC). 64 BC is a subunit of the bacterial acetyl-CoA carboxylase that uses adenosine triphosphate (ATP) to catalyze the first step of the reaction. The ATP binding site of BC has similarities to human kinases. Mochalkin et al. followed up on this target by using FBLD to discover potent leads with new scaffolds. 65 They employed a cascade of three screening experiments, summarized in Figure 1 . First, a biochemical assay that was available from their earlier work 64 was used to screen ~5000 fragments in mixes of 10. The use of two different, moderate screening concentrations probably helped them to recognize artifacts. About 20% of the fragment mixes showed inhibition of more than 25%. Then, to identify the active component(s) of each mix, their binding to BC was measured by STD NMR. This resulted in a set of 142 fragments (3% of the library). Finally, concentration-dependent inhibition was determined by titration of the single compounds in the original enzyme assay.
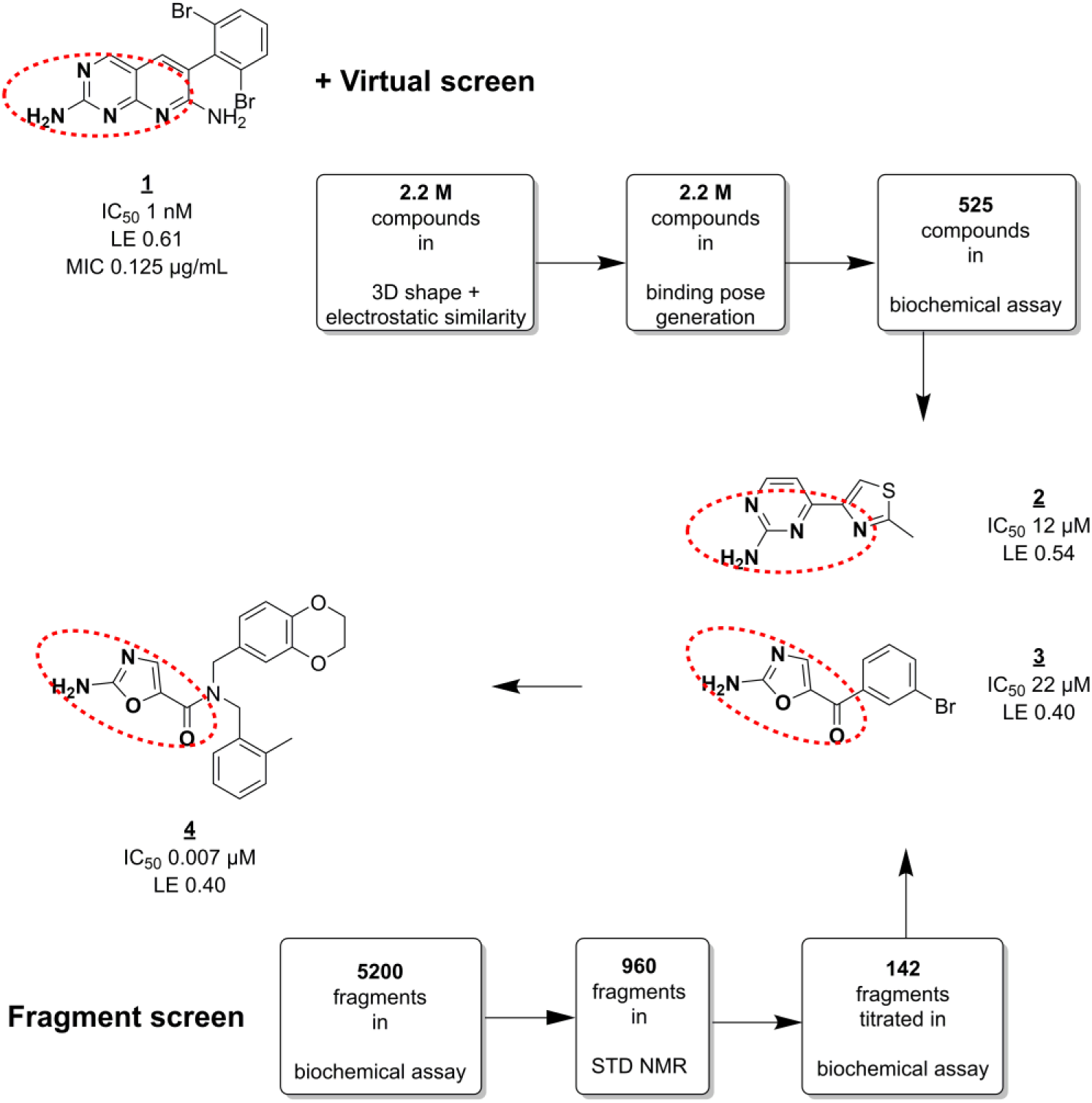
Screening against BC. Fragment and virtual screens conducted in parallel resulted in several highly ligand-efficient hits (
The fragment screen results were complemented by virtual screening. Out of a set of 2.2 million small organic in-house compounds (including fragments), a subset was selected for 3D similarity to previously identified HTS hit
Several of the most potent hits (such as
DNA Gyrase
Bacterial DNA gyrase, a type II topoisomerase, is a tetrameric complex that acts on topological isomers of DNA during replication and transcription to relieve positive supercoiling of double-stranded DNA. This is an essential process and proceeds via similar mechanisms in all domains of life. There are several classes of successful bacterial DNA gyrase inhibitors (e.g., quinolones, aminocoumarins, and novel bacterial type II topoisomerase inhibitors [NBTIs]) both on the market and under development, targeting either the ATPase domain (GyrB) or the DNA cleavage domain (GyrA).
There have been several fragment-based drug discovery campaigns against DNA gyrase. One of the earliest examples of FBLD
66
used a screening cascade similar to that described for BC. First, a selection of small fragments (then called “needles”) was made from a database of available compounds by pharmacophore fitting and docking into the GyrB ATP binding site. Then, this set of 3000 fragments was tested for biochemical activity at high (0.5 mM) concentration. Finally, a set of 150 hits divided over 14 compound classes was taken to the validation stage, which included a second biochemical assay, various biophysical binding experiments (including SPR and NMR), and SAR elaboration. Further structure-guided optimization yielded lead compounds (e.g.,
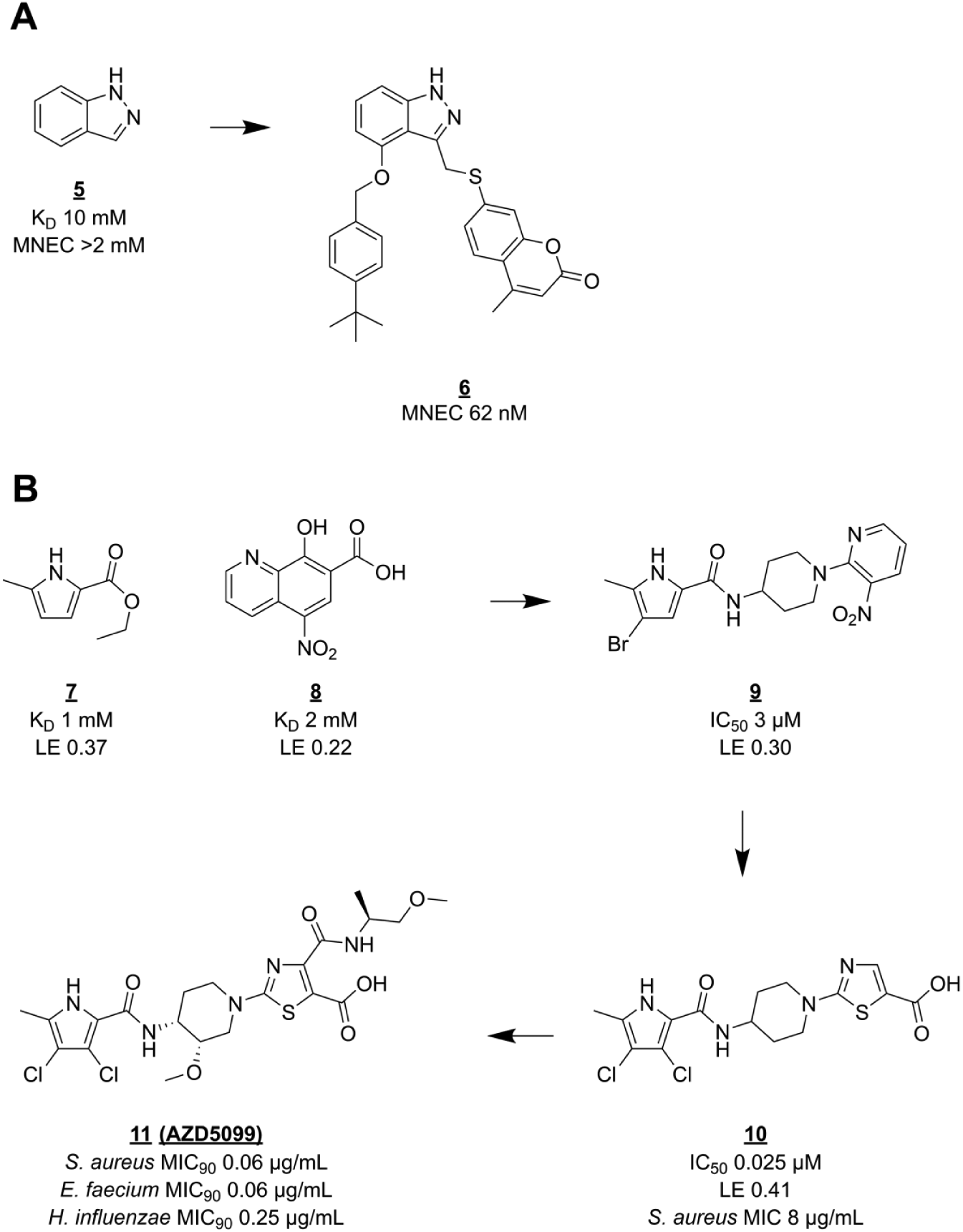
Hit-to-lead optimization of GyrB inhibitors. (
More recently, researchers at AstraZeneca reported the discovery
67
of pyrrolamide GyrB inhibitors and their development
68
into clinical phase. A low-affinity pyrrole carboxylate (
Currently, two new DNA gyrase inhibitors, both discovered during whole-cell screening, are being evaluated by Entasis Therapeutics and GlaxoSmithKline in late-stage clinical trials for Neisseria gonorrhoeae infections. 8
Cell Division Protein FtsZ
FtsZ plays an essential role in the separation of newly forming cells during bacterial replication. 70 It is homologous in structure to eukaryotic β-tubulin, a cancer target in humans. Like tubulin, FtsZ binds guanosine triphosphate to polymerize into strands, but FtsZ is only needed for cell division and not for chromosome separation. Inhibition of FtsZ does not directly stop growth but leads to formation of long filaments or large “balloons” that eventually lyse. Development of antibacterials targeting FtsZ has been ongoing for well over a decade. Most of the reported compounds act by preventing FtsZ polymerization, destabilizing polymers, or stabilizing polymers. 71
Shortly after the discovery of the function of FtsZ,
72
the protein was reported as the primary target for the fragment-like microbiological tool compound 3-methoxybenzamide (
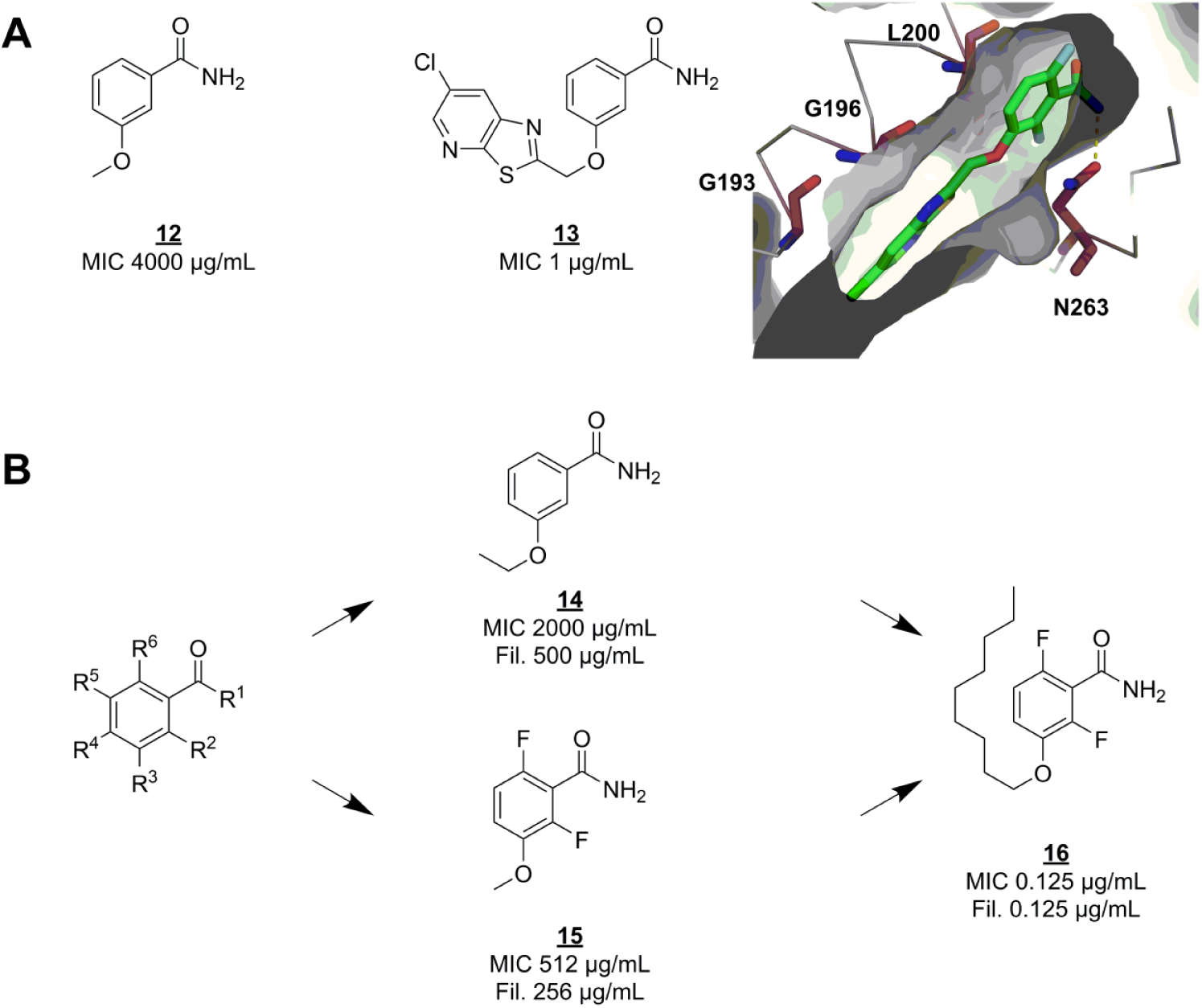
SAR exploration of the benzamide scaffold. (
This compound class is one of the most promising candidate antibiotics, even though it has a narrow spectrum (S. aureus, including drug-resistant strains) and has proven difficult to optimize in terms of pharmacokinetic properties. 78 It is currently under active preclinical development by Taxis Pharmaceuticals.
EthR
Isoniazid and ethionamide are anti-TB drugs targeting the same component of the mycolic acid synthesis pathway of mycobacteria. Because effective use of ethionamide requires high doses associated with liver toxicity, a way to lower the required dose would be an attractive therapy. Isoniazid and ethionamide have different resistance profiles because they are prodrugs activated by different bacterial enzymes. Ethionamide is activated by EthA. 79 The ethA gene is regulated by the transcriptional repressor EthR. 80 Therefore, several drug discovery projects have searched for EthR inhibitors as adjuvants for ethionamide. EthR is a relatively new anti-TB target with promising early inhibitors, as will be described. It will be interesting to see whether EthR inhibitors can be developed into therapies, as the role of EthA in ethionamide activation is not yet fully understood, as suggested by recent reports of redundant mechanisms.81–83
EthR has a deep, narrow, hydrophobic pocket that facilitates allosteric deactivation upon compound binding. Researchers at the Pasteur Institute found several active hits among a small set of compounds selected by pharmacophore modeling.
84
After attempting to expand the hits inside the binding pocket by in situ click chemistry,
85
they took one of the click reaction components,
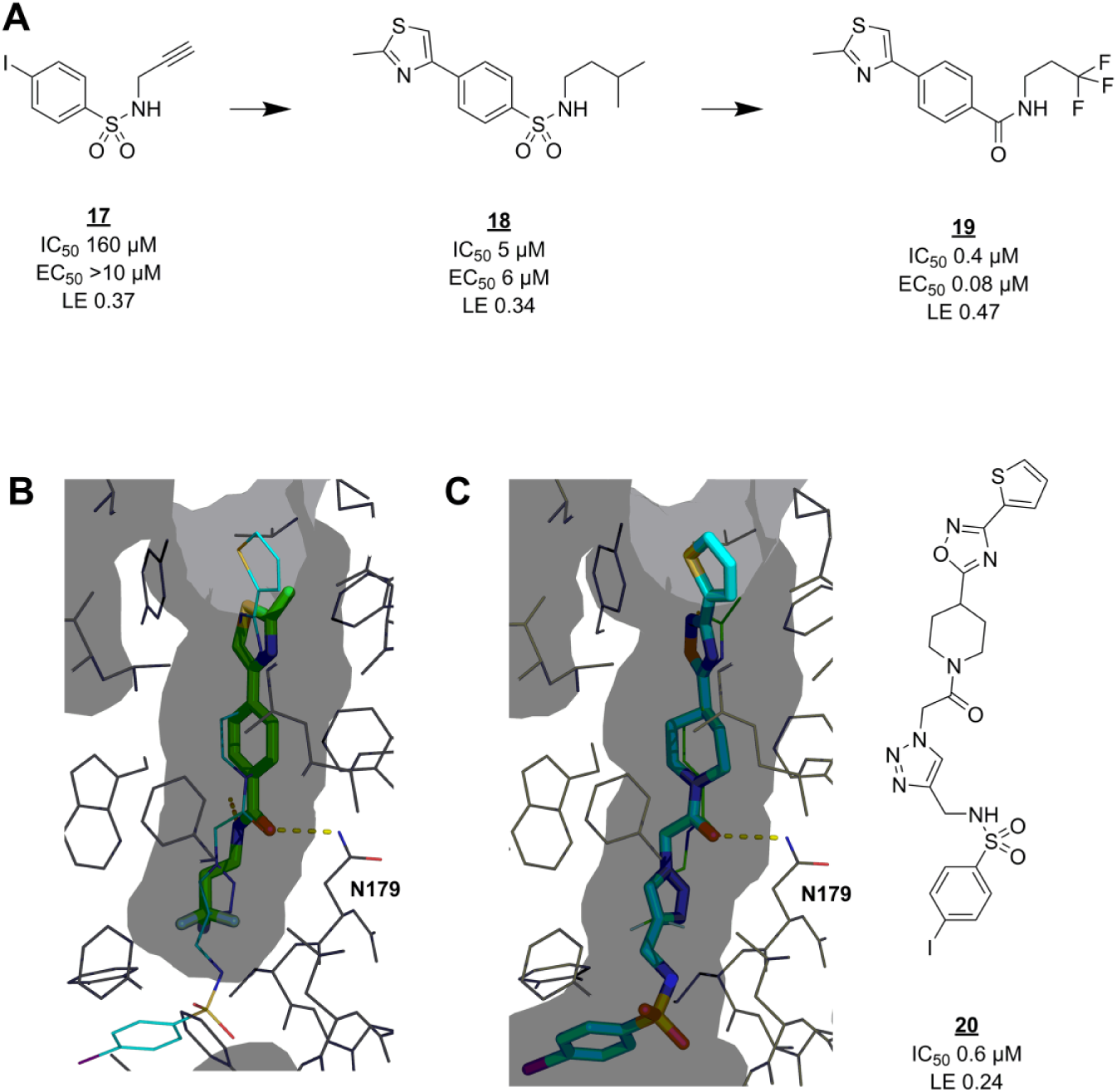
Fragmentation approach for EthR inhibitors. (
Fragment screening against EthR has produced another interesting series of compounds. Noting that the allosteric pocket of EthR is hydrophobic, Surade and colleagues used TSA to identify 86 hits from a library of 1250 fragments, some of which stabilize EthR by more than 5 °C.
87
The hits were validated using orthogonal biophysical techniques. As mentioned earlier, it is not trivial to select the best primary and secondary screening techniques and to interpret their combined data, as each technique has its own false-positive or false-negative rates and can be more suitable for certain screening subjects or libraries than others.
88
In this case, SPR validation data agreed well with the primary screen, with only 1 out of 45 negative controls (nonhits from TSA) showing up as an inhibitor in SPR. Only slightly more than half of the 86 TSA hits were identified as inhibitors, perhaps because of the large difference in assay concentrations (10 mM in TSA vs 0.5 mM in SPR). Several validated hits were soaked into EthR crystals to elucidate their binding modes. Interestingly, three hits contained similar arylsulfone scaffolds, as discussed in the previous example, with their sulfone groups interacting with N179. A fourth hit,
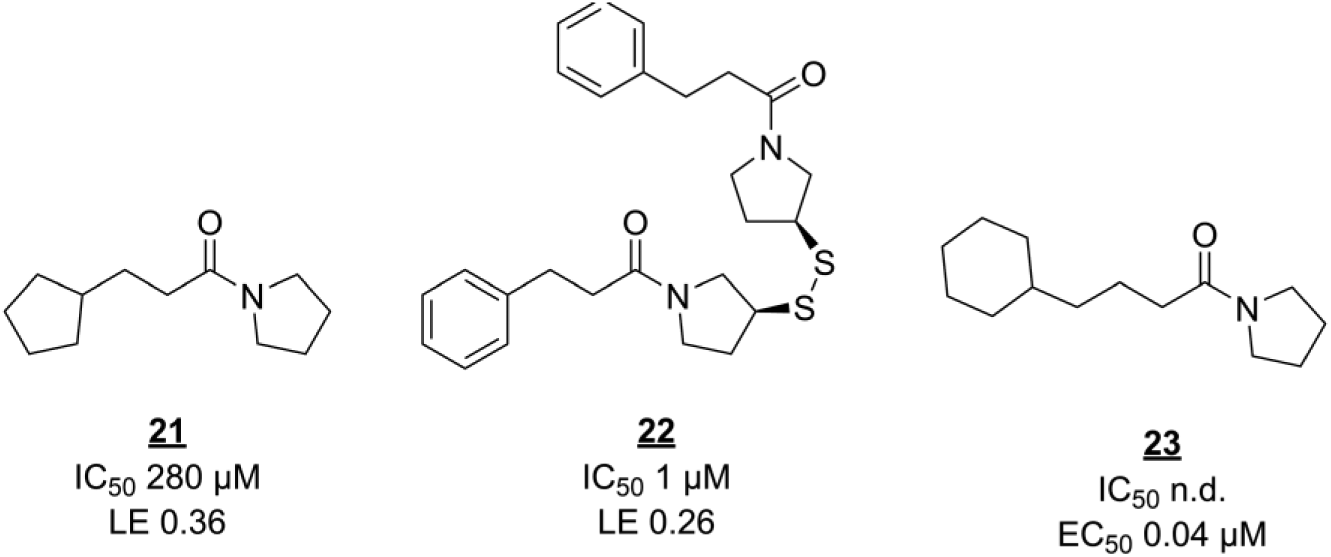
Fragment screening derived inhibitors of EthR. n.d. = not determined.
β-Lactamase
Evolution of plasmid-based β-lactamases to confer resistance to standard antibiotics was one of the first signs of a new era in healthcare. As the problem grew, inhibitors of β-lactamases (another example of adjuvants) were being used in hospital settings but not to great effect. 91 First-generation inhibitors like the natural product clavulanic acid, themselves β-lactams, quickly lost effectiveness due to resistance and narrow activity spectrum. Another class of β-lactamase inhibitors is boronic acids. Although their effects were noted since the 1980s and gained interest from researchers, no boronic acids entered clinical use until very recently, with the Food and Drug Administration’s (FDA) approval of a combination therapy of vaborbactam with a carbapenem for urinary tract infections.
Sulfonamide boronic acids are another recently developed class of β-lactamase inhibitors. As with vaborbactam,
92
early discovery and optimization of these molecules were guided by docking and modeling.93,94 Although several compounds in this series displayed high in vitro inhibitory activities, their effects on the antibacterial properties of β-lactamase-sensitive cephalosporins were only modest and did not follow the same trends. Reasoning that increasing β-lactamase affinities even further could solve the problem, the researchers made an in-depth analysis of possible enzyme–ligand interactions using results from virtual fragment screens and known inhibitor fragmentation approaches.
95
Bound fragments can give great insight into the characteristics of the interactions of larger ligands, because fragment binding modes are unconstrained by distant binding pocket geometries. Thus, with encouraging modeling results, lead inhibitor
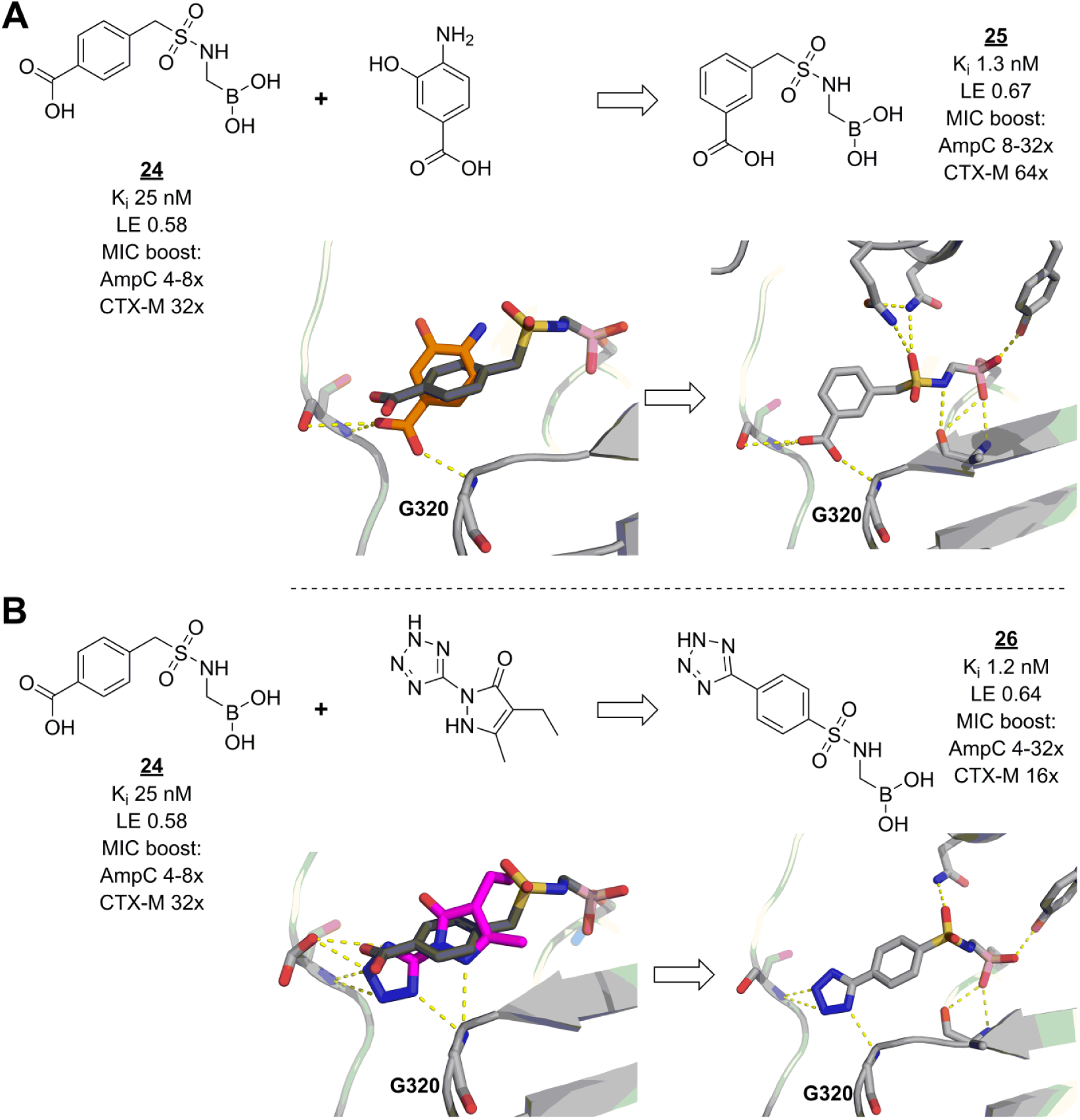
Fragments guide lead derivatization of β-lactamase inhibitors. (
Discussion
Not enough novel antibiotics have entered the market to mitigate the threat of AR. It appears that the challenge of developing antibiotics is a particularly difficult one, and there have been several explanations for this: biased screening libraries (both synthetic and organism-derived natural products) 96 and inadequate target selection, validation,11,97 or screening methods. 98 Apart from these, the fact remains that antibiotics development is more difficult because of the extra barriers that bacteria present between a drug and its target, such as additional and different (from human) cell membranes, drug efflux, and metabolism systems. Possible solutions to these problems have been proposed, though most of them too recently to influence the antibiotics pipeline yet. For example, important aspects of drug efflux mechanisms have been elucidated,99–101 allowing more rational design of inhibitors and other antibiotics. Similarly, past successful and failed antibiotics give insight into their properties that are important for membrane permeation.15,102,103 However, all these aspects still put extra constraints on a drug’s properties. Additionally, to address the risk of rapid development of resistance, antibiotics must have exceptional efficacy and low toxicity. A hit might be modified so heavily to address all these issues that some of the original properties are lost, or worse, the hit properties conveying efficacy are incompatible with those needed for good permeability, stability, and safety. Though this might seem exacerbated in the case of fragment-based hits, FBLD could offer an advantage by having a higher chance of generating other lead scaffolds (which is why we prefer the term fragment-based lead discovery). The process from lead to candidate is much the same, by whichever method a lead was found.
Recently, different approaches to screening for antibacterials have been explored, including exploration of natural products from uncultured bacteria
104
and target-based whole-cell screening.
98
Here various FBLD approaches in antibiotic lead discovery have been discussed. One of the principles of FBLD is that it is target based. Given a target, FBLD can be used to efficiently generate leads with control over desired properties, such as lipophilicity and selectivity. For example, for GyrB, fragment screening was a good choice because it offered a way of finding new compounds for a validated mechanism of action. The fragment hit
In the case of GyrB leads
In some cases, such as cell division protein target FtsZ, target-based whole-cell screening can even start from the fragment stage. The development of FtsZ inhibitor
There have recently been some exciting demonstrations of using fragments as probes for target identification in phenotypic screening. Off-target effects are expected because of the small size of the fragment, but it should have different affinities for the various targets, allowing the major targets to be identified. A recent example is where each of the fragments in the library is attached to a photoactivatable group—this covalently links the fragment to a target(s), which is then analyzed by mass spectrometry, together with methods of stable isotope labeling with amino acids in cells. 105 This could essentially be described as whole-cell screening with direct detection of target binding and has generated a lot of interest. It will be interesting to see if this idea of functional screening of fragment libraries is successful in identifying new targets for antibiotic drug discovery.
Footnotes
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: B.L. is supported by the European Union’s Horizon2020 MSCA Programme under grant agreement 675899 (FRAGNET); research in the group of R.E.H. is additionally supported by research grants from the BBSRC and institutional infrastructure support from funds provided by the Wellcome Trust and EPSRC.