
Abstract
Pathogenic fungi represent a growing threat to human health, with an increase in the frequency of drug-resistant fungal infections. Identifying targets from among the selected metabolic pathways that are unique to microbial species presents an opportunity to develop new antifungal agents against new and untested targets to combat this growth threat. Aspartate semialdehyde dehydrogenase (ASADH) catalyzes a key step in a uniquely microbial amino acid biosynthetic pathway and is essential for microbial viability. This enzyme, purified from four pathogenic fungal organisms (Candida albicans, Aspergillus fumigatus, Cryptococcus neoformans, and Blastomyces dermatitidis), has been screened against fragment libraries to identify initial enzyme inhibitors. The binding of structural analogs of the most promising lead compounds was measured against these fungal ASADHs to establish important structure–activity relationships among these different inhibitor classes. The most potent of these inhibitors have been docked into structures of this fungal enzyme target to identify important structural elements that serve as critical binding determinants. Several inhibitors with low micromolar inhibition constants have been identified that showed selectivity against these related enzymes from different fungal species. Subsequent screening against a library of drugs and drug candidates identified some additional inhibitors containing a consistent set of functional groups required for fungal ASADH inhibition. Additional elaboration of these core structures will likely lead to more potent and selective inhibitors.
Keywords
Introduction
Fungal infections pose serious threats to human life, ranging from superficial skin infections to life-threatening infections such as candidiasis and aspergillosis. The most common human fungal pathogens include Candida albicans (Cal; 20 to 40% mortality), 1 Cryptococcus neoformans (Cne; 20 to 70%), 2 and Aspergillus fumigatus (Afu; 50 to 90%), causing ~1.5 million life-threatening infections annually. 3 Spores released from these fungi can elicit several types of allergic reactions, depending on the taxonomic branch involved. 4 Fungi can infect a healthy human host, but the highest incidences of infections occur in immunocompromised populations such as neonates, cancer patients receiving chemotherapy, organ transplant patients, and patients suffering from HIV. 4 Recently, a significant increase in the frequency of fungal infections has been threatening to overwhelm our limited array of antifungal drugs, compared to the much wider range of antibacterial agents currently available. This antifungal drug deficiency is likely due to fungi being eukaryotic, with limited molecular targets available for selective antifungal development without cross-target effects in humans. Also, the growth of drug resistance in fungi is now paralleling the phenomenon already observed in bacteria. The US Centers for Disease Prevention and Control (CDC) has already classified Candida infections as a serious threat to human health, due to the dramatic increase in resistance to currently available antifungal drugs. 5
Despite extensive research aimed at developing new agents to combat fungal infections, there are only a handful of frontline antifungal agents. These agents, from four different molecular classes targeting three distinct metabolic pathways, are currently used in the treatment of essentially all systemic fungal infections; these classes are fluoropyrimidine analogs, polyenes, azoles, and newly discovered echinocandins. 4 There is clearly a growing need to develop new antifungals against novel targets to be available for use in clinical settings.
Selected amino acid biosynthetic pathways have the potential to yield attractive and relatively unexploited targets for development of antimicrobial agents, 6 primarily due to the absence of many of these pathways in mammals. 7 Aspartate β-semialdehyde dehydrogenase (ASADH), an enzyme that catalyzes the committed step in the aspartate biosynthetic pathway, is responsible for the biosynthesis of several essential amino acids and other cellular metabolites critical for the survival of microorganisms. ASADH, coded by the asd gene, catalyzes the reductive dephosphorylation of the substrate aspartyl phosphate to product aspartate semialdehyde (ASA). Previous studies have shown that deletion of the asd gene in Salmonella typhimurium has lethal consequences for microorganisms, 8 and the asd gene is on the list of the minimum set of genes required for survival of microorganisms such as Bacillus subtilis.9,10 Because this critical pathway is present only in plants and microbes but absent in mammals, disruption of the enzymes in this pathway should have minimum toxicity to the human host. Therefore, enzymes in this pathway are attractive and untested targets for antifungal drug development. In this study, we have identified some selective compounds that inhibit ASADHs from several pathogenic fungi at low micromolar levels. Selective and potent inhibitors of ASADH will be lethal to these fungi. Further development of these inhibitors can lead to potent antifungal agents.
Materials and Methods
Materials
Chemicals, including 4-(2-hydroxyethyl)-1-piperazine ethanesulfonic acid (HEPES), N-cyclohexyl-2-aminoethane-sulfonic acid (CHES), and ethylenediamine-tetraacetic acid (EDTA), were purchased from Chem Impex (Wood Dale, IL). Nicotinamide adenine dinucleotide phosphate (NADP), iodonitrotetrazolium chloride (INT), and phenazine methosulfate (PMS) were purchased from Sigma-Aldrich (St. Louis, MO), and dithiothreitol (DTT) was purchased from Gold Biotechnology (Olivette, MO). The majority of the enzyme inhibitors were purchased from either Oakwood Chemicals (Estill, SC) or Santa Cruz Biotechnology (Dallas, TX).
Enzyme Expression and Purification
Enzyme expression and purification of CalASADH, 11 CneASADH, 12 AfuASADH, 13 and SpnASADH 14 have been described previously. Blastomyces dermatitidis (Bde)ASADH was purified with a slight modification to the previous ASADH purification protocols. The gene encoding BdeASADH was cloned into pET-28a(+) vector and transformed into an Escherichia coli BL21(DE3) cell line. Protein expression was induced using 0.1 mM IPTG once cell growth reached an A600 from 0.6 to 0.8. The recombinant enzyme was initially purified using a Ni-NTA (nitrilotriacetic acid) IMAC (immobilized metal affinity chromatography) column, followed by a Source 30Q anion exchange column for the final purification step. The enzyme was dialyzed into a final buffer containing 20 mM HEPES, pH 7.0, 50 mM NaCl, 0.5 mM EDTA, and 1 mM DTT. The enzyme was estimated to be at least 95% pure from SDS-PAGE (sodium dodecyl sulfate–polyacrylamide gel electrophoresis) analysis, and it was flash frozen in liquid nitrogen and stored at −80 °C. The substrate ASA was synthesized as described previously 15 and stored in 4 M HCl at −80 °C. The final working solution of ASA was neutralized with 4 N NaOH immediately prior to use due to the instability of ASA under basic conditions.
Enzyme Activity Assay
ASADH catalyzes the reductive dephosphorylation of the substrate aspartyl phosphate to produce ASA using the reductive power of NADPH. Due to the instability of the substrate aspartyl phosphate, the enzymatic activity assay was carried out in the non-physiological direction by measuring the increase in absorbance of NADPH at 340 nm (e340 = 6220 M−1 cm−1) using a Molecular Devices SpectraMax Plus microtiter plate reader at 25 °C. The assay was conducted in 96-well clear ultraviolet (UV) plates (Costar) at 25 °C in a 200 µL final volume using a reaction buffer containing 20 mM phosphate, 1.5 mM NADP, 0.3 mM ASA, 120 mM CHES buffer, pH 8.6, and 200 mM KCl. The assay mixture was incubated in these plates for 30 s at 25 °C, and the reaction was initiated by adding purified ASADH at a final enzyme concentration of 0.05 mg/ml . Plates were shaken for 20 s before reading, with a background rate measured using a buffer-only control. The absorption at 340 nm was continuously monitored for 20 min, with the slope of UV absorption versus time for the first 600 s calculated by linear regression.
Coupled Enzymatic Assay and Enzyme Inhibition Studies
Because the enzymatic activity was initially monitored at a lower wavelength (340 nm), a set of screened compounds showed background absorbance at this wavelength that interfered with the rate measurement. To avoid this interference, an alternative assay was developed to shift the wavelength to the visible region by using a coupled enzyme assay in which phenazine methosulfate (PMS), an electron carrier from NADPH, will reduce iodonitrotetrazolium (INT), an internal dye, to a wine red–colored compound called formazan that absorbs at 500 nm with a screening assay quality coefficient (Z’ factor)
16
of 0.82. This assay was conducted in a CHES buffer (pH 8.6) containing 200 mM KCl, 20 mM phosphate, 1.5 mM NADP, 0.3 mM ASA, 0.5 mM INT, and 0.1 mM PMS. The enzymatic assay was initiated by adding 30 µL of 0.05 mg/ml ASADH in 200 µL total reaction mixture and monitoring the increase in absorbance of formazan at 500 nm. A schematic of the details of the coupled assay is shown in
Water- and DMSO-Soluble Fragment Library Preparation
The water-soluble and DMSO-soluble libraries were assembled using commercially available low-molecular-weight compounds dissolved in either water or DMSO, with some pH adjustment as needed to solubilize acids or bases. The water-soluble library consists of 384 compounds from four different classes: amino acids and derivatives, metabolites and analogs, carbohydrates and bases, and water-soluble organics and aromatics. Similarly, the DMSO-soluble organic fragment library also consists of 384 compounds from four different classes: benzene derivatives, five-membered heterocycles, six-membered heterocycles, and fused or multiple-ring systems. 18
ASADH was found to retain at least 70% of its enzymatic activity at DMSO concentrations as high as 50%, allowing the screening of organic compounds with low water solubility in a 40% DMSO–water mixture. The average molecular weight of the compounds in these libraries is ~160 Da, with 90% of the compounds between 90 and 260 Da. The majority of the compounds obey the general fragment library rules: 19 (1) heavy atoms count ≤22; (2) clogP <3; (3) number of H-bond donors ≤3; (4) number of H-bond acceptors ≤3; and (5) number of rotatable bonds ≤5, with only a few exceptions.
Constrained Analog and Drug Library Preparation
The constrained analog library was compiled using aspartic acid–based analogs that mimic the core ASA structure. This library contains 83 compounds consisting of 5–6-membered heterocycles, isoxazoles, amino acid analogs, halo acids, pyrazoles, and pyrrolidine derivatives with an average molecular weight of 173 Da. Compounds were dissolved in DMSO at 200 mM stock concentrations and tested at 1 mM initial concentrations. Many of the compounds in this library contain novel chemical scaffolds when compared to our fragment libraries, increasing the probability of finding novel starting points. The National Institutes of Health (NIH) Clinical Collection (NCC) library consists of 726 compounds supplied in 96-well plates prepared in DMSO solution at a stock concentration of 10 mM. The Prestwick Chemical Library consists of 1320 drugs and drug candidates supplied in 96-well plates dissolved in 50% DMSO–water at a concentration of 1 mM. From 10 to 20 µl of each inhibitor was transferred to 200 µl solution of assay recipe to give a final concentration of 100 to 500 µM for each compound.
Ligand Preparation for Docking Studies
All inhibitors were prepared with Ligprep software, which generates low-energy tautomers and enumerates realistic protonation states at physiological pH. Each ligand was optimized for equilibrium geometry in vacuum using the HF/6-31+G* level basis set. Proper charges were assigned when necessary before optimization. Calculations were carried out using the Spartan’14 (Wavefunction, Irvine, CA). The minimized ligands were prepared for docking using the AutoDock Tools (ADT) by adding polar hydrogen atoms, merging nonpolar hydrogen atoms, adding atom-type parameters, and defining rotatable bonds.
Protein Structure Preparation for Docking Studies
Four structures of different fungal ASADHs are currently available in the Protein Data Bank (PDB). 20 The CalASADH structure has the best resolution (PDB id: 3hsk, 2.2 Å), and molecular docking studies were performed with the best inhibitors into the active site of CalASADH using AutoDock Vina. 21 Water molecules were removed, and coordinates of NADP were transferred into the enzyme structure in its binding site. The grid box was set to 30×28×30 Å with grid spacing of 1.00 Å. The box was centered in the active site region, ensuring coverage of the residues involved in substrate and cofactor binding. AutoDock Vina was performed at exhaustiveness level 16 with the number of binding modes set to 20. The top 20 docking poses were generated, with the lowest energy pose of each docked ligand used to analyze the interactions of the ligand at the active site of the enzyme. The Python scripts in the MGL tools package were used to analyze the docking results, and images were generated using Pymol. 22
High-Throughput Docking
AutoDock Vina runs much faster 23 and is capable of docking larger ligands more accurately than the previous software versions. For virtual high-throughput screening (VHTS), the ZINC library 24 was selected because it contains a large collection of drug-like molecules in pdbqt format and also allows the purchase of library compounds. A subset of about 70,000 compounds was screened, selected from the “Full_nci_ALLTAUTOMERS” and “NCI_DiversitySet2” (http://zinc.docking.org/pdbqt/) folders for VHTS. VHTS was downloaded using VSDK 25 codes with slight modifications, and Cygwin was downloaded from www.cygwin.com. VS01.bash of VSDK codes was modified to perform iterative docking for several ligands and extract binding energy into binary files. The lowest binding energy conformation in the first cluster was considered as the most favorable docking pose. This was further validated by analyzing the ligand interactions by using the program LIGPLOT. 26
Results and Discussion
Fragment Library Screening
Fragment-based drug discovery has gained significant popularity in the field of drug design in the past decade. 27 Several marketed drugs, such as ABT-263 (an inhibitor of B cell lymphoma-2) and ABT-518 (a matrix metalloprotease inhibitor) from Abbott Laboratories (Lake Bluff, IL), AT7519 (a cyclin-dependent kinase-2 inhibitor) and AT9283 (a serine–threonine kinase inhibitor) from Astex Therapeutics (Cambridge, UK), and LP261 (a tubulin inhibitor) from Locus (Blue Bell, PA), were each developed from compounds initially identified through a fragment-based drug-screening approach. 28
Traditional compound library screening relies on very large library sizes to increase the likelihood that compounds that were developed against specific drug targets will show some affinity against a new target. Given the vastness of chemical space and the narrow diversity of most compound libraries, even the largest libraries will be able to sample only a miniscule fraction of that space. By contrast, the fragment-based drug development strategy uses low-molecular-weight compounds as a starting point for lead compound generation, with the goal of identifying subsites where these fragments will bind with at least moderate affinities. There are significantly fewer possible chemical structures as the number of atoms is decreased. So, despite the much smaller library sizes, fragment libraries can lead to improved coverage of their chemical space. A limitation of this approach is that the fragment library components are relatively small compounds possessing few functional groups and limited drug-like properties. These hits can, however, be rapidly expanded and optimized to produce effective lead compounds. Compound optimization, either through the coupling of fragments that bind in adjacent sites or by the elaboration of these core structures, is then used to develop these initial hits into advanced lead compounds. Any hits obtained from fragment library screening will typically have low binding affinities (mid- to low millimolar), but their selection for further development is based on their high ligand efficiency value, 29 which is defined (Eq. 1) as the free energy of binding of a ligand (ΔG in kcal/mol) per ligand-heavy (nonhydrogen) atom.
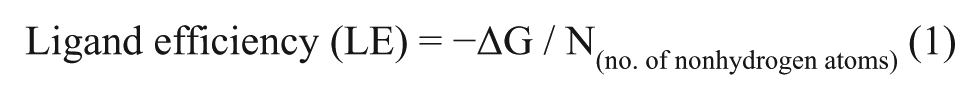
Generally, for a hit to be a useful starting point for drug development, its ligand efficiency should be 0.3 or higher, with the goal of maintaining this value during inhibitor optimization. This ligand efficiency metric is used as the selection criterion to guide the development of initial inhibitors into advanced lead compounds.
Previous studies have developed inhibitors against bacterial ASADH30–32 as potential antibacterial agents, but the development of antifungal inhibitors against this target has not yet been investigated. Two focused fragment libraries with chemical class diversity were screened to identify initial inhibitors against representative members of the fungal ASADH family. A water-soluble and a DMSO-soluble library were screened in 96 four-compound cocktails at 2 mM initial concentrations. Cocktails showing ≤20% initial velocity compared to the control reaction were classified as containing strong enzyme inhibitors, those showing 20–50% as moderate, and those with >50% as weak or noninhibitors. These libraries were initially screened against four fungal enzyme forms (CalASADH, AfuASADH, CneASADH, and BdeASADH) and one bacterial form (SpnASADH) as a selectivity control. Five of the water-soluble fragment library cocktails showed strong inhibition and five showed moderate inhibition (
Inhibitor Identification from Library Screening
Compounds from each cocktail hit were then screened separately, yielding five compounds from the water-soluble library and 13 from the DMSO-soluble library that showed good inhibition against several fungal ASADHs, resulting in overall hit rates of 1.3% and 3.4%, respectively. Because these library compounds can bind with low selectively to different enzyme targets, the hit rates are higher than is typically seen for high-throughput screening (HTS) of libraries. An aspartic acid analog library composed of 83 rotationally constrained substrate analogs was also assembled and tested. Screening of this analog library yielded three strong hits and three moderate hits against the C. albicans form of ASADH.
Selectivity of Initial Fungal ASADH Inhibitors
Inhibition constants (Ki values) were determined for each inhibitory compound, initially against the C. albicans form of ASADH, to analyze their potency. Compounds with Ki values lower than 1 mM and with high LE values were considered as promising starting points for further elaboration. To evaluate fungal ASADH selectivity, inhibitor potency was next evaluated against four different fungal orthologs and also checked against a representative bacterial ASADH. These fungal ASADHs share ~55% sequence similarity, but they share only about ~25% sequence similarity with their bacterial counterparts. Despite the highly conserved catalytic residues throughout the ASADH family, and nearly identical active site architecture, several inhibitors show selectivity between the bacterial and the fungal species, and some compounds also showed selectivity between the ASADHs from different fungal species. Some compounds are nonselective inhibitors; for example, catechol, benzene-1,3,5-triol, and cyclohexyl iodide show similar potency with both the fungal and bacterial enzymes ( Table 1 , section A). Several fungal inhibitors have little inhibitory activity against the bacterial ASADH, but show fourfold or greater potencies against each of the fungal enzymes ( Table 1 , section B). Some compounds show preferential inhibitor selectivity for one of the fungal ASADHs, with twofold to sevenfold greater potency relative to the other fungal enzyme orthologs ( Table 1 , section C, bold entries). These results suggest that subtle differences in the nature of the substrate binding pocket may lead to differences in potency and selectivity of these inhibitors between these different enzyme forms. Although these selectivities are still fairly modest, these lead compounds will be used as starting points for the optimization of species-selective inhibitors that, after further development, could provide novel compounds against this untested drug target.
ASADH Inhibitors from Fragment Library Screening.
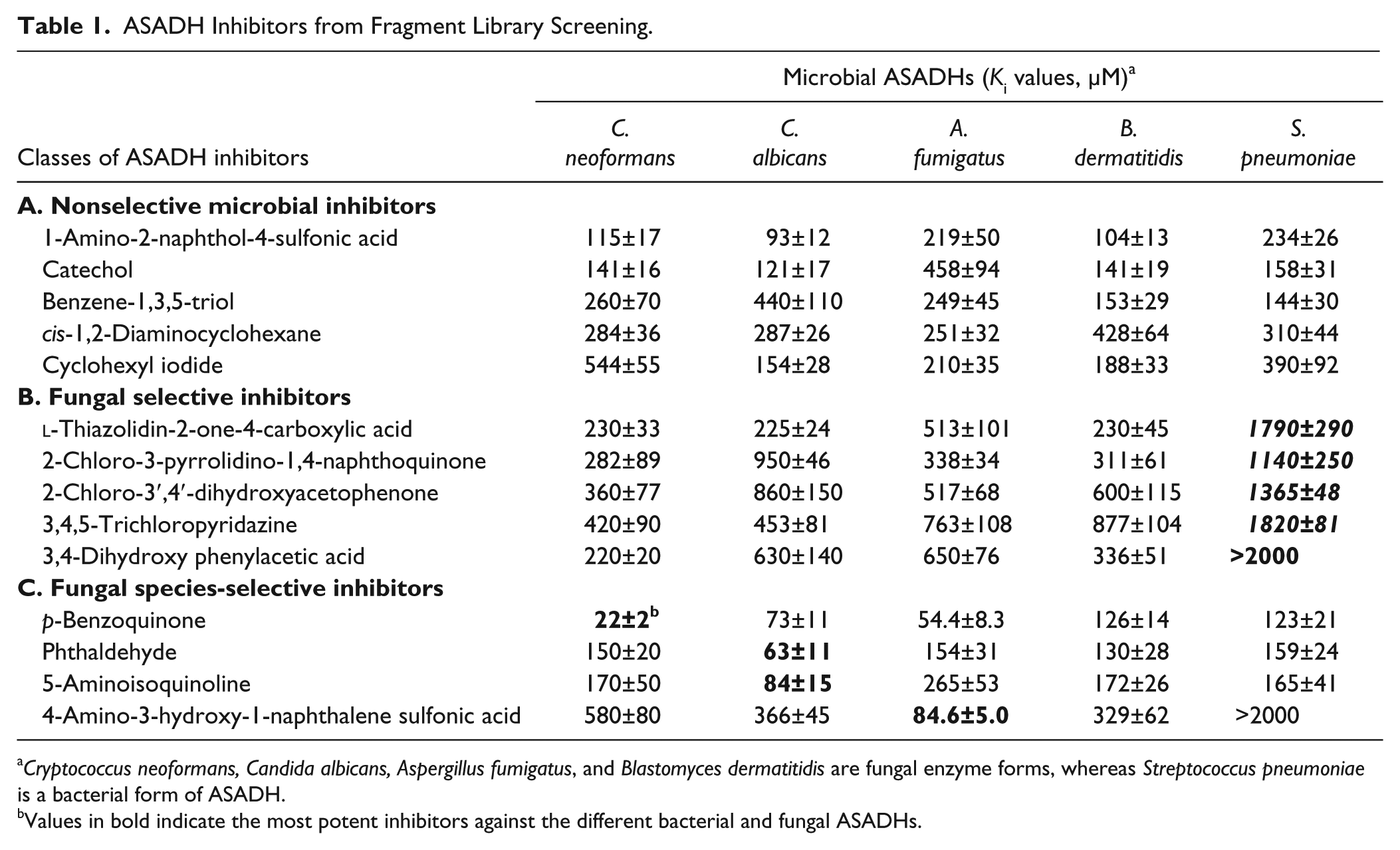
Cryptococcus neoformans, Candida albicans, Aspergillus fumigatus, and Blastomyces dermatitidis are fungal enzyme forms, whereas Streptococcus pneumoniae is a bacterial form of ASADH.
Values in bold indicate the most potent inhibitors against the different bacterial and fungal ASADHs.
From our initial fragment screening of about 850 compounds, a total of 24 fungal enzyme inhibitors were identified, 14 of which showed good inhibition against multiple ASADHs ( Table 1 ). These initial hits display a range of binding affinities, with the best inhibitors possessing Ki values in the low micromolar range against fungal ASADHs. Several of the inhibitors identified were already shown to inhibit other metabolic enzymes, and they have been used as a starting point for the development of more potent inhibitors against these potential drug targets.33–34 Similarly, further elaboration of these initial ASADH inhibitors will be needed to develop their selectivity for this enzyme as a drug target.
Establishing the Mode of ASADH Inhibition
Many of our hits from library screening contain a quinone moiety, which could potentially act as a redox recycling agent on the PMS assay coupling step or by generating hydrogen peroxide through reaction with a strong reducing agent such as DTT. 35 To test for the possible occurrence of a non-enzymatic redox reaction, the assay was carried out by including two quinone-containing inhibitors, p-benzoquinone (200 µM) or 2,3-dichloro-1,4-naphthoquinone (20 µM), in the absence of ASADH. Alternatively, the complete coupling assay with ASADH and those inhibitors was tested without the substrate ASA. In each case, no activity was detected, confirming the absence of quinone-induced redox cycling and any false-positive hits from redox active compounds. To examine the potential reaction between our quinone inhibitors and a strong reductant, ASADH was dialyzed to replace DTT with a weaker reducing agent (β-mercaptoethanol) and also in the absence of any reducing agents. In both cases, the enzyme remained active, and the same inhibition was observed with these two quinone-containing inhibitors, confirming that these compounds are targeting ASADH and that the observed inhibition is not due to redox artifacts.
Because the enzyme active site contains a cysteine nucleophile, it is possible that some of these inhibitors could function by covalently modifying the active site thiol group, thereby leading to an inactivated enzyme. To test this possible mode of action, the enzyme was incubated with a high concentration (500 µM) of either phthaldehyde or p-benzoquinone, two potentially reactive inhibitors, for 1 h and then dialyzed against buffer overnight to remove excess unbound inhibitor. The dialyzed enzyme remained active despite extensive exposure to these potentially reactive compounds. The redox inhibitor-incubated enzyme was also diluted into the assay mixture without dialysis. This 10-fold dilution lowers the final concentration of the inhibitor to lower than its Ki value, resulting in a rate that is only slightly lower than the control in the absence of inhibitor, confirming that the observed inhibition by these compounds is freely reversible.
To examine the type of reversible inhibition, the concentration of each inhibitor was varied at different ASA concentrations, and the kinetic data were fit to different models of reversible inhibition. p-Benzoquinone was found to be a competitive inhibitor versus ASA, confirming that this class of inhibitors bind at the active site of ASADH. Phthaldehyde inhibits fungal ASADHs by binding at the subunit interface and causing dissociation into dimers (Dahal & Viola, unpublished results). Consistent with this mode of inhibition, phthaldehyde was found to be noncompetitive with respect to ASA.
Structure–Activity Relationship (SAR) Studies
For inhibitor optimization and SAR studies, CalASADH was used as a model enzyme, due to the ease of crystallization of this enzyme form, enzyme stability, a higher yield during purification, and, most importantly, because C. albicans infections are considered a serious threat against humans. All of the compounds identified to date as inhibitors against CalASADH have also shown reasonably good inhibitory activity against the other fungal ASADHs.
Structural analogs of four different chemical scaffolds identified from fragment library screening—phthaldehyde and 5-aminoisoquinoline (CalASADH selective inhibitors), p-benzoquinone (a CneASADH selective inhibitor), and cyclohexyl iodide (a nonselective inhibitor)—were chosen for evaluation of their SAR properties. In the available crystal structures,12,13,36 the active site of fungal ASADHs is shallow and partially open, with a central cavity that is fairly solvent accessible. The features of this accessible active site can be exploited to generate inhibitors encompassing the full active-site environment. Also, the fungal forms are missing a small helical subdomain found in bacterial ASADHs, and these fungal ASADHs are found to be tetrameric 36 in contrast to dimers found in bacterial species. This discrepancy can be used to develop selective inhibitors to potentially disrupt this tetrameric interface, which would present a novel route to develop inhibitors with a unique mode of action.
A characteristic feature of phthaldehyde is the presence of aldehyde functional groups in the 1,2 positions of the ring. Examination of a series of phthaldehyde-related compounds as potential CalASADH inhibitors revealed the importance of both the position and nature of functional groups. Removal of one of the aldehyde groups (benzaldehyde) leads to complete loss of potency, and changing the relative positions of the aldehyde groups to the 1,3 position (isophthaldehyde) or 1,4 position (terephthaldehyde) has a similar effect. Oxidizing phthaldehyde to phthalic acid also leads to a loss of potency; in contrast, reduction to the dialcohol (catechol, Ki = 121±17 µM) causes only a twofold reduction of potency. Replacing one of the aldehyde groups with a nitro group (2-nitrobenzaldehyde, Ki = 180±19 µM) results in a threefold loss of potency, whereas extending the benzene ring to a naphthalene (Ki = 45±8 µM) leads to a slight improvement in potency. Each of these CalASADH inhibitors possesses very good ligand efficiencies that are used to select fragment library hits for further structural elaboration. Examination of nearly 20 additional benzene or naphthalene structural derivatives, with either alcohol, thiol, nitro, or carboxyl groups in various combinations and in various positions, did not identify any additional fungal ASADH inhibitors.
A series of benzoquinone (
Fig. 1
, cpd.
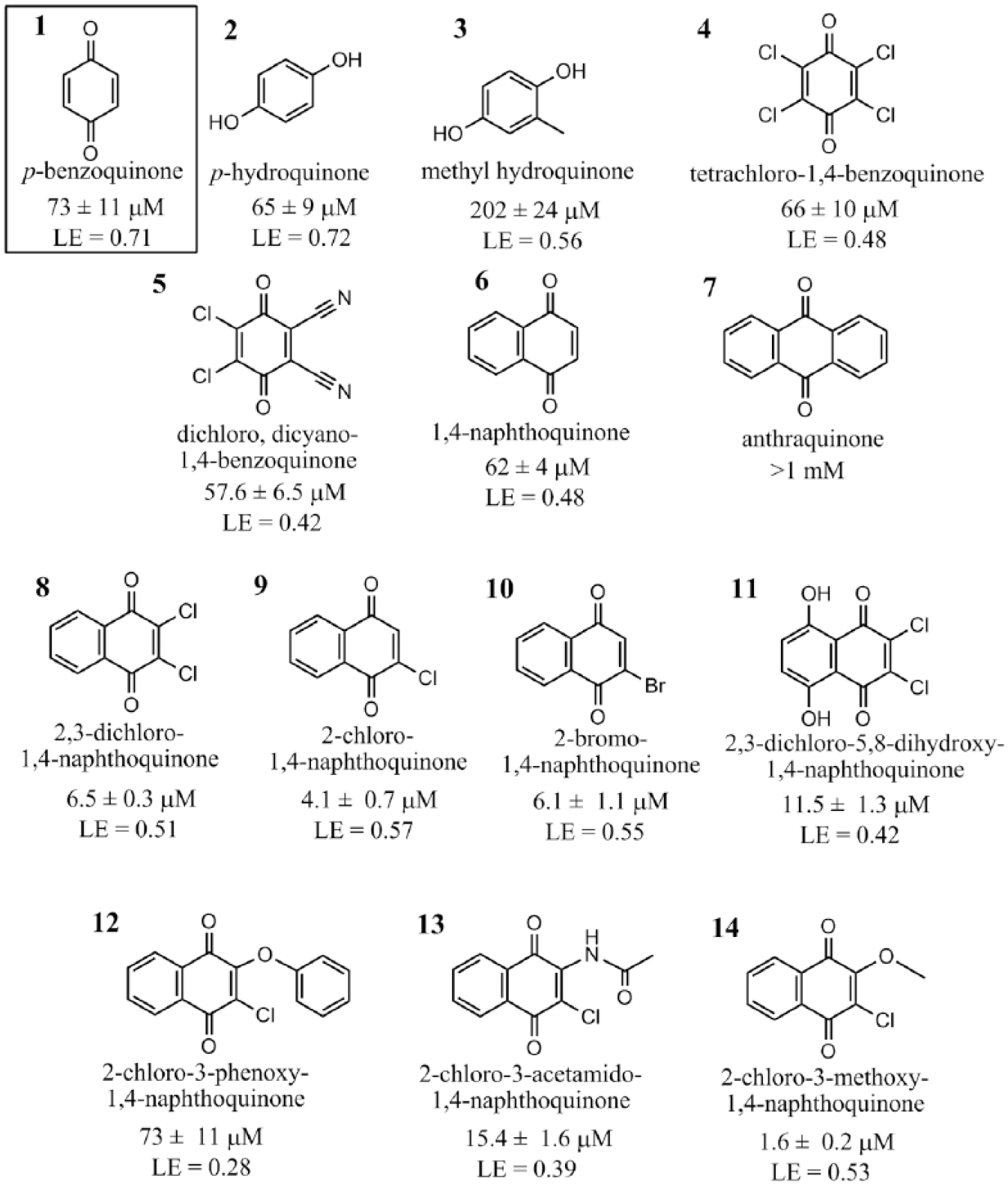
p-Benzoquinone structural analogs screened as potential inhibitors against CalASADH (Cal: Candida albicans; ASADH: aspartate β-semialdehyde dehydrogenase). The most potent of these inhibitors, based on a naphthoquinone core structure, have inhibition constants in the very low micromolar range and excellent ligand efficiency (LE) values.
For the initial 5-aminoisoquinoline inhibitor, moving the position of the amino group or replacing the amino group with a different potential hydrogen bond donor/acceptor group (hydroxyl) at those different positions resulted in a complete loss of potency. Similarly, modifications made to the cyclohexyl iodide inhibitor, such as replacement of the iodide with other halides or with an amine group, caused a loss of potency. The 1,2-diaminocyclohexane derivative is a moderate inhibitor (Ki = 251±32 µM) when these functional groups are oriented in a cis-geometry, but the trans-diamine derivative does not show measurable inhibition against CalASADH.
Inhibitor Docking and Binding Affinities
To explore the binding mode of the more potent inhibitors of CalASADH, and to provide guidance for additional inhibitor optimization, a comprehensive molecular docking study was performed to identify the most plausible and accurate pose of these ligands bound at the active site of fungal ASADHs. The binding orientation of the most potent quinone derivative, 2-chloro-3-methoxynaphthoquinone (
Fig. 1
, cpd.
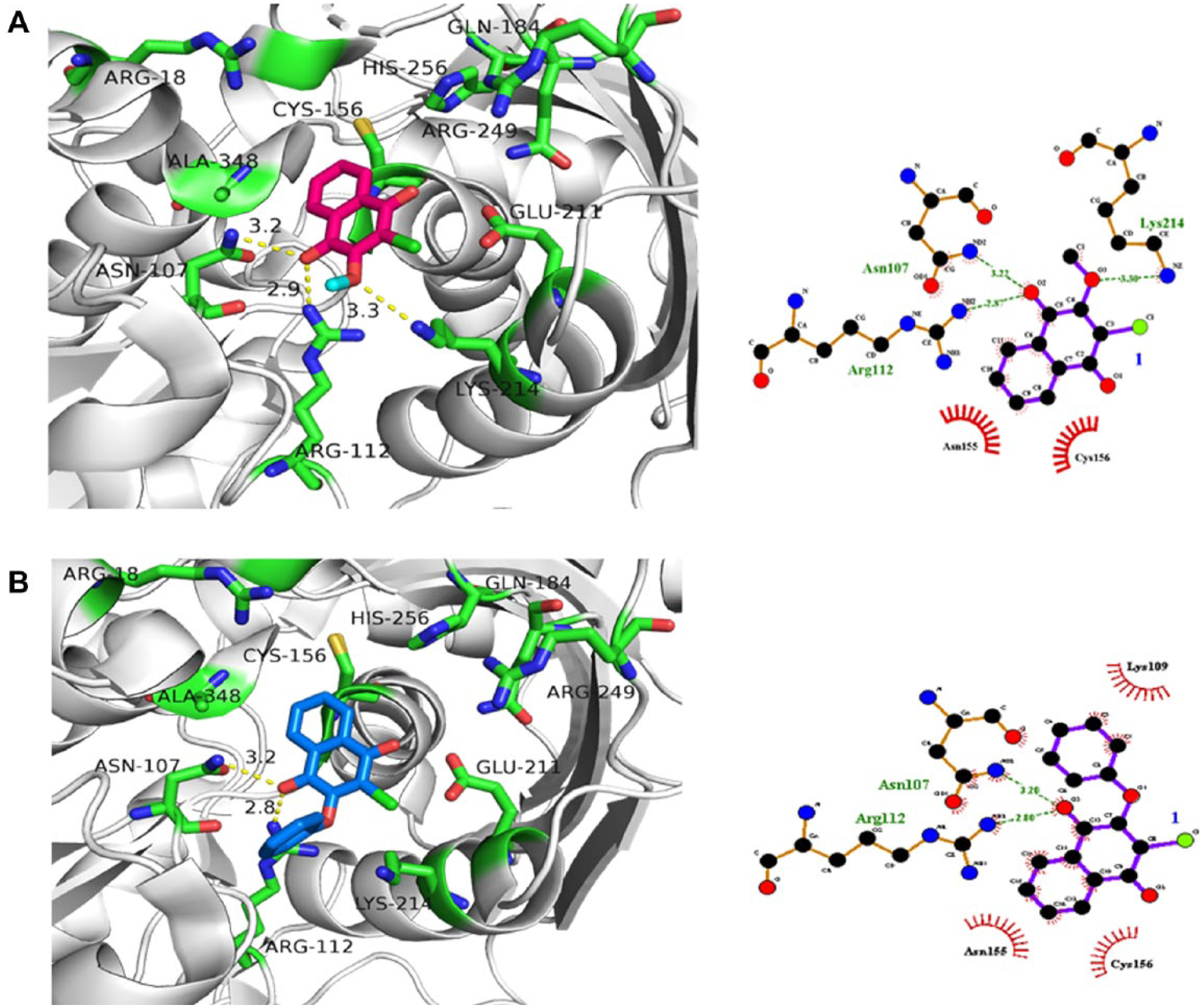
Predicted binding mode of the most potent 1,4-naphthoquinone derivatives binding in the active site of CalASADH (Cal: Candida albicans; ASADH: aspartate β-semialdehyde dehydrogenase). Residues in the active site of CalASADH are shown as sticks, hydrogen bonds are indicated by dashed lines with bond distances, active site residues involved in hydrogen bonding interactions are labeled, and each type of interaction is also shown on the LIGPLOT diagram (right). These docked structures suggest an essential binding role for the carbonyl oxygen moiety. (
Some additional naphthoquinone derivatives, with various hydrophobic and hydrophilic functional group substitutions at positions 2 and 3, were also found to be inhibitors against CalASADH. The 45-fold decrease in potency when the 3-methoxy group in compound
Although there is not an exact correlation between the calculated binding energies and the experimental Ki values for a range of CalASADH inhibitors from different structural classes, and there are a few outliers, a general trend is observed in which the most potent inhibitors (average Ki of 28 µM for nine compounds) are predicted to have stronger binding affinities, whereas the weakest inhibitors examined (average Ki of 250 µM for seven compounds) have the lowest predicted binding affinities (
Drug Library Screening
The more traditional approach to compound screening uses larger chemical libraries containing drug-like compounds that have been optimized against previously identified targets to assess any potential cross-reactivity against new potential drug targets. The NCC is a library of more than 700 drug and drug-like compounds, whereas the Prestwick Library is a University of Toledo collection of more than 1300 drugs and drug candidates. The NCC library was screened at 500 µM, with these screening experiments performed in duplicate using the coupled enzymatic assay for primary screening. Due to limited compound availability, screening was initially performed only with CalASADH. From this initial screening, a total of 14 compounds showed good enzyme inhibition, with the percent inhibition ranging from 70% to as high as 97%. The overall hit rate for the NCC library was about 2%, which is quite high compared to other chemical library screens. Once again, however, it is not surprising to have these higher hit rates considering that the majority of these drugs have gone through multiple selections from numerous libraries and have been optimized for binding to a range of different drug targets.
The compounds showing 70% or more decrease in enzyme rates compared to controls were selected as potential inhibitors for Ki value determination. The compounds examined each inhibited CalASADH in the lower micromolar range, with Ki values ranging from ~200 µM down to 42 µM. Some of these best NCC library hits possess similar pharmacophoric features as those of the better fragment library hits ( Fig. 3 , highlighted in bold).
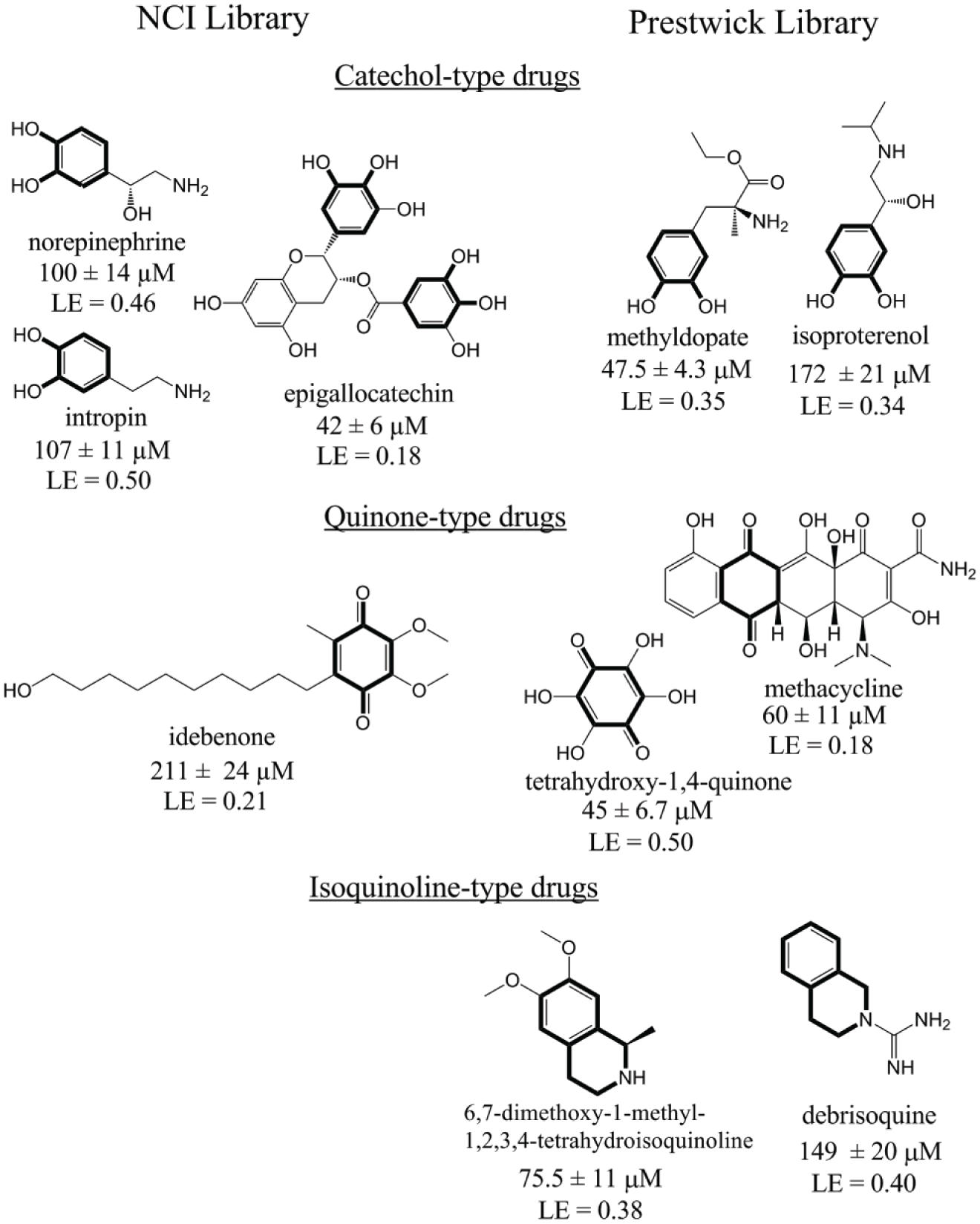
The most potent inhibitors against CalASADH (Cal: Candida albicans; ASADH: aspartate β-semialdehyde dehydrogenase) identified from the National Institutes of Health (NIH) Clinical Collection library (left) and the Prestwick Library (right). The essential core substructures are highlighted in bold, showing the common catechol, quinone, and isoquinoline moieties that are also found in the inhibitors that were identified from fragment library screening.
To test the importance of these pharmacophores in binding to fungal ASADHs, a set of about 70 compounds were selected from the Prestwick drug library for testing, based on the presence of their potentially related substructures. Screening of these compounds identified an additional 15 inhibitors of CalASADH, an unusually high hit rate of 20% that serves to validate the importance of these substructures in recognition by the fungal ASADHs. The six most potent of these inhibitors have Ki values between ~50 and 200 µM, and they included compounds with catechol, quinone, and isoquinoline-type substructures ( Fig. 3 ). The majority of these drugs still have quite high ligand efficiency values, except for those with more extensive structures relative to the fragment library hit structures. The failure of the clinical drugs to bind with significantly higher affinities than that seen for the simpler structural components confirms that these compounds have already been optimized for selectivity against their designated targets. A similar optimization scheme will be needed to generate potent and selectivity inhibitors against the fungal ASADHs.
ZINC Library Virtual Screening
To expand the coverage of chemical space diversity, AutoDock Vina was used to virtually screen about 70,000 commercially available ZINC library compounds to identify various chemotypes that are potent and specific to our fungal target. The result of this screening is a ranked list of library molecules from highest to lowest predicted affinity scores. Because the difference in docking scores among the top-ranked molecules fell in a narrow range, a final list of compounds was selected by visual inspection of their structures, using productive interactions made by these compounds at the ASADH active site, and availability for purchase as the selection criteria. High-throughput docking of this subset of selected ZINC library compounds yielded promising hits showing predicted binding energy as high as −8.9 kcal/mol. Ultimately, a set of these compounds were selected for experimental testing (Ki determination), with the compounds obtained from the National Cancer Institute (NCI) Developmental Therapeutics Program (DTP) repository. Some compounds were eliminated as potential candidates due to limited solubility, but of the eight compounds tested, three were non-inhibitors and five showed Ki values ranging from about 450 µM to as low as 4 µM (
Inhibitor Species Selectivity
A number of different naphthoquinone derivatives show enhanced affinity to CalASADH relative to the parent benzoquinone compound, with as much as a factor of nearly 50 for the most potent derivative (
Fig. 1
). An examination of these compounds as inhibitors of the other fungal ASADHs shows similar improvements. The parent p-benzoquinone binds to AfuASADH with comparable affinity to that against CalASADH, and with an IC50 value that is similar to its inhibition constant (
Fig. 4
). This inhibitor shows some ortholog specificity, with a threefold greater affinity in binding to CneASADH (
Table 2
). Each of the halo-naphthoquinones, however, shows the greatest affinity with AfuASADH, with improvements ranging from twofold to sevenfold for these derivatives relative to that against CalASADH. The 2,3-dichloro derivative (cpd.
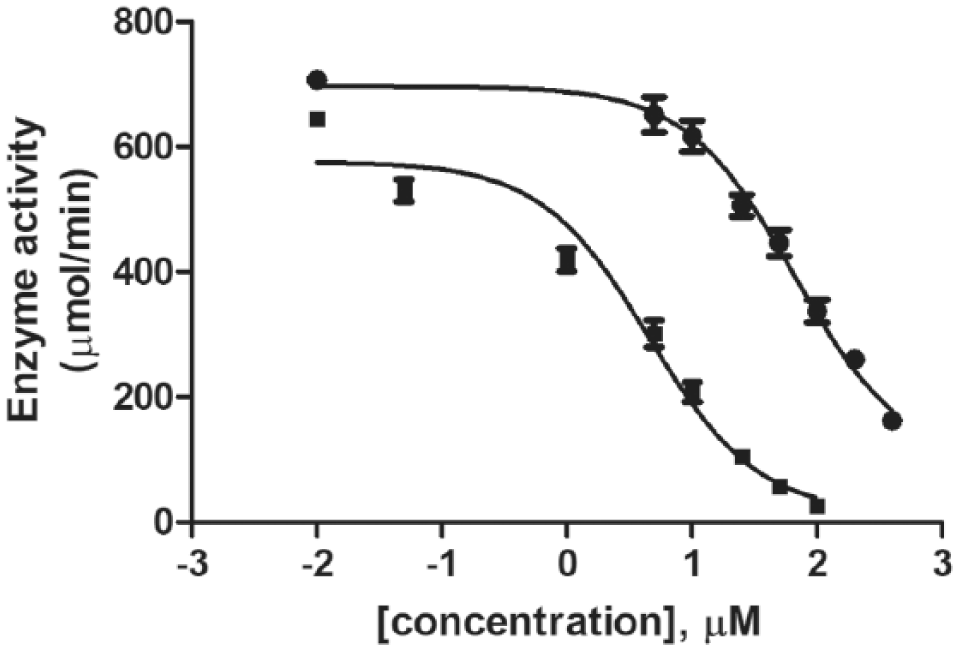
Representative dose–response curves for 2-chloro-1,4-naphthoquinone (■), a strong inhibitor (IC50 = 0.6 µM), and for p-benzoquinone (●), a moderate inhibitor (IC50 = 50 µM), interacting with AfuASADH (Afu: Aspergillus fumigatus; ASADH: aspartate β-semialdehyde dehydrogenase). Each rate was determined in triplicate, with the error bars shown on each data point.
Selectivity of Naphthoquinone Derivatives against Fungal ASADHs.
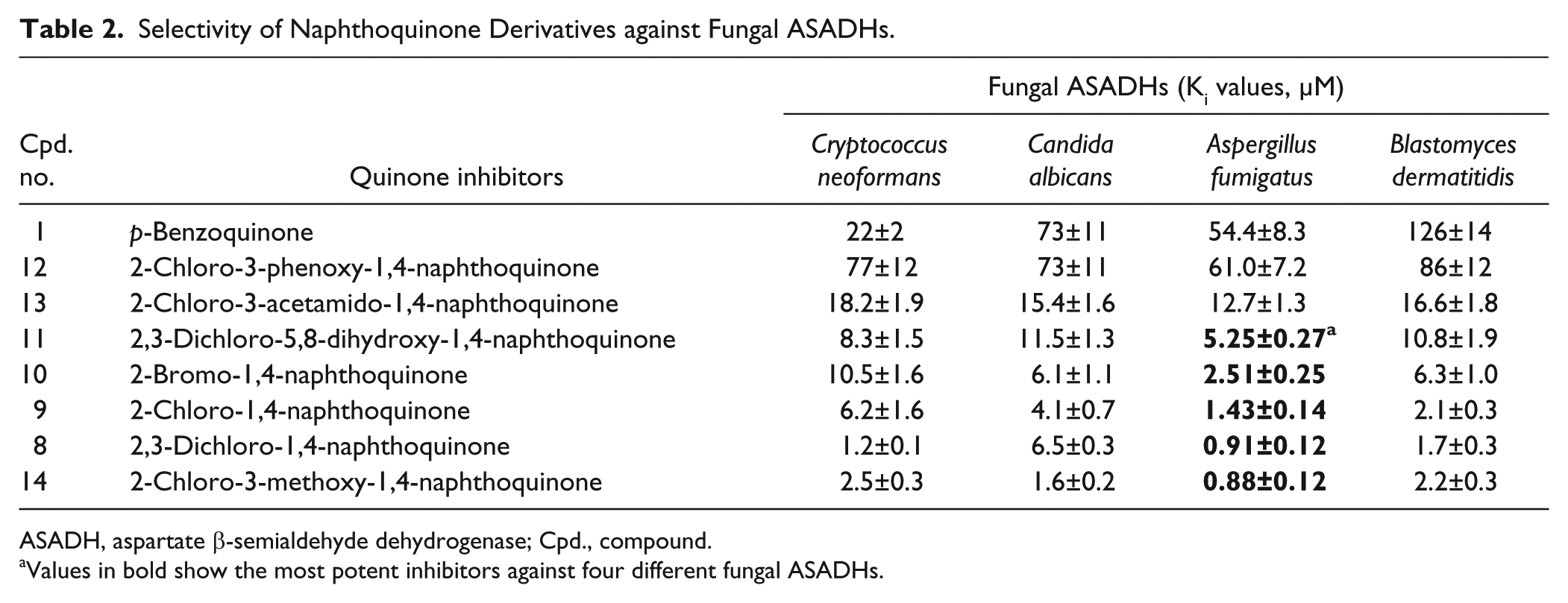
ASADH, aspartate β-semialdehyde dehydrogenase; Cpd., compound.
Values in bold show the most potent inhibitors against four different fungal ASADHs.
Future Inhibitor Optimization
Most of the productive interactions identified between these fungal ASADHs and the more potent inhibitors are proposed to involve just two functional groups on the naphthoquinone ring ( Fig. 2 ). Additional functionalization and elaboration of this core structure have the potential to make new interactions with an array of active site functional groups. The proposed interactions with the docked compounds from virtual library screening will be tested by the incorporation of these and related functional groups into the inhibitor structures. As structures of these inhibitors bound to our target enzyme become available, this information will be incorporated into our inhibitor optimization scheme. This structure-guided approach will likely lead to significantly more potent and selective inhibitors of this essential fungal enzyme.
Supplemental Material
DS_DISC767844 – Supplemental material for A Fragment Library Screening Approach to Identify Selective Inhibitors against an Essential Fungal Enzyme
Supplemental material, DS_DISC767844 for A Fragment Library Screening Approach to Identify Selective Inhibitors against an Essential Fungal Enzyme by Gopal P. Dahal and Ronald E. Viola in SLAS Discovery
Footnotes
Acknowledgements
The authors thank Dr. Stephen Thomas (Turing Pharmaceuticals) for designing and assembling the constrained analog library, Dr. Stephen White (National Cancer Institute) for providing the ZINC library compounds, and Dr. Travis Taylor (University of Toledo College of Medicine) for providing the Prestwick drug library.
Supplementary material is available online with this article.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.