
Abstract
Pseudomonas infections are common among hospitalized, immunocompromised, and chronic lung disease patients. These infections are recalcitrant to common antibacterial therapies due to inherent antibiotic resistance. To meet the need of new anti-Pseudomonas drugs, a sensitive, homogenous, and robust assay was developed with the aim of identifying inhibitors of acyl–coenzyme A synthetases (ACSs) from Pseudomonas. Given the importance of fatty acids for in vivo nutrition of Pseudomonas, such inhibitors might have the potential to reduce the bacterial fitness during infection. The assay, based on a coupled reaction between the Pseudomonas spp. ACS and the firefly luciferase, allowed the identification of three classes of inhibitors by screening of a diverse compound collection. These compounds were confirmed to reversibly bind ACS with potencies in the micromolar range. Two classes were found to compete with acyl–coenzyme A, while the third one was competitive with fatty acid binding. Although these compounds inhibit the bacterial ACS in cell-free assays, they show modest or no effect on Pseudomonas growth in vitro.
Introduction
Infectious diseases lead to approximately 30% of deaths worldwide, and antibiotic therapy represents the main treatment for bacterial infections. However, the emergence and dissemination of resistance among bacterial pathogens is narrowing treatment options to a few last-resort drugs. In addition, these molecules are losing efficacy, thus opening a worrisome clinical scenario on global scale. 1 The shortage of effective antibiotics in the industrial pipeline highlights the need for novel antibacterial drug discovery initiatives. To this aim, the screening of drug-like and lead-like compound libraries constitutes a viable strategy.
Among the drug discovery approaches to search for new antimicrobial agents, high-throughput screening (HTS) seems to be the most productive. This approach consists in testing extensive libraries of chemical or natural compounds against specific microbial targets, with the aim of finding novel antibacterial drugs. 2 In this approach, (1) the chemical diversity of the compounds within the library, (2) the relevance of the target in bacterial infection, and (3) the suitability of the assay performed to test the compounds are crucial.2,3 Indeed, the assays used in HTS campaigns must be simple, reproducible, and cost-effective. 4 HTS approaches have been widely used to identify broad-spectrum antibiotics in vivo. However, the incessant failures in the discovery of broad-spectrum antibacterial agents have led scientists to a targeted approach aimed at identifying compounds that specifically inhibit a crucial metabolic process and/or the virulence of a given pathogen. 3
Among the most worrying pathogens, the Gram-negative bacterium Pseudomonas aeruginosa represents a major cause of hospital-acquired infections. 5 P. aeruginosa is also responsible for morbidity and mortality in patients suffering from chronic lung disease, like chronic obstructive pulmonary disease and cystic fibrosis (CF).6,7 P. aeruginosa is able to uptake and metabolize environmental fatty acids (FAs) using fatty acid degradation (Fad) enzymes, which also play a role in the fitness of this pathogen during lung infection. The lung surfactant phosphatidylcholine (PC) represents an abundant reservoir of FAs that can be metabolized by P. aeruginosa as carbon sources. 8 The major nutritional component of PC is represented by the two highly reduced long-chain FAs linked to choline. In the lung of CF patients, P. aeruginosa expresses and secretes phospholipases and lipases, which enable the degradation of PC to choline and FAs, thereby providing substrates to support bacterial replication in vivo. 9 In order to convert FAs derived from PC degradation to a usable carbon source, FAs must be transported inside the cells by the membrane transporter FadL. 10 Once inside the bacterial cell, and before entering the β-oxidation pathway, FAs are activated by a class of enzymes, named fatty acyl–coenzyme A synthetases (ACSs). 9 Although P. aeruginosa expresses six different ACSs, growth studies performed with different ACS mutants suggest that FadD1, FadD2, and FadD4 represent the main contributors to FA metabolism. 8 Among them, the best characterized are FadD1 and FadD2. The expression of FadD1 is mainly induced by the presence of long-chain FAs in the medium, while FadD2 is specifically induced by short- to medium-chain FAs. 9 It has been demonstrated that FadD genes are upregulated in P. aeruginosa during the lung infection in CF patients. 11 In vivo studies demonstrated a key role of these enzymes in the virulence and fitness of P. aeruginosa in a mouse model of lung infection. 9 The importance of FA degradation during lung infections makes the enzymes involved in β-oxidation suitable targets to undermine P. aeruginosa fitness in vivo.
Although targeting ACSs may represent a viable approach to reduce P. aeruginosa fitness, no compounds able to inhibit P. aeruginosa ACS have been identified to date. The aim of the present work was to set up an assay to identify small molecules with the potential of inhibiting ACS in vitro. We have developed and optimized an ACS activity assay for HTS, based on a luminescence readout, which was then successfully used to screen a library of more than 60,000 compounds, and allowed the validation of three classes of bona fide ACS activity inhibitors.
Material and Methods
ACSs Activity Assay
Pure ACS from Pseudomonas sp. (EC 6.2.1.3), coenzyme A (CoASH), sodium oleate, ATP, DTT, and FA-free bovine serum albumin (BSA) were purchased from Sigma-Aldrich (St. Louis, MO); ATP Kinase-Glo kit was purchased from Promega (Madison, WI). The ACS luminescent assay was performed in 384-well black plates (Greiner Bio One, Frickenhausen, Germany). Briefly, compounds from 10 mM DMSO stock solutions were transferred to assay plates by acoustic transfer (EDC Biosystems, Milmont, CA). The following components were then added to the reaction in a final volume of 40 µL: 100 mM HEPES (pH 8.1), 10 mM MgCl2, 0.4% Triton X-100, 1 mM DTT, 0.01% FA-free BSA, 25 µM CoASH, 10 µM ATP, 5 µM sodium oleate, and 5 µU ACS. After 90 min incubation, the residual amount of ATP was detected by the Kinase-Glo kit as per the manufacturer’s protocol. The luminescent signal was acquired by a ViewLux plate reader (PerkinElmer, Waltham, MA).
The screening results were analyzed using the Dotmatics suite (Dotmatics, Bioshops Stortford, UK). The half-maximal inhibitory concentration (IC50) values were calculated by fitting the dose–response curve with a four-parameter logistic regression.
Competition Assays
Competition assays were performed at two concentrations (16 and 64 µM) of competing compounds. The reaction buffer and components were identical to those used for the ACS assay described above, with the exception of 25 µU of ACS being used together with a serial dilution of either oleate or CoASH. After 30 min incubation at 22 °C, the amount of ATP was revealed using the Kinase-Glo kit as per the manufacturer’s protocol. The luminescent signal was measured using the ViewLux plate reader (PerkinElmer). IC50, Vmax, and Km values were calculated using the Prism software (GraphPad Software, San Diego, CA).
Compound Similarity Search
Compound similarity searches were performed by Tanimoto similarity 12 using the extended fingerprint method implemented in KNIME (KNIME.COM, Zurich, Switzerland) 13 through the CDK toolkit14,15 An extended fingerprint was calculated for all molecules within the search collection; then, by means of the KNIME similarity search node, a subset of nearest neighbors of each query molecule was selected, setting the appropriate Tanimoto similarity threshold.
Binding Analysis by Surface Plasmon Resonance
Surface plasmon resonance (SPR) interaction analysis was performed using a Biacore T200 (GE Healthcare, Uppsala, Sweden). ACS was immobilized on a CM5 chip by amine coupling according to the manufacturer’s protocol (Amine Coupling Kit, GE Healthcare). Briefly, the surface of the sensor chip was activated for 7 min using a mixture of 0.1 M N-hydroxy succinimide (NHS) and 0.4 M N-ethyl-N′-[3-dimethyl-aminopropyl] carbodiimide (EDC); then, 30 µg/mL of ACS in 10 mM sodium acetate (pH 4.5) was injected for 420 s at 10 µL/min. Finally, residual activated groups on the surface were blocked by injection of 1 M ethanolamine (pH 8.5) for 7 min. A reference channel for background subtraction was prepared by activation with a EDC/NHS mixture (0.1 M/0.4 M as per ligand immobilization), followed by blocking with 1 M ethanolamine. The binding of the selected hits to the immobilized ligand was evaluated by a multicycle kinetic procedure in PBS-P buffer (GE Healthcare) supplemented with 5% DMSO. The analyte was injected for 2 min at 30 µL/min until equilibrium, and the dissociation was monitored for 3 min. Biomolecular binding events were reported as changes of resonance units (RUs) over time. Reference channel subtracted sensorgrams were analyzed by the Biacore T200 evaluation software. Compound binding affinity was evaluated by plotting the response at the equilibrium versus the concentration of the analyte.
Bacterial Strains, Media, and Growth Inhibition Assays
P. aeruginosa PAO1 (ATCC 15692) and Pseudomonas fragi (DSM 3456) were routinely grown in Luria-Bertani (LB) medium at 37 and 30 °C, respectively. For growth inhibition assays, overnight cultures were washed twice in M9 minimal medium and inoculated at OD600 = 0.01 in 96-well microtiter plates containing 200 µL of M9 plus 1% BriJ-35 and either 1% casamino acid (CAA) or 0.2% FA (adipic or oleic acid) as carbon sources. Bacterial growth was monitored by OD600 measurements over time using a Victor3V plate reader (PerkinElmer). FAs were dissolved at 3% in 1% Brij-35, as previously described. 16 ACS inhibitors were solubilized in DMSO at 100 mM and used at a 100 μM final concentration.
Results
Assay Development and Optimization
With the aim of developing a new, reliable, and cost-effective luminescent assay, suitable to measure the activity of ATP-dependent microbial enzymes, the commercially available ACS enzyme from Pseudomonas spp. was chosen. In order to obtain a detectable luminescence signal in response to the activity of the ACS, the residual ATP after conversion of oleate to oleyl-CoA was quantified by a luciferase enzyme ( Fig. 1 ). The assay optimization was carried out in a 384-well plate in order to ensure the seamless application of the optimized protocol to an HTS campaign.
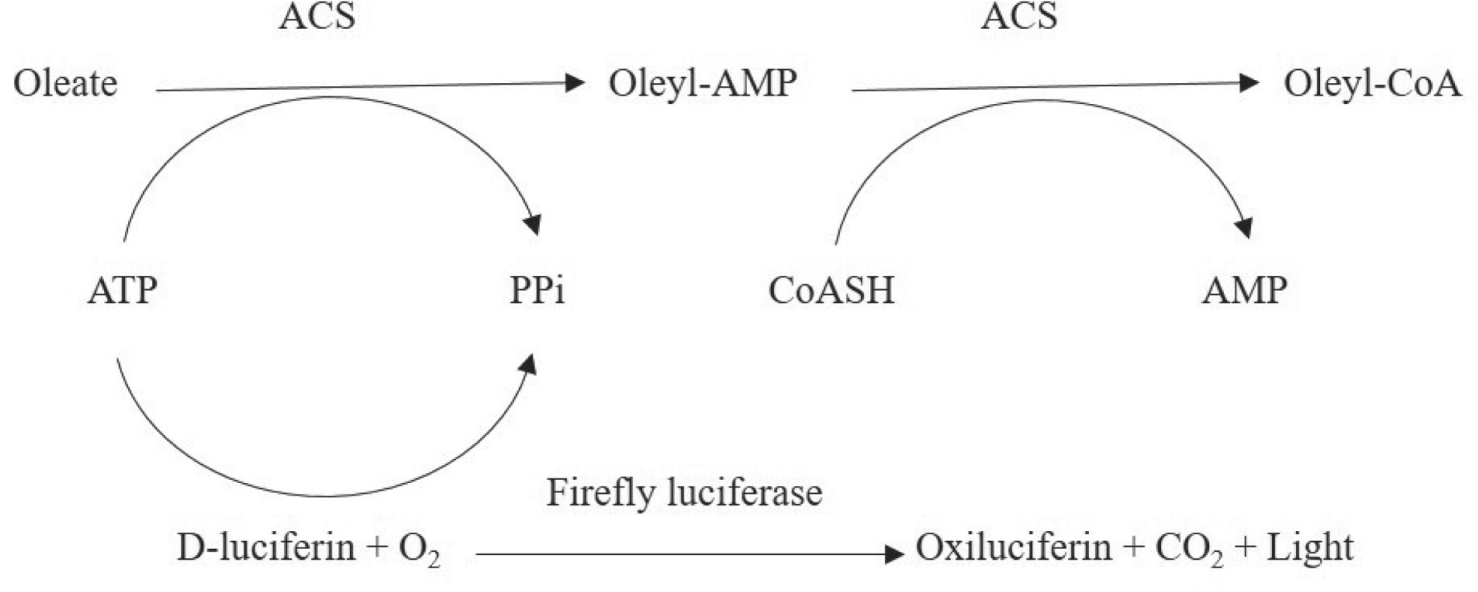
Schematic of the ACS assay reaction. The lucifarase activity is dependent on the residual ATP concentration after the two-step ACS reaction. As a consequence, the luminescent signal is inversly proportional to the ACS activity.
An initial attempt aimed at grossly proving the feasibility of the experimental setup was made by varying the concentration of the enzyme and the oleate substrate. In order to better appreciate a reduction of ATP due to the ACS activity, the concentration of ATP was set to 10 µM. This concentration is close to the upper limit of quantification of the Kinase-Glo assay (i.e., the luciferase reaction kit used in the present work). The CoASH concentration was initially set to 50 µM in order to have it in molar excess with respect to ATP. The measured residual ATP was dependent on both the enzyme and oleate concentration ( Fig. 2A ), proving that the assay format is suited to the aim of the present work. Following preliminary demonstration of the assay feasibility, the Km values of substrates (oleate and CoASH) were determined in order to optimize their concentration in the assay. To this purpose, alternate serial dilutions of either oleate or CoASH were tested in the presence of an excess of ACS enzymatic activity (50 µU) and 10 µM ATP, as previously described. The luminescence signal was recorded during the first 15 min for each concentration of the two substrates. The V0 calculated for all the conditions allowed us to establish that both substrates are compliant with the Michaelis–Menten kinetic ( Fig. 2B , C ), with the oleate Km being 5 µM and the CoASH 25 µM.
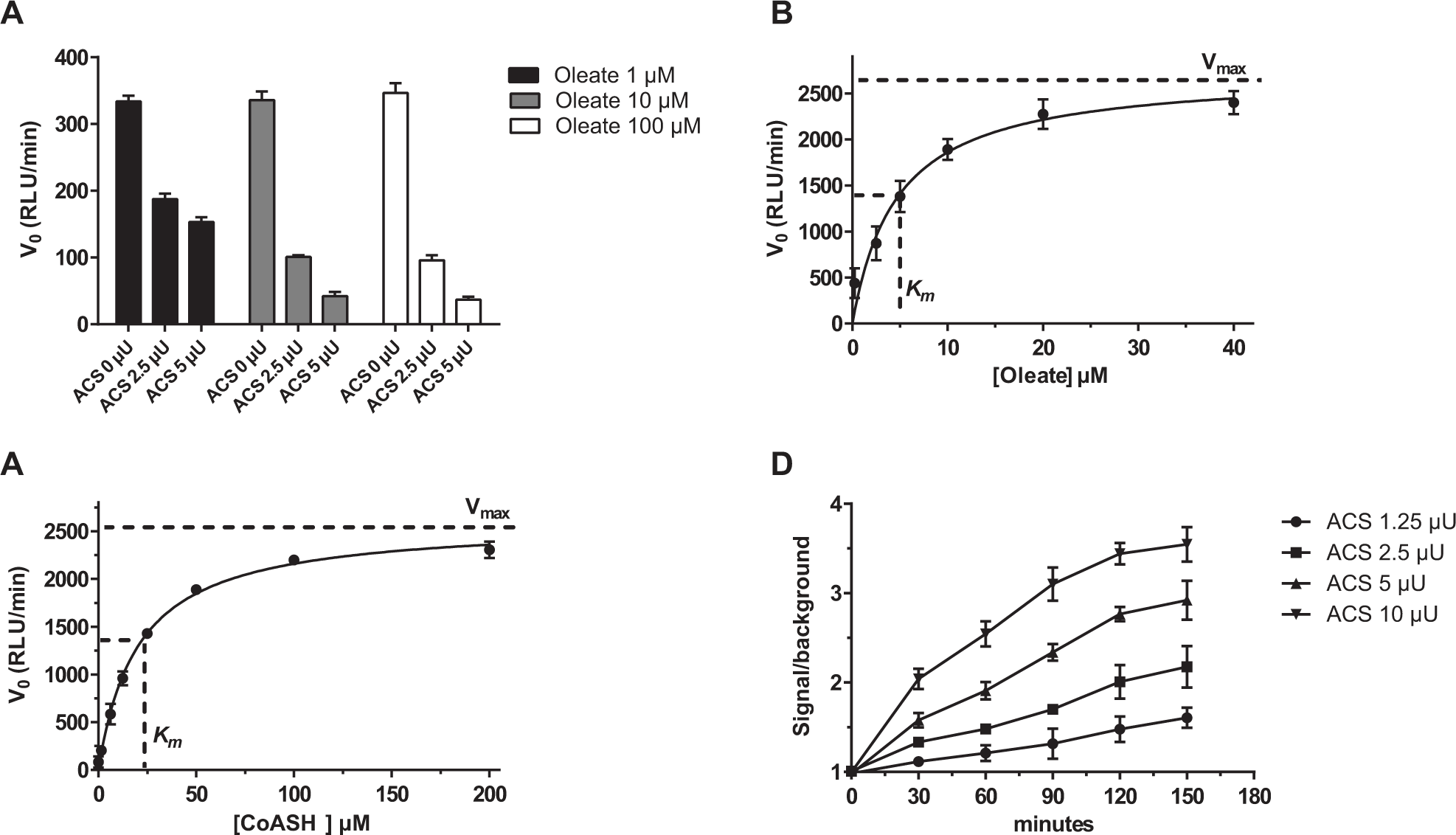
Development of the ACS reaction in a 384-well plate format. (
To further optimize the reaction protocol, a time course experiment was carried out in which different concentrations of the ACS enzyme, ranging from 1 to 10 µU, were used, while the oleate and the CoASH concentrations were fixed at the Km values. The ATP concentration was set to 10 µM, as reported above. The aim of this optimization step was to identify the proper enzyme concentration and reaction time in order to obtain a feasible compromise among (1) the lowest possible enzyme concentration to reduce the screening cost and (2) a time point at which a clear signal is detected over the background and (3) where the rate of the substrate consumption is still constant (i.e., a linear trend is appreciable between signal and time). Five microunits and 90 min incubation at 22 °C were identified as the most suitable enzyme concentration and assay duration. In fact, these are the lowest enzyme concentration and the shortest time point at which the signal to background is acceptable, while the linearity between the reaction signal and time is maintained ( Fig. 2D ). The percentage ATP conversion in the optimized assay conditions was determined to be 62% ( Suppl. Fig. S1 ).
Hit Identification
Using the above-obtained conditions, a stability study was carried out to determine the reagent stability during the HTS run. In the absence of a known enzyme inhibitor, the positive control was set to be the “no-enzyme” reaction. The ACS enzyme, ATP, and CoASH stock solutions were kept at 4 °C, while the oleate and Kinase-Glo reagents were kept at 22 °C. The results of the reagent stability test confirmed that the assay readout is consistent up to 300 min under the test conditions. In fact, the Z′ 17 values calculated between the reaction and the no-enzyme controls were greater than 0.5 at all the tested time points ( Fig. 3A ). A minor Z′ decrease trend with time was appreciated, which was not considered to be relevant. As a consequence, the optimal size of a single batch of assay plates was set in order not to exceed 5 h of total operation time.
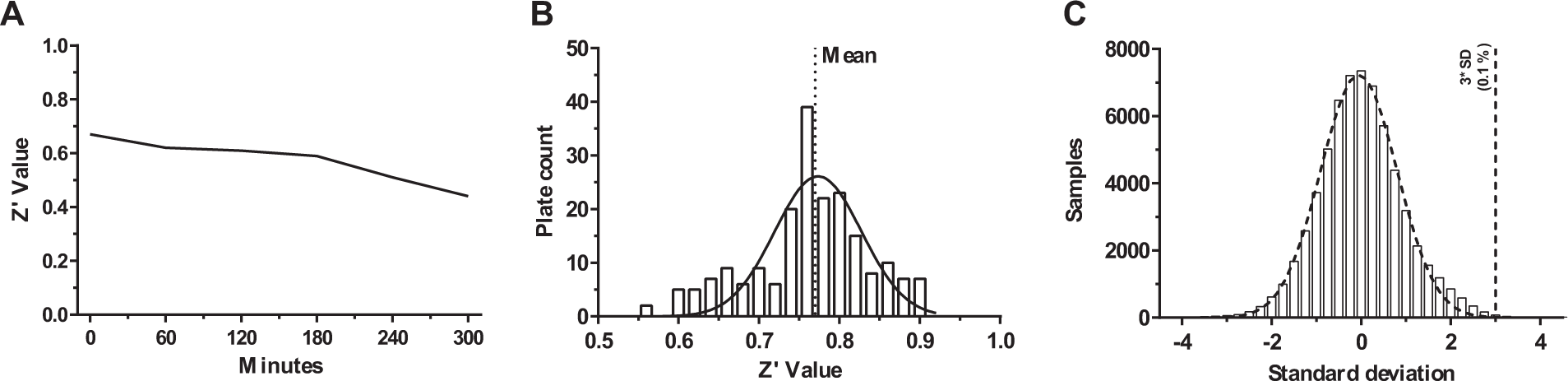
(
A collection of around 64,000 compounds was then screened using the above-described ACS inhibition assay at a fixed concentration of 5 µM. The collection contains a mixture of druglike and leadlike structures, with more than 90% of the compounds having a molecular weight below 500 Da (the mean molecular weight is 348 Da). The total polar surface area (TPSA), as well as the Lipinski rule of five features, is in the suitable range for orally bioavailable compounds; the average number of rotatable bonds is 5. When screened for diversity by both MACCS166 fingerprints 18 and methods measuring the maximum common substructure of the central scaffolds, the collection clusters into ca. 4500 distinct clusters, of which more than 1000 are singletons. The average number of molecules per cluster is around 6. The Z′ values of all the tested plates were found to be greater than 0.5, with an average of 0.77 ( Fig. 3B ). The activity of each compound was calculated as the ratio between the compound signal and the sum of the whole compound average plus one standard deviation. The distribution of the compound activity was found to be Gaussian ( Fig. 3C ); as a consequence, compounds with an activity above the sample average plus three standard deviations were considered hit compounds. In fact, the probability of these compounds to be identified as active by chance is less than 0.1%. Seventy-nine compounds, 0.12% of the total, were identified as active in the primary screening and subjected to the confirmation assays. The number of hits was found modest, likely because the assay design did not allow the identification of false positives. In fact, in most assays, false positives are caused by compounds that decrease the assay signal; thus, they are confounded with inhibitors. In our screening, compounds interfering with the luciferase activity produce a low signal, similarly to compounds not active on the ACS enzyme, decreasing the probability to spot false positives.
Hit Confirmation and Validation
The selected molecules were confirmed in a dose–response manner starting from a concentration of 50 µM. The confirmation process was carried out by means of the same assay used in the hit identification phase. Eleven compounds out of the 79 identified by the primary screen were confirmed to be active in the ACS assay, with IC50 potencies ranging between 5 and 15 µM. As anticipated, the design of the primary assay did not require running a luciferase inhibition counterscreen. Therefore, all selected compounds were further characterized in order to identify those acting by competing with either the substrate (oleate) or the cofactor (CoASH).
To this aim, the influence of the 11 selected compounds on the apparent Km (Kmapp) of either oleate or CoASH was determined. Alternate saturating dilutions of either oleate or CoASH were assayed on the ACS enzyme in the presence of vehicle plus 16 or 64 µM hit molecules. Compounds that could be considered competitive inhibitors were those causing a right shift of the Kmapp, either for the oleate or the CoASH, without affecting the Vmax.
19
Two compounds,
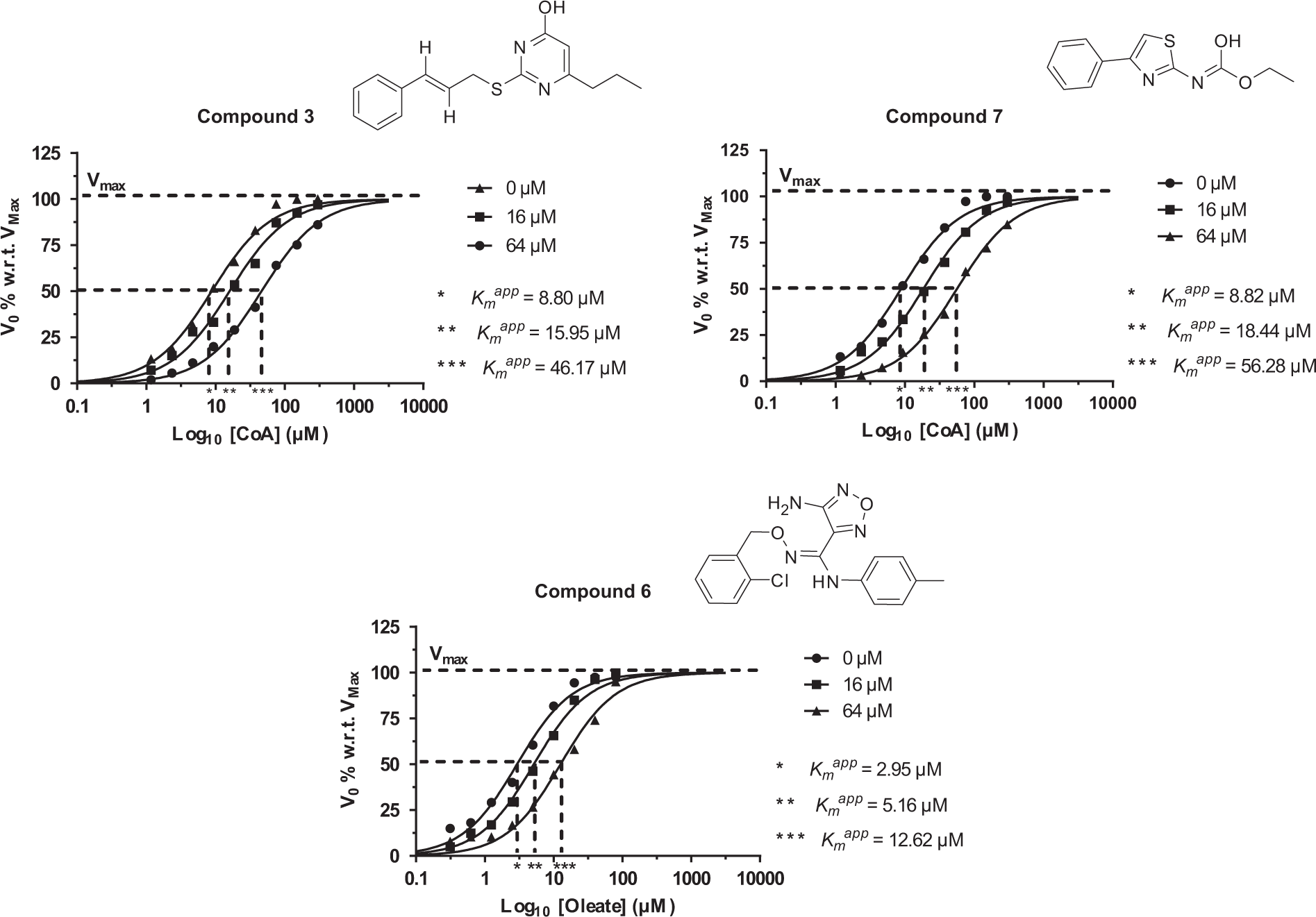
Hit compound competition for either CoASH or oleate binding to ACS. Either CoASH or oleate was titrated alone or against two hit concentrations (16 and 64 µM). The ACS assay was performed using 25 µU ACS and 20 µM ATP. Reactions were incubated for 30 min at room temperature.
Hit Compounds Binding to ACS
To further characterize the molecular interaction between ACS and compounds
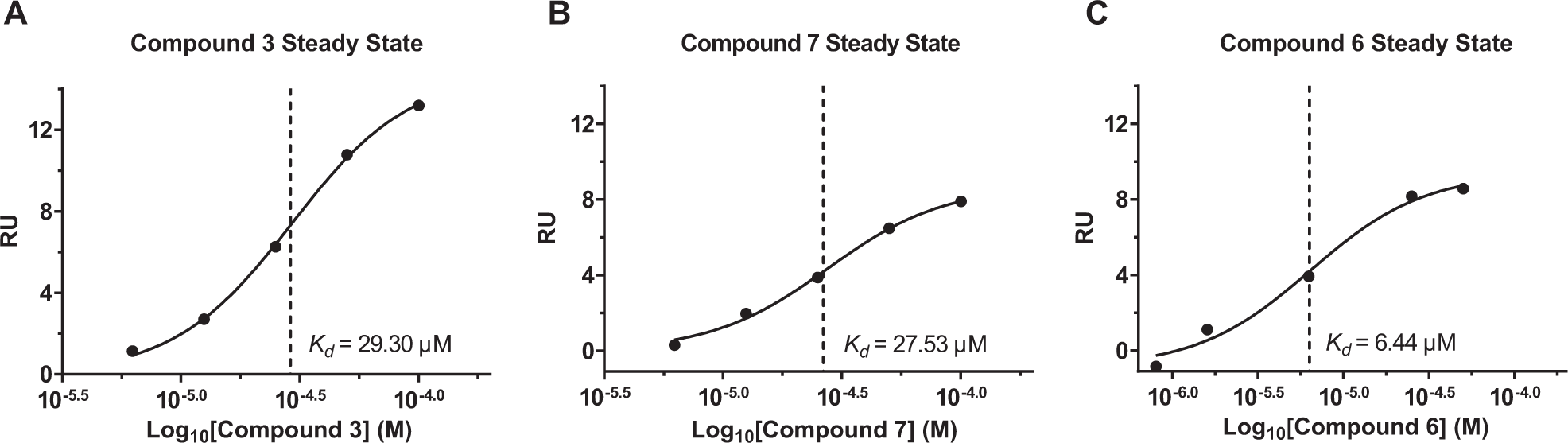
ACS binding assay by SPR. Compounds
Bacterial Growth Inhibition
The expression of Fad enzymes, the targets of the hit compounds identified by the HTS campaign, is known to be induced in P. aeruginosa by the presence of FAs as sole carbon sources.
8
Therefore, the ability of P. aeruginosa to grow in the presence of FAs as the sole carbon source was investigated. In particular, P. aeruginosa was grown in M9 minimal medium supplemented with 1% Brij-35 and either adipic or oleic acid. Oleic acid is a long-chain FA that may induce the expression of FadD1, while adipic acid is a short-chain FA that induces the expression of FadD2.
9
Growth in CAAs, as well as in oleic acid, was unaffected by the presence of any of the three hit compounds (
Fig. 6A
,
B
). Instead, a moderate growth inhibition (20%–30%, depending on the growth stage) was observed with either compound
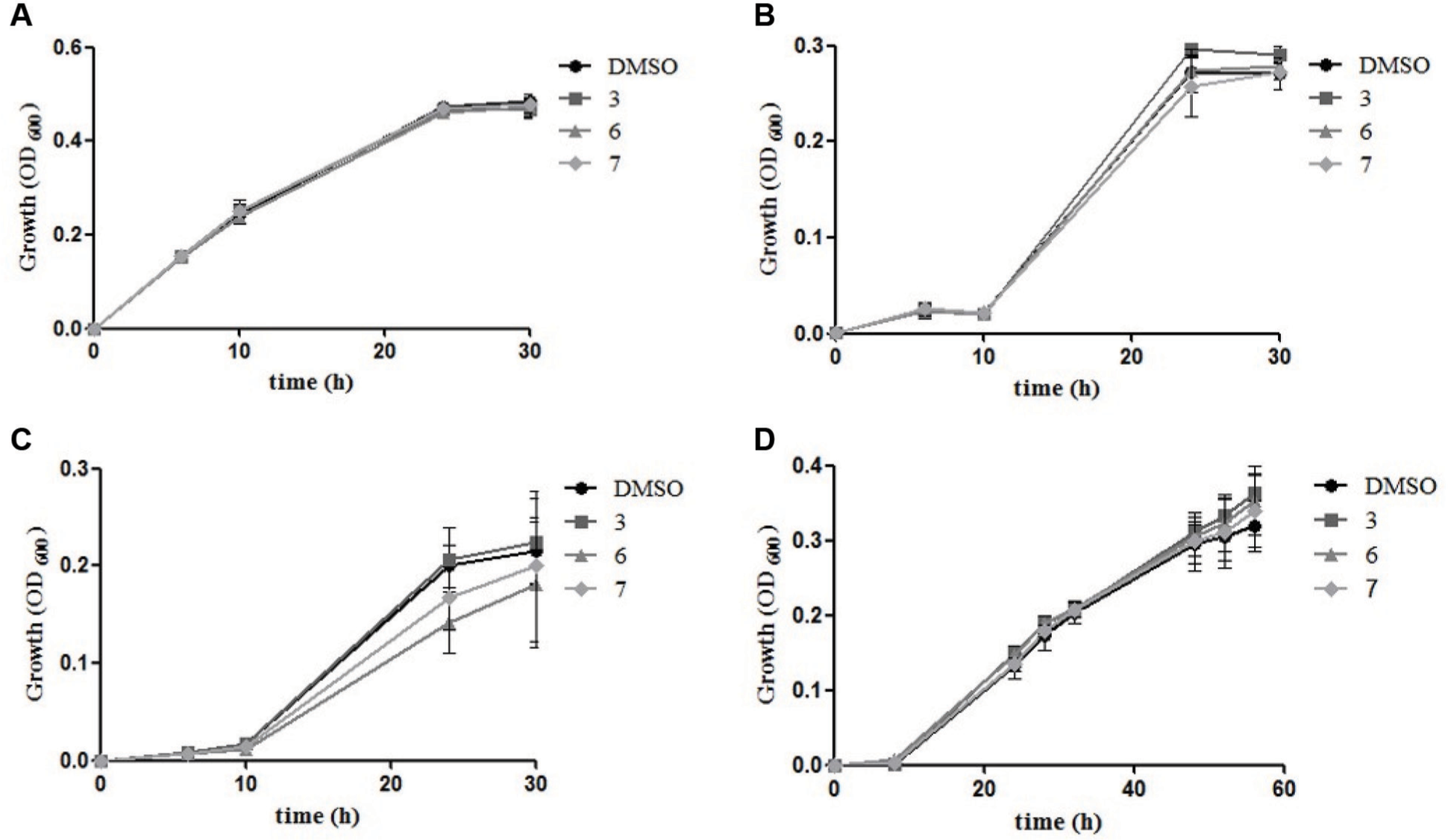
Effect of the hit inhibitory compounds on Pseudomonas growth. Growth of P. aeruginosa in M9 minimal medium supplemented with CAAs (
Discussion
The primary aim of the present work was the identification of Pseudomonas ACS inhibitors by screening of a large compound collection. To this aim, a new assay with a luminescence readout was developed that is able to measure ACS activity. The advantages of the presented design are the high reproducibility of the luminescence signal, together with a reduced risk of compound interference. In addition, given that the readout is high when the ACS enzyme is inhibited, there is no risk of selecting compounds that inhibit the luciferase reaction rather than the ACS enzyme. By this approach, three compounds able to inhibit the ACS enzyme in a low micromolar range were found by screening a diverse collection of ca. 60,000 compounds. To expand on the three hits, analogue molecules were selected that proved to be active in a concentration range similar to the one of the parent structures, holding promise for future investigation of structure–activity relationships (SARs). Interestingly, when the mode of ACS inhibition of the three classes was investigated, two were found to be CoASH competitive (compounds
Despite the significant inhibitory activity of the hit compounds in vitro, no pronounced effect on the bacterial growth was observed in vivo. Hit compounds
Target accessibility is a crucial point for an explanation of this phenomenon. The cytoplasmic localization of the ACS target enzyme implicates that the selected hit compounds have to pass both the outer and the inner membrane of the bacterial cell in order to reach their target. Despite the small molecular size of the selected compounds, it is possible that they do not cross the bacterial membranes. In addition, the low outer membrane permeability of P. aeruginosa is a well-known characteristic of this pathogen, providing an effective barrier to the penetration of antimicrobial compounds. 22 Moreover, the slow intake of antimicrobial compounds may also favor the activity of secondary intrinsic resistance mechanisms, such as efflux or compound-degrading enzymes.20,23 In our hands, medium supplementation with the membrane-permeabilizing agent EDTA did not increase the growth inhibitory activity of any hit compounds on P. aeruginosa (data not shown). We might therefore speculate that hit compounds might be pumped out or degraded upon entrance in the bacterial cells.
Low target accessibility is not the only plausible explanation of the inactivity or low activity of the hit compounds against P. aeruginosa. We investigated whether the poor sequence identities of all P. aeruginosa ACS homologues, to the commercially available ACS, may account for the low effect of the hit compounds. However, no growth inhibitory effect was observed for P. fragi, a bacterium encoding an ACS enzyme with nearly complete sequence identity to the one used in the HTS. This observation suggests that the moderate effects of the hit compounds on P. aeruginosa growth were not dependent on the poor similarity between P. aeruginosa ACS and the enzyme used in the HTS campaign. Of note, P. aeruginosa possesses six different enzymes endowed with ACS activity; therefore, there is the chance that, to exert a growth inhibitory activity, the hit compounds should act on multiple targets simultaneously. This target redundancy may also account for the poor or no inhibition observed for all three hits.
Although further studies are required to understand the reasons that impair the activity of the identified hit compounds in vivo, this work provides a new, cost-effective, highly reproducible method to assess the activity of ACS enzymes and other enzymes that require ATP as a cofactor.
Footnotes
Supplementary material is available online with this article.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The work was supported by a grant from Regione Lazio to P.V. for the project L13/2008 “Piattaforma integrata per lo screening di nuovi farmaci antimicrobici.” IRBM is also supported by CNCCS s.c.a.r.l.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.