
Abstract
There has been much debate around the success rates of various screening strategies to identify starting points for drug discovery. Although high-throughput target-based and phenotypic screening has been the focus of this debate, techniques such as fragment screening, virtual screening, and DNA-encoded library screening are also increasingly reported as a source of new chemical equity. Here, we provide examples in which integration of more than one screening approach has improved the campaign outcome and discuss how strengths and weaknesses of various methods can be used to build a complementary toolbox of approaches, giving researchers the greatest probability of successfully identifying leads. Among others, we highlight case studies for receptor-interacting serine/threonine-protein kinase 1 and the bromo- and extra-terminal domain family of bromodomains. In each example, the unique insight or chemistries individual approaches provided are described, emphasizing the synergy of information obtained from the various tactics employed and the particular question each tactic was employed to answer. We conclude with a short prospective discussing how screening strategies are evolving, what this screening toolbox might look like in the future, how to maximize success through integration of multiple tactics, and scenarios that drive selection of one combination of tactics over another.
Introduction
Since the early- to mid-1990s, high-throughput screening (HTS) has been a mainstay discovery tactic for most pharma and biotech companies and more recently has also expanded into academia. Screening of tens or hundreds of thousands of chemical entities against various extra- and intracellular molecular targets to find compounds with the desired mode of action is now a mature and well-embedded discipline. 1 The degree to which HTS has been successful in finding new starting points for lead discovery, which then translate into successful drug candidates, is a topic of widespread debate. Some estimate HTS success rates as about 50%, 2 whereas others consider this to be an underestimate. 3 The most active debate around success rates of screening tactics is the comparison between target-based HTS, in which a specific molecular target is screened against a very large diverse compound library, and phenotypic screening, in which compounds are tested in a target agnostic assay monitoring functional responses. Often, phenotypic screening is conducted on smaller compound libraries because of the complexities associated with the more physiologically relevant cells and frequently higher-content multiparametric readouts, although this is not always the case. Several publications suggest that phenotypic screening is the more successful of the two approaches in yielding first-in-class medicines.4,5 However, some argue that this is in part due to the long time frame of drug discovery and development and the relatively recent introduction of target-based approaches in that context. Eder et al. conducted an analysis of the origins of all 113 first-in-class drugs approved by the U.S. Food and Drug Administration between 1999 and 2013, showing that the majority (78) were discovered through target-based approaches. 6 An additional layer of complexity in these analyses arises from the fact that target-based drug discovery actually incorporates a wide range of different lead discovery tactics, all of which may have their own individual success rates at delivering first-in-class molecules. One of the reasons it is so difficult to define the success rate for HTS is that HTS is just one of several tactics employed early in the lifetime of a drug discovery project to try and identify new chemical matter, the outputs of which are integrated during a lead optimization campaign to ultimately deliver the candidate molecule.
In addition to HTS, fragment-based drug discovery (FBDD) has also become an increasingly mature lead discovery tactic in cases in which there is a target hypothesis. Rather than screening millions of compounds to find drug-sized starting points, FBDD begins with collections of low-molecular-weight (<250 Da) compounds, with most fragment libraries containing just a few thousand molecules, about three orders of magnitude smaller than a typical HTS collection. 7 Erlanson et al. 8 provide a comprehensive review of the outputs from FBDD, which include many marketed drugs such as vemurafenib, an inhibitor of mutant B-Raf proto-oncogene serine/threonine kinase (BRAF), and the first fragment-derived drug on the market.
Another notable target-based screening tactic that is increasingly applied in lead discovery is DNA-encoded library screening technology (ELT). DNA encoding involves the conjugation of chemical compounds to DNA barcodes.9,10 These libraries can then be screened via affinity selection, typically on an immobilized protein target. The concept of DNA encoding was first described by Brenner and Lerner in 1992 11 and enables the exploration of deeper chemical space than traditional HTS methods, as smaller quantities of protein (and compounds) can be used to screen libraries of many billions of molecules. The number of publications reporting successful use of ELT as a source of novel chemical matter has increased rapidly over the past 4 years and there are now numerous examples of the technique identifying high-affinity hits for biological targets relevant to drug discovery.12,13
It is not only experimental screening tactics that have yielded novel chemical starting points. Computational methods are now a well-established part of the lead identification toolbox. In silico compound libraries, far in excess of those accessible experimentally, can be rapidly evaluated against a computational model to prioritize molecules more likely to be active in experimental screens. 14 This approach is generally known as virtual screening 15 and became available in the 1980s.16–19 There are broadly two forms of virtual screening: ligand based and structure based. Ligand-based methods are best suited to hit generation studies in which there are known inhibitors or substrates, as they look for new templates with similar chemical or pharmacophoric features. In contrast, structure-based approaches simply require the availability of either an X-ray structure or homology model of the target protein, 20 and no chemical starting point is necessary. By the mid-1990s, some methods, particularly ligand-based methods such as pharmacophore searching, 21 were established as useful tools, especially in the “lead-hopping” use case, in which known ligands are used to define database-searching queries that identify novel chemotypes. 22 , 23 These techniques often out-performed fledgling HTS technologies, leading to debates on the cost-benefit of HTS investment. 24 As availability of structural information and computing power has increased, so have the number of examples where in silico approaches have successfully yielded chemical equity.14,25–28
Although there are examples of each of the aforementioned lead-generation tactics, both target-based and phenotypic, being successfully used in isolation, many organizations now recognize the power of their synergistic use to yield even greater success ( Fig. 1 ). A clear example of this is the use of phenotypic screening to identify a target, which is then prosecuted via a target-based approach to identify potent and selective starting points. 29 Phenotypic screening remains a challenging endeavor, particularly the ability to translate a screening hit into a clinical drug candidate. In an excellent recent review of the challenges and opportunities in phenotypic screening, Moffatt et al. 30 concluded that phenotypic and target-based drug discovery should be seen as complementary approaches, rather than an either/or scenario. The same can be proposed for the various target-based screening tactics: HTS outputs can inform on strategies for growth of fragment starting points, whereas the output of a fragment screen can provide information on the minimal pharmacophore within an HTS hit. Likewise, DNA-encoded library screening can supplement HTS hits with molecules with alternate binding modes and more depth of chemical space, and virtual screening is often used in combination with the other approaches described.
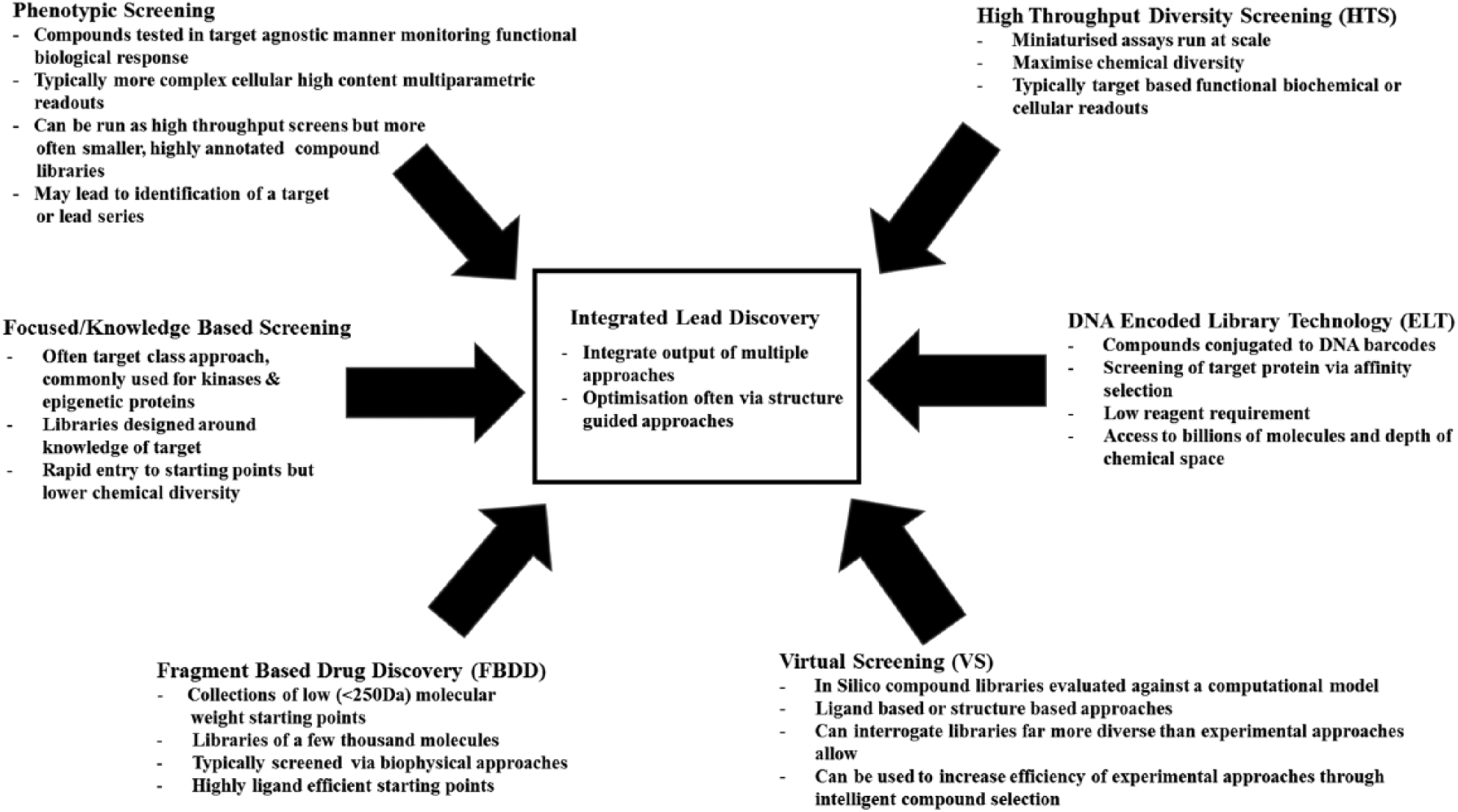
The current toolbox of hit discovery tactics employed in drug discovery and their key features; high-throughput diversity screening (HTS) is still the cornerstone of many hit identification campaigns and designed to maximize access to chemical diversity. DNA-encoded library screening technology (ELT), whereby chemical compounds are conjugated to short DNA fragment barcodes and then screened via affinity selection, typically on an immobilized protein target, is increasingly being deployed both in its own right and in conjunction with HTS. Fragment-based drug discovery, in which collections of thousands of low-molecular-weight compounds are screened typically using biophysical approaches in conjunction with structure-guided design to identify highly ligand efficient starting points, has precedent in the identification of several marketed drugs. Focused screening, typically using a target class approach based on existing knowledge, can be a rapid entry point to novel chemical matter. Structural and computational methods complement all the aforementioned screening tactics both to inform and guide structure-activity relationships during hit-to-lead campaigns but also frequently to increase efficiency of screening and modeling experiments that would be prohibitively expensive in the lab.
In this article, we illustrate the benefit of combining screening tactics to find lead series, highlighting examples in which ELT has been successfully deployed in combination with HTS and in which phenotypic screening can identify novel targets that are then successfully prosecuted via a combination of target-based screening tactics. We will focus in particular, although not exclusively, on case studies for two targets: receptor-interacting serine/threonine-protein kinase 1 (RIP1) kinase and the bromo- and extra-terminal domain (BET) family of bromodomains. In each example, we highlight the unique insight or chemistries individual approaches provided, emphasizing the synergy of information obtained from the various tactics employed. We end with a discussion on the evolution of screening strategies and technologies, and how that might alter the future lead discovery toolbox. Finally, we propose a series of scenarios which might drive selection of different combinations of lead discovery tactics, and illustrate how the overall lead discovery strategy should be tailored to the specific scientific question at hand, and based on the relative balance of risk and reward. We conclude that the likelihood of success will probably be highest when more than one approach is employed in a complementary manner, factoring in the strengths and weaknesses of each.
Deploying DNA-Encoded Library Technology in Combination with HTS and Other Screening Tactics
Both HTS and ELT are now well-established platforms for small-molecule drug discovery. Although they are often used independently, the examples below highlight synergies derived from coordinated campaigns performed in parallel. This parallel approach can maximize the respective strengths and offset the weaknesses of each platform, to improve overall quality and success rates. ELT provides access to deep chemical diversity in a rapid and efficient manner. Large numbers of unique compounds (millions to billions) can rapidly be assembled through split and pool chemistry. The diversity of chemical space that can be accessed in these collections is limited to diversity of available building blocks and the available DNA-compatible reaction space. However, significant progress continues to be made in reducing the historical issues with limited numbers of on-DNA–compatible chemical reactions and higher-molecular-weight DNA-encoded libraries (DELs). 31 In the past decade, rapid expansion in the numbers of building blocks and on-DNA–compatible reactions12,31–33 has enabled the generation of far more leadlike DELs, and ELT is now commonly deployed by several pharma companies alongside, and in some cases instead of, HTS as part of lead-generation campaigns, providing a highly complementary output. HTS itself has gone through many development phases since its inception, with a focus progressively evolving through a quest for high well counts, large collections, miniaturization, and large-scale automation. The platform is now relatively mature, and accordingly, the focus has correctly shifted to quality: high-quality collections of molecules with improved molecular properties, high-quality targets with strong target validation, and assays that more faithfully represent the relevant biology. This maturity, along with the understood effectiveness in generating leads, means that for most large pharma companies, HTS is a routine lead-generation approach for new projects. 34 When deployed in parallel, however, HTS and ELT provide a powerful and highly complementary lead discovery strategy, which offers a more comprehensive output than either technique alone.
One notable example that highlights the synergistic use of ELT and HTS is the campaign that led to the eventual identification of a first-in-class RIP1 kinase inhibitor clinical candidate. 35 The initial discovery efforts at GlaxoSmithKline (GSK) used a florescence polarization (FP)–based assay to screen an in-house focused kinase set of approximately 40 000 compounds. The assay used an adenosine triphosphate (ATP)–competitive FP probe, so inhibitors would be expected to demonstrate that mechanism. Multiple potent, but nonselective, series were identified from this screen, and exemplars from the lead optimization effort demonstrated in vivo activity in mouse models. 36 Although the molecules helped validate the confidence in the target, they were terminated based on a challenging developability profile, with a propensity toward high molecular weight, high lipophilicity, and a range of off-target kinase activities.
To identify more optimal chemical matter, a binding assay–based ELT screen against a pool of 45 libraries containing >7.7 billion encoded compounds and an ADP-Glo–based activity assay HTS were performed against RIP1 kinase. The coordination of the campaigns helped leverage the full value of each platform and yielded higher-quality hits from each. The affinity-based ELT screening provided access to a significantly larger collection of greater depth than the ~2 million compound HTS campaign. The low reagent requirement and ease of multiplexing ELT selections also enabled multiple screening conditions to be run in parallel (for example, against selectivity targets or multiple constructs/mutants of the same target). Because ELT should detect any binding mode, the parallel experiments can generate mechanistic information around the selected chemotypes.12,37–39 The HTS campaign provided access to a broad and diverse collection of compounds, with optimal molecular properties. The use of a functional assay also enabled identification of more inhibitor modalities as compared with the FP assay and ensured that qualified hits would inhibit in relevant biology. In contrast to ELT, running an HTS is a relatively large endeavor and is therefore typically limited to a single, optimized condition. For RIP1, both tactics were successful, and each yielded a broad range of qualified hits.40,41 In both cases, the majority of the hits demonstrated high similarity to known kinase hinge binding motifs. For RIP1, the breakthrough in identifying molecules with the desired activity and selectivity profiles derived from one on the key advantages of ELT: selections are affinity based and therefore mechanism and binding site agnostic. This allows the identification of chemical series with unique binding modes and activity.39,42–45 Indeed, one of the series identified from the RIP1 kinase ELT selections was structurally distinct from known kinase inhibitors and was uniquely selected by RIP1.
At GSK, all observed target DEL ligand interactions from every affinity selection are maintained in a searchable database; in 2015 alone, 90 000 selections (library/protein construct binding assays) were run. 31 Comparison of the primary hits for the target of interest (in this case, RIP1) against all prior screens allowed for the identification of chemotypes that were uniquely selected from the DEL pool by that target (this process is described in ref. 38). In this instance, a benzoxazepinone series of compounds was synthesized off-DNA ( Fig. 2A ), and molecules in the series showed excitingly potent biochemical activity, cellular activity, and, even as an unoptimized hit, good oral exposure in rodents. Most noteworthy, the series further showed remarkable kinase selectivity. One exemplar (GSK’481) was tested in commercial kinase panels in both binding and activity modes and showed no activity other than RIP1 versus any of the >300 kinases tested ( Fig. 2B ). Mechanistic studies and structural biology on the series revealed the basis for the exquisite selectivity: the molecule had the same binding mode as Necrostatins,46–48 binding in a hydrophobic pocket between the N-terminal and C-terminal domains. Human RIP1 needs to undergo substantial shifts and refolding to form this pocket and accommodate the compound ( Fig. 2C ). The molecule was ATP-competitive but made no contacts to the hinge region.35,41
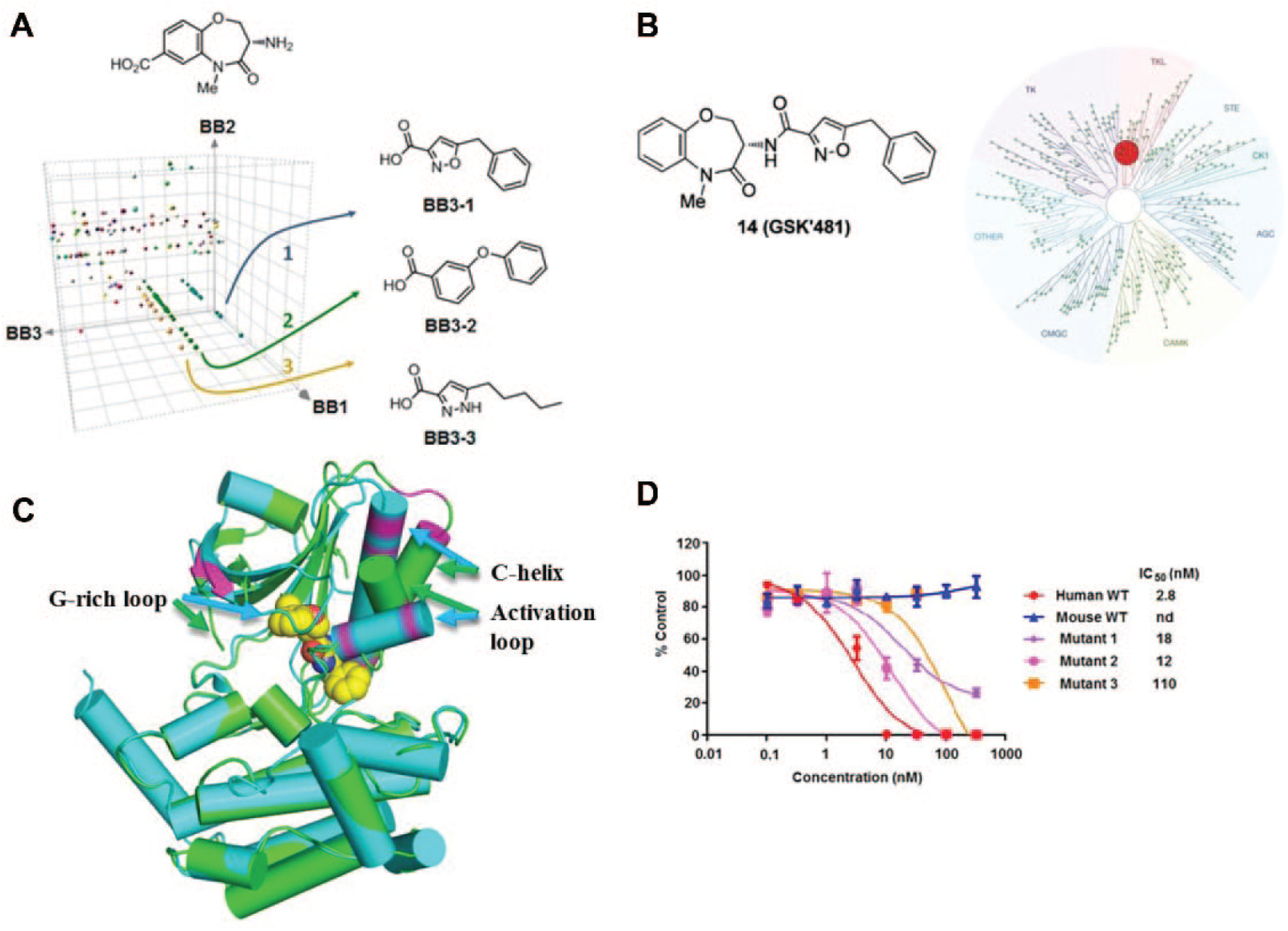
(
Further profiling of the ELT inhibitors against RIP1 orthologues demonstrated that the compound’s unique selectivity further extended to nonprimate orthologs of RIP1 kinase, where compounds showed >100-fold selectivity against rodent, rabbit, minipig, and dog RIP1 kinase. Sequence analysis led to identification of the key residue differences that were associated with ortholog selectivity and a prediction that the residues in the nonprimate RIP1 restricted flexibility needed for the activation loop to accommodate inhibitor binding ( Fig. 2C ). Mutation of these residues in the mouse protein restored potency of the inhibitors and supported the hypothesis ( Fig. 2D ). 41 The mechanistic investigation of these ELT molecules now provided one possible framework to triage for other molecules with this unique mechanism. The RIP1 HTS yielded an abundance of qualified hits, including what was later shown to be molecules with the same binding mode to the ELT series. Although molecules with this mechanism were inhibitors in the functional HTS assay, there was nothing appreciably different in the activity data that would distinguish the unique mechanism and, prior to the ELT, no reason to suspect that these should exist. Thus, the ELT campaign provided not only these key assets in their own right but also both the intellectual basis and the framework to retriage the HTS output for additional molecules with this mechanism. Indeed, multiple novel chemotypes with this mechanism were found in the HTS from this analysis. These HTS series demonstrated similar mechanism and kinase selectivity of the original ELT series and provided the project with an expanded array of chemotypes to progress.
This example highlights how ELT and HTS can be used in a coordinated and complementary manner that builds on the strengths of each platform. In this case, the ELT identified hits with a mechanism that might have been missed by other technologies and the route to exploit it. The HTS built on those learnings to expand the breadth in chemistry and opportunities for the project. One could argue that in this case, performing the ELT up front and then using the learning to inform the mechanistic design of the HTS experiment could have been more efficient than having to re-mine. However, this argument largely benefits from hindsight, might not apply to all cases, and would have increased overall cycle time.
Numerous other examples of the greater chemical and mechanistic diversity that can be achieved by combining activity- and affinity-based screening exist. A related kinase, receptor-interacting serine/threonine-protein kinase 3, is one such example, in which deploying both HTS and ELT screens enabled the identification of three distinct chemical series. 49 Another is the enzyme human mitochondrial branched-chain aminotransferase, in which HTS, FBDD, and ELT were applied to identify four distinct series.50–53 In both examples, the chemical diversity achieved from the complementary screening approaches was greater than that which would have been achieved from one tactic alone. 7-Transmembrane receptor targets have also shown to be tractable to ELT as well as HTS. Examples of ELT hits have come from selection against receptors expressed in intact cells as well as purified receptors.44,54 AstraZeneca in collaboration with X-Chem used a combination of HTS and ELT screening to identify protease activated receptor 2 agonists and antagonists with distinct binding modes and mechanisms of action.55,56
Beyond providing chemical or mechanistic diversity, different screening techniques can also turn over shared pharmacophores, with the combined structure-activity relationships (SAR) accelerating hit to lead chemistry. In the case of protein phosphatase 1D, parallel activity- and affinity-based ELT screens led to identification of a shared pharmacophore. The combined SAR from the two screens led to the identification of a potent allosteric inhibitor. 42 Complementary SAR was also combined to engineer a selective receptor-interacting serine/threonine-protein kinase 2 (RIPK2) inhibitor and RIPK2 proteolysis targeting chimera (PROTAC).57,58 In this case, HTS identified a potent active pharmacophore. The same hit was also selected by ELT, increasing confidence in the HTS primary hits. Examination of the primary hits from the ELT selections and comparison against historical ELT kinase screens provided additional SAR and identified modifications that provided improved selectivity. For bivalent molecules such as PROTACs, using ELT hits confers the additional benefit of having a predefined point of conjugation at the location of the DNA tag. As PROTACs continue to emerge as an alternate drug discovery modality, 59 the use of affinity-based methods such as ELT to identify binders becomes increasingly attractive.
In summary, although both HTS and ELT are powerful lead discovery methods in their own right, the combination of activity-based and affinity-based screening also provides a highly complementary output. Whether through the identification of novel binding modes and mechanisms, shared pharmacophores to accelerate SAR, or simply increased diversity of starting points with a common mechanism, this combination of approaches has become a core component of today’s lead discovery toolbox.
Phenotypic Screening followed by Target-Based Lead Discovery
Another route of integration of lead discovery approaches is the complementary use of phenotypic and target-based screening. A case study that nicely highlights the strength of phenotypic screening to identify novel targets, which in turn can be prosecuted using a combination of synergistic target based approaches, is that of the identification of the BET family of bromodomains. In the early 2000s, GSK began to investigate apolipoprotein A1 (ApoA1) up-regulation as a target for dyslipidemia. Although it was known that ApoA1 up-regulation was associated with protection from atherosclerosis progression and with anti-inflammatory effects, no mechanism by which up-regulation could be achieved with a small molecule was known. This situation, whereby a strong biological hypothesis exists but the mechanism is unknown or intractable to small-molecule intervention, is one in which phenotypic screening can be a particularly powerful tool. In this case, a stable human HepG2 cell line containing an ApoA1 luciferase reporter was used to screen for activators without target bias ( Fig. 3A ). A multitude of chemical templates were found that showed specific induction of the ApoA1 reporter gene but had no effect on other similarly configured reporter systems or on cell viability. 60 Potent nanomolar compounds ( Fig. 3B ) derived from these initial hits included isoxazoles such as I-BET151, 61 tetrahydroquinolines such as I-BET726, 62 and benzodiazepines (BZP) such as GSK’s clinical candidate, I-BET762.60,63
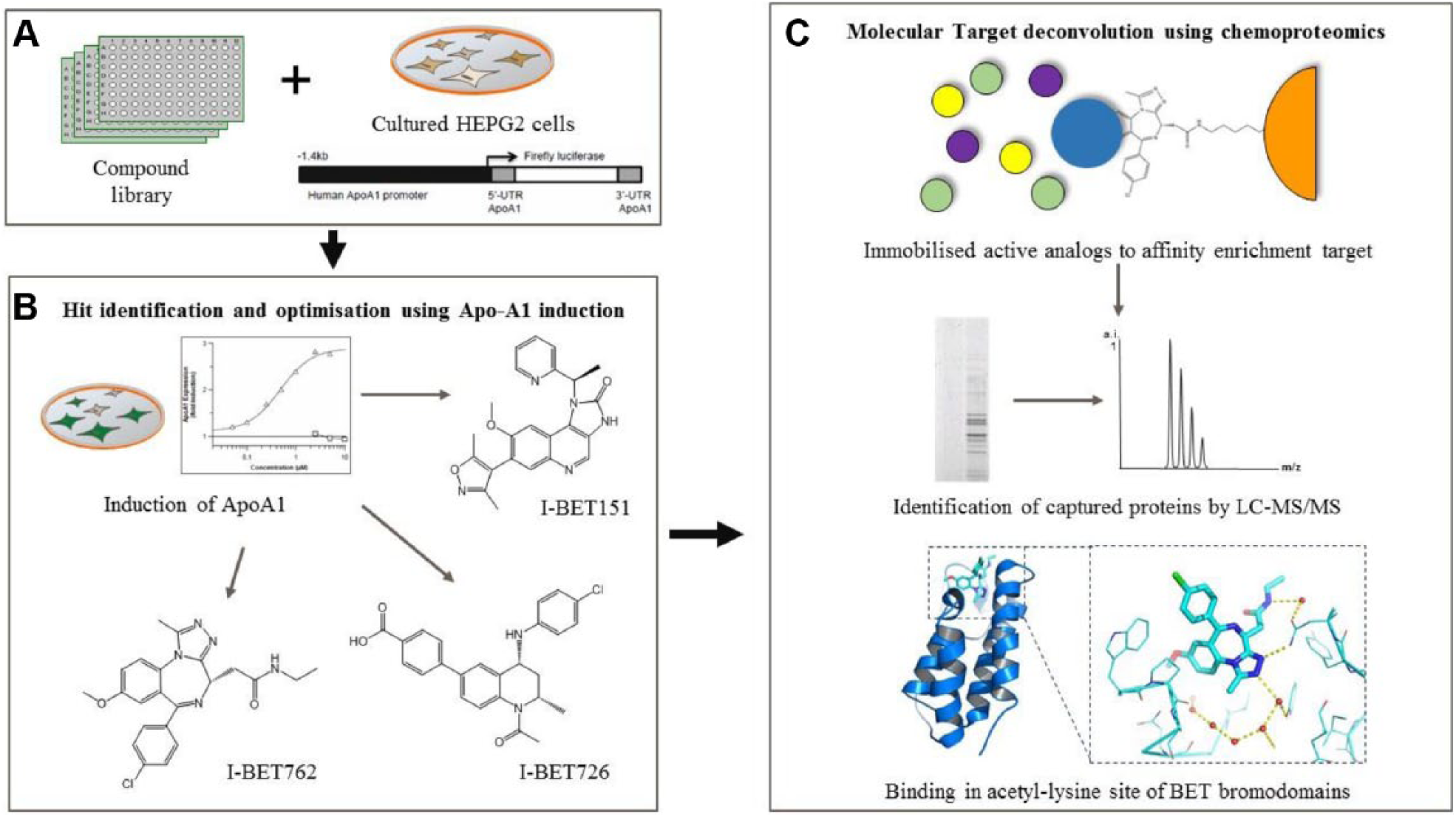
(
A second notable example of deploying phenotypic screening to find small molecules to achieve a biological response where the molecular mechanism is complex and may not be easily identified through more traditional target-based approaches is proprotein convertase subtilisin/kexin type 9 (PCSK9). Genetic linkage of PCSK9 to control circulating cholesterol levels provided a strong rationale for the pursuit of inhibitors of this target as a treatment for cardiovascular indications. 64 Despite extensive attempts by many institutions to find direct small-molecule inhibitors of protein-protein interaction of PCSK9 with its target receptor, the low-density lipoprotein receptor (LDLR), successful inhibition was initially achieved only via a phenotypic screen. Researchers at Pfizer identified an inhibitor of PCSK9 secretion, R-IMPP, which was demonstrated to stimulate uptake of low-density lipoprotein in hepatoma cells by increasing LDLR levels but without altering levels of secreted transferrin. 65 Detailed mode of action studies revealed R-IMPP binds to 80S ribosome to cause transcript-dependent inhibition of PCSK9 translation. This unprecedented mechanism opens up opportunities for the development of drugs that modulate the activity of target proteins by inhibition of eukaryotic translation, a mechanism most would have believed intractable without the discovery of R-IMPP, although clinical success for PCSK9 has yet been achieved only with antibodies that directly antagonize LDLR binding. 66
One of the challenges associated with phenotypic screening can be the optimization of hit series, especially where the phenotypic assay is a more complex cellular system. In the ApoA1 example, however, the use of a cellular assay and the unknown molecular target were not barriers in optimizing several series to the point where they had good potency and reasonable pharmacokinetic properties. 60 However, further challenges of a phenotypic approach were evident at every stage of the ApoA1 up-regulator program. Although multiple chemical starting points had been identified, without a target hypothesis, the choice of the “best” series to pursue for lead optimization was compromised. Diverse chemical templates had the potential for diverse molecular targets, diverse modes of action, and diverse selectivity profiles equating to importantly different therapeutic opportunities and safety liabilities. Chemical optimization of the chosen starting points was also more challenging, as target affinity, cellular permeability, and on/off target cell functions and toxicities were convoluted into a small number of functional cellular readouts focused on the primary ApoA1 cell activity and cell viability. Based on these challenges, which are common to phenotypic screening in general in our experience, although knowledge of the target is not essential to initiate a lead optimization campaign, it is often the case that target deconvolution and subsequent target-based drug discovery approaches represent the best path forward following a phenotypic screen. In the ApoA1 example, the molecular target was uncovered by a chemoproteomics approach. Using immobilized modified BZP compounds, members of the BET subfamily of bromodomain proteins were consistently recovered from cell lysates. This was a surprise because bromodomains bind acetylated-lysine (AcK) on histones, and potential protein-protein inhibitors of this target class were unknown at the time, as was the involvement of BET proteins in ApoA1 regulation. Biophysical and crystallographic studies added the final details to this novel molecular mode of action. ApoA1 activators were found to be pan-BET molecules, able to bind within the AcK pocket of both bromodomain 1(BD1) and bromodomain 2 (BD2) of each of the four BET family members BRD2/3/4/T 60 ( Fig. 3C ).
After successful target deconvolution, chemical starting points identified through phenotypic screening can be prosecuted using the available toolbox of target-based approaches, and indeed, it is often the case that the reagents and assays generated to aid target deconvolution lend themselves perfectly to initiation of a target-based drug discovery approach, therefore minimizing duplication of resource and facilitating rapid entry into the target-based effort. In the case of BET, the purified tandem and single bromodomain BET truncated proteins produced for in vitro validation studies provided an ideal platform for further target- and structure-based hit identification approaches, enabled by high-throughput biochemical fluorescent-based competition assays and biophysical technologies such as surface plasmon resonance and thermal shift. Groups have since used a wide spectrum of integrated, target-based screening approaches 67 but, most notably, computational and structure-based methods, which are especially accessible and efficient for these small and crystallographically amenable BET proteins.
Beyond the BET bromodomain story, another area of drug discovery in which phenotypic screening is proving successful in identifying new targets is for parasitic diseases such as tuberculosis 68 and malaria, 69 in which the adoption of whole-organism phenotypic screens to find new targets has reemerged as an attractive option. This reemergence has been driven by the increased ability to deconvolute to the protein target, facilitated by modern experimental tools such as gene editing, chemoproteomics, and whole-genome sequencing as well as increasingly powerful computational tools such as chemogenomics. An interesting example of the evolution from phenotypic to targeted approaches is that of enoyl-acyl carrier protein reductase (InhA) a well-known anti-tuberculosis (TB) target. Almost six decades ago, first- and second-line TB drugs, isoniazid and ethionamide, were discovered by whole-cell phenotypic screening. Unknown at the time, both of these drugs are prodrugs that require metabolic activation to be active, by the heme enzyme catalase peroxidase for isoniazid, and flavin adenine dinucleotide–containing monooxygenase (EthA) for ethionamide. Clinical resistance to both drugs is often observed not due to mutations on InhA itself but via mutations that regulate or alter the efficiency of drug activation. Therefore, although neither of these compounds would have been discovered within a purified enzymatic InhA assay, identification of direct InhA inhibitors may have benefit. 70 An alternative target-based screening strategy has been to find compounds that rescue or enhance the activation pathways, which would not only circumvent some of the resistance mechanisms but also reduce the required dosage, inhibitors of helix-turn-helix-type transcriptional regulator EthR, for example, that would result in up-regulation of EthA. Indeed, only very recently have inhibitors of both EthR and InhA been identified, not experimentally, but using predictive in silico methods, and definitively confirmed through biophysical methods, biochemical assays, and X-ray crystallography. 71
It is clear that phenotypic screening remains a challenging endeavor but one that can open the door to targets and mechanisms that would simply not otherwise be found. Rather than viewing phenotypic and target-based drug discovery as mutually exclusive approaches and debating their relative success rates, the examples described here, and our own experience, leads us to agree wholeheartedly with Moffat et al.’s 30 argument that target-based and phenotypic approaches are highly complementary and integrated drug discovery tactics, which should not be discussed in an “either/or” context, as one naturally leads and provides rapid entry to the other.
Integration of Knowledge-Based Lead Discovery and Diversity Screening Approaches
Returning to those projects for which there is a specific target hypothesis: underlying the selection of appropriate target-based lead discovery tactic is often the question of diversity versus knowledge. Focused libraries have the advantage of increased hit rates, whereas diverse sets are more likely to yield unexpected sites and binding modes. Selection of the appropriate strategy is therefore very much dependent on project timelines, objectives, and the target in question. In both the RIP1 and BET case studies discussed above, this balance is well exemplified. For RIP1, knowledge-based, kinase-focused set screening based on classical ATP competitive inhibition was rapidly able to identify potent, and in vivo active, hits. 36 Should rapid discovery of tool molecules have been the overall aim of this project, this would have been ideal. However, these molecules were suboptimal for progression as lead series. Diversity screening, using the combination of HTS and ELT described above, was ultimately able to identify novel and progressible lead series.
In the BET example, having identified the target from diversity-based phenotypic screening, groups have deployed fragment-based screening using more focused libraries. 72 In one example, information from historical work on BET bromodomain inhibitors was used to assemble a knowledge-based fragment library of AcK mimetics ( Fig. 4A , B ). Of the 1376 compounds tested, 132 showed >30% displacement of the fluorogenic BZP-ligand from at least one of the tandem BET proteins (BRD2/3/4). 73 These compounds were retested at full curve in a BZP-ligand fluorescence anisotropy assay and in an orthogonal time-resolved fluorescence resonance energy transfer peptide-displacement assay, prior to validation using crystallography. More than 40 crystal structures were solved with both familiar (eg, isoxazoles) and new AcK warheads (eg, pyridones) providing a rich starting point for chemistry, not only for the BET bromodomain family but also for the wider target class that shares the common pharmacophoric features of the AcK pocket. This is well illustrated by the fragment growth of these warheads into potent and selective inhibitors of non-BET bromodomains such as CREBBP (SGC-CBP30 74 ), BAZ2B 75 ( Fig. 4C ), BRPF1/2 (GSK6853, OF-1), 76 and their presence in BET clinical candidates. 63 Such examples readily demonstrate the power of knowledge to rapidly lead hop between related targets.
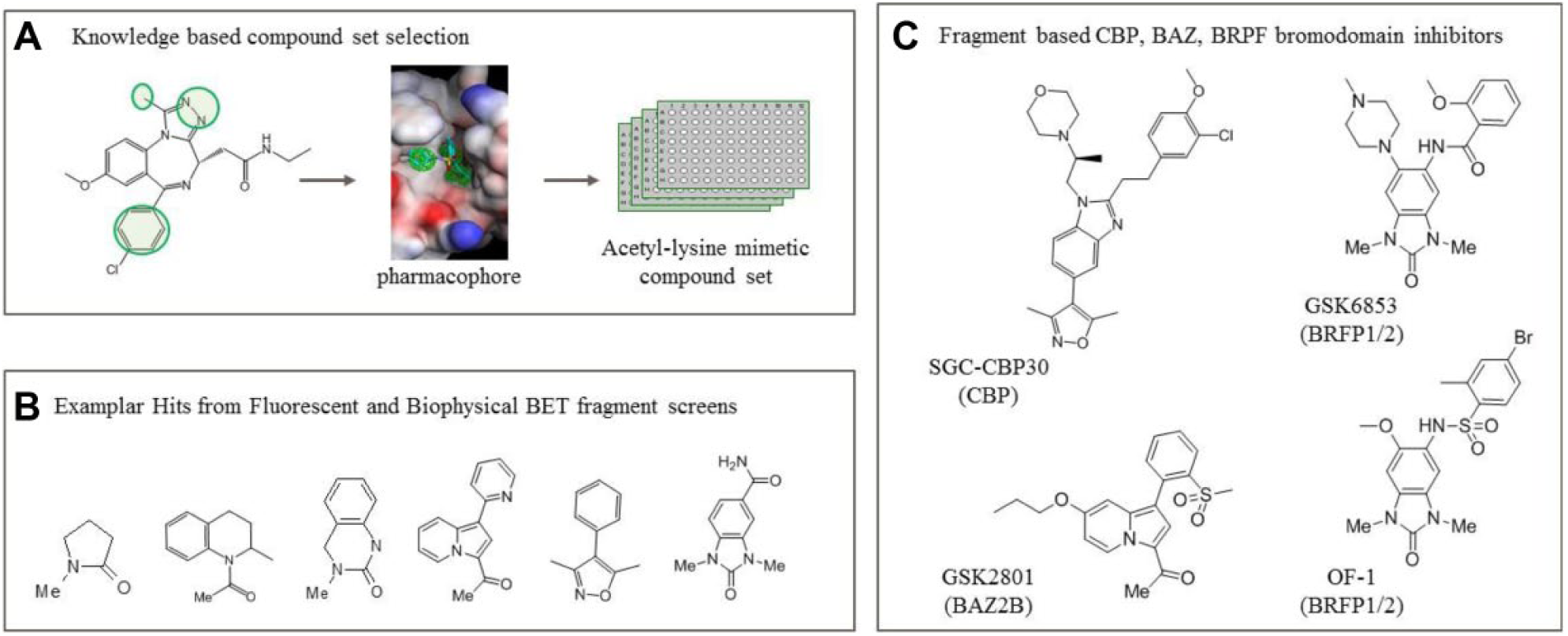
(
Ligand-based virtual screening is another powerful knowledge-based approach that is often integrated with other approaches in the lead discovery toolbox. This is exemplified by the work of a team at Pfizer, who were interested in compounds that are modulators of the high-affinity choline transporter (CHT), 77 to enable testing of the hypothesis that positive modulation of CHT-mediated transport would enhance activity-dependent cholinergic signaling and therefore be an attractive therapeutic target. This project had a common scenario, with a small number of literature compounds (orthosteric inhibitors, negative allosteric modulators, and putative positive modulators), none of which were quite what the project needed. As part of a small screening campaign, a virtual screen was conducted to find novel molecules that could be used as tools and as possible start points for further medicinal chemistry. The team used a ligand-based approach, the FieldScreen technique, 78 which compares the molecular fields (electrostatic and hydrophobic) of seed molecules (in this case, the literature molecules) to those of the Pfizer corporate database. Virtual screens were conducted to identify compounds with similar molecular fields to the CHT-positive allosteric modulator MKC-351 79 and CHT-negative allosteric modulator ML-352. 80 The top-scoring 500 molecules were selected from each screen, yielding a final set of 887 compounds for test after further filtering (availability and removal of chemically unattractive groups). 81 Both virtual screens yielded novel chemotypes: three positive allosteric modulators and two negative allosteric modulators ( Table 1 ). The results illustrate the power of virtual screening to efficiently identify novel chemotypes based on a limited amount of information, which can then be tested experimentally.
Choline Transporter (CHT)–Positive Allosteric Modulators (PAM) 79 and CHT-Negative Allosteric Modulators (NAM) Identified from Virtual Screening.
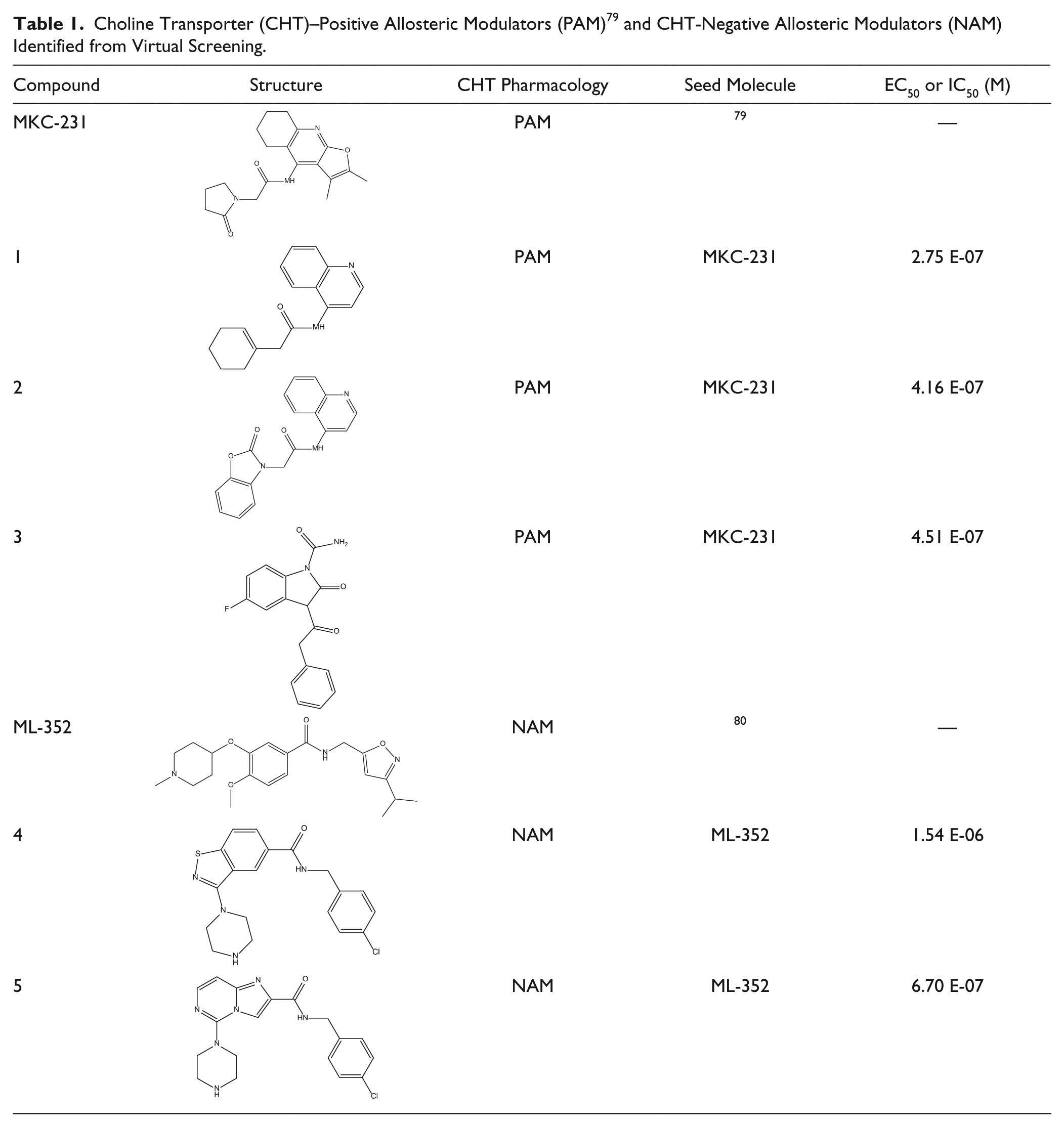
Overall, the RIP1, BET, and CHT examples demonstrate how knowledge-based computational and focused set screening are powerful lead discovery tools for related proteins within a target class or for targets for which some literature information is available. This approach can lead to rapid identification of tool molecules and chemical starting points without the need for investment in full-scale HTS. HTS, although a mainstay lead discovery tactic, is a costly and time-consuming endeavor, and in examples such as those above, it may be an unnecessary investment when knowledge-based approaches can rapidly generate starting points in a more resource-efficient manner. Of course, knowledge-based approaches by their very nature are more likely to identify starting points with precedented mechanisms of action. The decision on how to integrate knowledge and diversity approaches therefore very much depends on the desired outcome, level of investment, and time scale of the project in question, a discussion we will return to in the summary section of this article.
Where more novel and differentiated binding modes or mechanisms of action are desired or beneficial, diversity screening approaches still likely represent the greatest opportunity. Here again, the BET bromodomains provide us with an illustrative example. Despite a high level of activity, thus far, most reported BET ligands have been pan-BET compounds that show similar activities against both BD1 and BD2 domains of all the BRD2/BRD3/BRD4/BRDT isoforms. Given the high-sequence homology between these protein domains, the lack of selectivity is unsurprising, but diversity screening approaches have now led to the identification of domain-selective hit compounds, which display a degree of differential BD1 and BD2 activity, with 10-fold differences accessible in many templates. This includes RVX-208, a BD2-biased phenotypic screening–derived clinical candidate for atherosclerosis, acute coronary syndrome, and Alzheimer’s disease. 82 Interestingly, this compound was being progressed without an understanding of its mode of action before emerging BET literature prompted the testing of this compound for BET activity. Domain-selective starting points have also been found using an ELT diversity screening approach by Nuevolution, who have reported ligands with novel binding modes that exploit a unique switch in the Psi angle of a conserved proline in BD1, which is a histidine in BD2. Leveraging structural insights, Nue7770, a nanomolar BD1-selective (>64-fold) compound, was found to have distinct pharmacology over pan-BET or BD2-biased compounds. BioMap 83 profiles confirmed it preserved the strong anti-inflammatory activities in B- and T-cells, as with pan-BET molecules, but lacked their antiproliferation properties, making it attractive for autoimmune and anti-inflammatory diseases. Indeed, this compound has shown dose-dependent inhibition in murine models of collagen-induced arthritis and pristine lupus. Novel ELT approaches have also delivered covalent BET inhibitors.84,85
Evolution of Screening Strategies
The case studies presented in this article demonstrate how the full toolbox of lead discovery approaches can be employed and integrated successfully in a variety of combinations and indeed how combining approaches gives unique insights and often an increased probability of success than deploying one tactic alone. As new screening methodologies emerge, they will continue to add to and evolve the nature of this toolbox, as was the case for ELT over the past decade and HTS before that. It is therefore worth spending a portion of this article reviewing what the future tool box might look like and which areas of science are most likely to shape it in the near future. Rapid developments in two distinct yet related areas of science, virtual screening coupled with active learning algorithms, and complex cellular models are, in our opinion, likely to have the greatest impact on the screening landscape in the coming years.
Evolution of Cellular Models
Several technologies are now becoming embedded into the drug discovery process, which allow the development of more affordable, scalable, and accessible disease-relevant cellular models. Horvath et al. 86 published a thoughtful overview of the limitations of traditional cellular screening approaches, due to cell type, culture conditions, and lack of relevant cellular context (for example, extracellular matrix), which result in an inability to recapitulate in vivo physiology and therefore a lack of translation of screening hits into viable drug candidates. They propose some defining principles for disease-relevant assays that capitalize on the rapid developments in new cell-based models such as induced pluripotent stem cell technology (iPSC), three-dimensional (3D), and co-culture systems and organ-on-a-chip. iPSC technology facilitates more scalable assay development than primary cells and removes some of the challenges associated with donor-to-donor variability that can compromise screen robustness but additionally enables access to cell types that are otherwise difficult to obtain, such as neurons. This technology can also enable access to patient-derived cells, which recapitulate disease phenotypes. 87 Likewise, there is some evidence that 3D cultures, in the form of spheroids, scaffolds, and organoids, better mimic in vivo physiology. 88 Each of these technologies has seen rapid development and uptake over the past decade, predominantly in very focused areas of drug discovery such as tumor spheroids in oncology. Only now, though, are they becoming truly embedded into hit identification screening approaches more broadly in industry, as they reach the necessary scale, cost, and scientific maturity to do so. With concomitant development of higher-content detection technologies such as confocal imaging, mass spectrometry imaging, and single-cell transcriptomics, it is our expectation that we will increasingly see phenotypic screens employing these models as a primary driver of hit identification.
Having said this, even with increasing use of complex cellular systems, in our view it is unlikely that we will see a dramatic shift away from target-based screening approaches. It is our belief that target-based and phenotypic approaches will continue to be highly complementary. Indeed, many of the cellular technologies are also very enabling for target validation studies and so may in fact strengthen the argument for prosecuting one target over another, which is then best progressed using a target-based approach. Although these advances address some of the previously mentioned challenges of phenotypic screening (for example, variability in complex primary cell assays for hit-to-lead SAR campaigns due to donor-to-donor variability), they cannot address them all, and therefore target deconvolution will likely remain a natural next step in many cases.
In fact, the increasing availability of techniques and cellular models to identify and validate targets, whether it be, for example, pooled CRISPR-Cas9 genome editing screens in primary cells or genome-wide association studies, is likely to lead to a new screening challenge: how to screen for the best target to take forward for a full discovery campaign out of a long list of tens, or even hundreds, of putative drug targets for a given indication. When in silico approaches cannot be applied, technologies historically used for hit identification can be repurposed to prioritize targets based on their chemical tractability. ELT is an experimental approach that is particularly amenable to this given the ability to multiplex numerous targets in parallel in a single selection experiment very rapidly and efficiently.38,89 Similar to ligandability assessment by fragment screening, 90 historical experience at GSK has indicated a correlation between the number of enriched chemotypes from an affinity-based ELT selection against a specific target and that target’s overall small-molecule tractability to other screening methods. The combination of ease of multiplexing, low reagent requirements, and lower cost for ELT compared with many other methods provides a means of rapidly profiling panels of genetically validated targets or a pathway from a phenotypic screen to identify the most chemically tractable, allowing downstream reagent production and assay development to focus on the targets with the highest likelihood of success.31,38
Evolution of Virtual Screening
The second area of rapid evolution in the lead discovery space is in virtual screening. HTS in whatever form, be it target-based or phenotypic, has become established as the gold standard method for generating hits not only because of its ability to find starting points for novel targets using a wide variety of assay types but also because virtual screening techniques are not yet good enough to eliminate the false-negative problem (true hits that were not predicted) either due to the approximations inherent in the computational technique or to unexpected binding modes, protein movement, or allostery. A systematic evaluation of this is provided by Shoichet et al. 91 An HTS of 197 861 molecules was performed against the Cruzain cysteine protease, a target for Chagas disease. In parallel, a virtual screen was carried out using the DOCK molecular docking system. 92 Of the five chemotypes followed up, two were not identified by the virtual screen; however, two of these series (identified by both techniques) were followed up only due to the information gleaned from the docking (i.e., would not have been followed up without the mechanism of action hypothesis provided by the virtual screen). This study exemplifies both the strength and weakness of virtual screening: good hits can be found by screening only a fraction of the compound collection, but it is unlikely to predict all hits. The conclusions from this study, that both HTS and virtual screening have value but are best used in a synergistic way, explain the differential use of virtual screening by large pharmaceutical companies (running virtual screens alongside HTS to find the very best available starting points) and small companies or academic groups (using virtual screens to find a starting point quickly and cheaply). So, if the false-negative issue prevents the replacement of experimental screening approaches by virtual screening, what is the future for computational methods in this space? There are two clear use cases that are growing in popularity: making the screening process more efficient and performing experiments that are prohibitively expensive in the laboratory.
Making Screening More Efficient
A small but steady stream of methods using computational models to increase the efficiency of HTS have been proposed. Sequential or iterative screening protocols were implemented 93 when HTS screens were often in 96-well plates and much more expensive on compounds/reagents. Typically, a small diverse set of molecules is screened to generate initial data, which is used to build a predictive model (in this case, a decision tree). The model then picks the compounds to be screened next, and this process is repeated until enough hits are found or screening is no longer deemed productive/affordable. The general result is similar to that of a virtual screen: hits are found with more efficiency, but not all possible hits are found. Nonetheless, this technique still has advocates 94 and has the advantage over a pure virtual screen of combining serendipity with prediction.
These early sequential screening strategies have now been superseded by a class of machine learning techniques known as active learning. 95 This was first proposed for drug discovery by Warmuth, 96 but it has taken some time before the benefits of the technique have become apparent. In active learning, the selection of the next batches of molecules for screening not only is made to find new active molecules but also includes those that will yield data that will improve the predictive model. In active learning terminology, this is the balance between exploit (use the model to find new actives) and explore (probe areas of structure-activity space that give information the model needs). Applications of active learning to drug discovery are now becoming mainstream. 97 The combination of computational selections with screening logistics can be difficult—the volume of cherry-picks from compound stores can reduce the efficiency of the whole process. Therefore, it is attractive to consider automation systems that combine screening, compound handling, and machine learning. One prominent example of such a system is University of Manchester’s EVE system, 98 which combines familiar technology such as acoustic dispensing, a shaking incubator, and plate readers and imagers with a sophisticated software management system.
Performing Experiments That Are Prohibitively Expensive in the Laboratory
The great advantage of computational methods is that they can be applied to molecules that have not been synthesized, assays that have not been developed, or combinations of compounds and assays that would require prohibitive laboratory resources. Many companies and compound suppliers can define very large numbers of compounds that they believe can be made quickly. 99 These very large collections of molecules can be used for virtual screening, thus neatly passing on the false-negative disadvantage to the HTS alternative: if you do not have the compound, you cannot screen it, and it is not physically possible to synthesize, store, or assay (even with DNA-encoded libraries) all the diversity of chemical structures that can be envisaged by simple combinations of reagents and known reactions. The Reymond group 100 has set out to systematically enumerate every possible chemical structure, and groups are looking to virtually screen these incredibly large numbers of molecules. 101 Virtual screens do not need to be restricted to single targets and can be run across the proteome, although the effectiveness of this remains uncertain. 102 More realistic is the use of protein families, so-called chemogenomics strategies, 103 where the virtual screens are focused on similar proteins to find hits that are predicted to be selective. 104 These techniques can be made more efficient by combination with active learning strategies. 105 Going beyond target-based discovery, an intriguing possibility is the use of such methods to aid phenotypic pathway discovery without first knowing the full set of possible phenotypic responses. The Murphy lab 106 demonstrated the ability to predict the effect of 48 molecules on the subcellular localization of 48 proteins by performing only 29% of the experiments compared with the full matrix screen.
The power of these evolving approaches in both the cellular model and virtual screening space is to use them in combination. In reality, complex cellular models, even in miniaturized format and with all recent advances, are costly and time-consuming to develop, and so it may be unrealistic to drive them to the scale of a 2 million compound HTS. However, when coupled with an active learning approach it may be possible to achieve the same or similar success rate by screening far lower compound numbers. This is a somewhat circular argument as it is likely that the rate of uptake of the complex cellular systems will increase proportionally with the rate of adoption of these prediction-guided screening approaches.
Summary: Integration to Maximize Success with an Evolving Toolbox
Overall, the nature of the lead discovery toolbox is evolving, but in our opinion, and as shown by the examples throughout this article, the likelihood of success is still going to be highest when more than one approach is employed in a complementary manner. Whilst it could be argued that if you are likely to succeed with one approach you would be likely to succeed with any, the examples in this article highlight the complementarity of information provided by the various approaches taken, and the different situations under which each tactic is most advantageous. Whether it be target based diversity driven experimental approaches like ELT coupled to HTS, target family based focused screening coupled with ligand based Virtual Screening, or phenotypic screening in complex cell models coupled with Active Learning based computational approaches. It is the integration of more than one different hit discovery approach to address specific biological questions over the lifetime of a drug discovery project, that in the examples shown, has led to the greatest probability of success. Whether these different approaches are deployed concurrently or iteratively, and the choice of approaches deployed, depends on a multitude of factors, including whether it is a tool molecule or lead that is required, the degree of information available on the target or biology of interest and the strength of overall target validation, the timelines of the project, and the degree to which the specific molecular mechanism is understood and tractable.
In the case of a more speculative or higher-risk target, a more iterative process may be preferable, in which one deploys one tactic to answer an initial question, be it, “Can I find a tool to address the biological validation of my target?” or “Is this target small-molecule tractable?” and then, having progressed one’s understanding of that initial question and built further confidence in the target, returns to apply additional screening approaches to answer the next question and so on. This more circular, iterative approach obviously brings with it a risk of extending cycle times in discovery. However, this can be addressed through clarity in the questions being answered at each stage, and by each approach, and clear decision gates at each stage of the process that lead either to initiation of the next approach or to stop. In fact, this approach has potential to reduce overall cycle times and attrition rates for less well-characterized targets, for example, by preventing one committing to a full HTS with an assay format that does not truly answer the biological question of interest or committing to a target that is unlikely to be tractable in the long term. In bigger picture terms, this integrated and more iterative approach has potential to increase the overall success from discovery into the clinic by ensuring that all key biological and chemical questions have been thoroughly addressed along the way and that risk and reward are appropriately balanced.
In contrast, for a target with very strong genetic target validation, well-understood link to disease biology, and good tractability, one may prefer to prosecute multiple approaches concurrently to maximize probability of successfully identifying multiple high-quality starting points in the shortest time frame. This of course brings with it a resource and cost consideration, and the priority of the target in the overall portfolio against other opportunities will have some bearing on the number and choice of tactics deployed in parallel as well as, of course, technical feasibility. Figure 5 illustrates some of these considerations and how they may drive decisions on which tactics to employ and in what way, although these scenarios are by no means exhaustive.
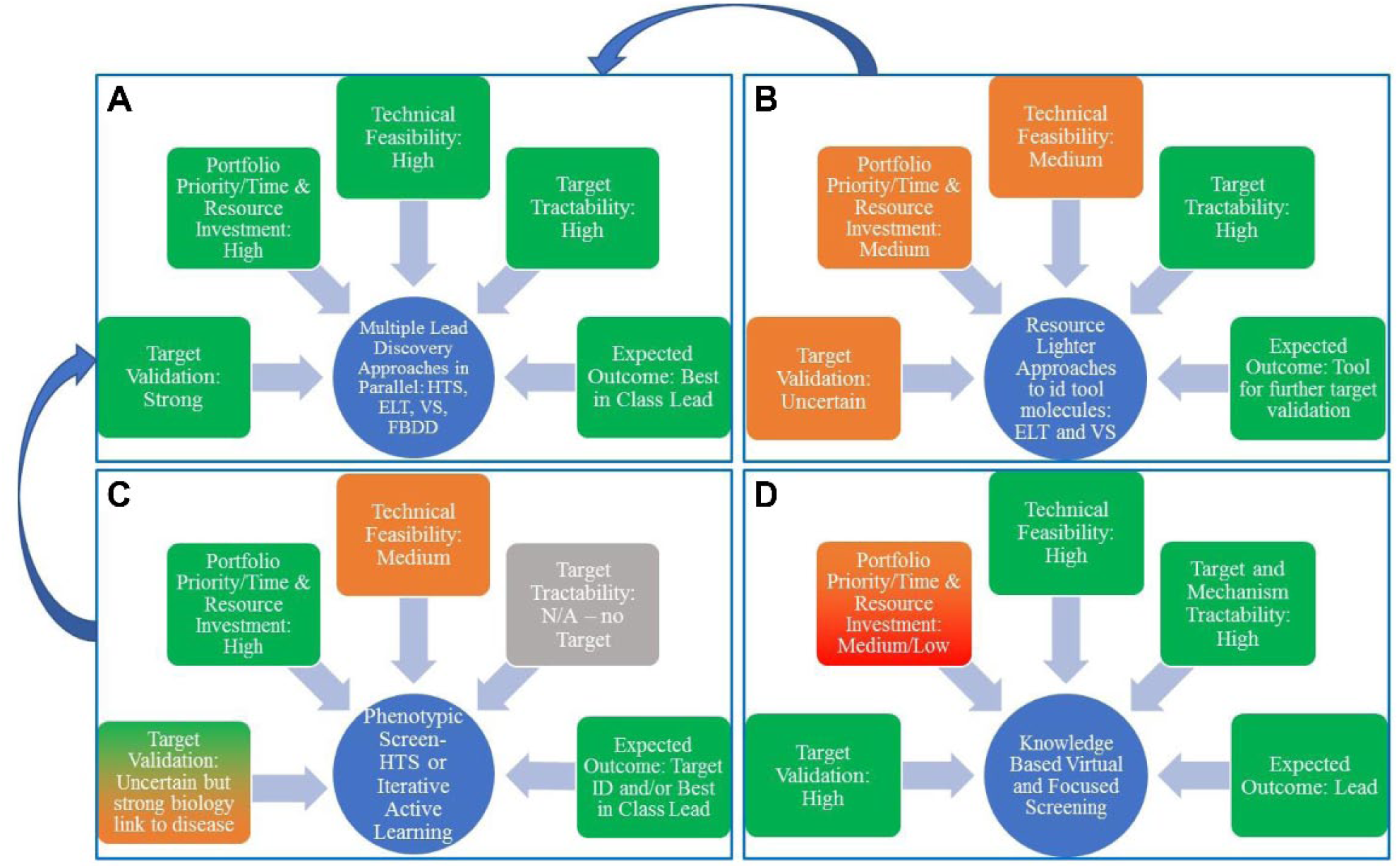
Illustration of considerations that drive decisions on choice of lead discovery tactics
to employ and in what combination. Four example scenarios are shown in which factors
including strength of target validation, priority of the project in the portfolio,
availability of resource, and desired outcome are considered to determine the
combination of tactics selected. In many cases, these may represent staged investments,
whereby one scenario feeds another, such as in the case of tool identification to
strengthen target validation, which then prompts more exhaustive screening.
(
In conclusion, lead discovery strategies should be tailored to the scientific question and based on the relative balance of risk and reward in each case. However, it is important that the full tool box of potential strategies, both those available now and newer emerging strategies, be considered at the start of a project and the pros and cons of each weighed up, as one size clearly does not fit all and there are many possible approaches at our disposal.
Footnotes
Acknowledgements
The authors thank Christine Donahue, Eric Manas, and Phil Harris for their review of the manuscript. They also thank John Bertin and Rab Prinjha for their support on the RIP1 and BET case studies.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: All work was completed as work-for-hire for the employer GlaxoSmithKline.