
Abstract
The microbial threat to human health is growing due to the dramatic increase in the number of multidrug-resistant organisms. The decline in effective antibiotics available to treat these growing threats has provided greater urgency to the search for new antibiotics. Clearly, new approaches must be developed against novel targets to control these resistant infectious organisms. The screening of low molecular weight compounds against new protein targets provides an opportunity to identify novel inhibitors as starting points for the development of new antibiotics. Custom fragment libraries have been assembled and screened against 3 representative forms of a key enzyme in an essential microbial biosynthetic pathway. Although each of these aspartate semialdehyde dehydrogenases (ASADHs) catalyzes the same reaction and each shares identical active site functional groups, subtle differences in enzyme structures have led to different binding selectivity among the initial hits from these fragment libraries. Amino acid analogues have been identified that show selectivity for either the gram-negative or gram-positive bacterial enzyme forms. A series of benzophenone analogues selectively inhibit the gram-negative ASADH, whereas some haloacids and substituted aromatic acids have been found to inhibit only the fungal form of ASADH. Each of these low molecular weight compounds possesses high ligand binding efficiency for their target enzyme forms. These results support the goal of designing lead compounds that will selectively target ASADHs from different microbial species.
Keywords
Introduction
W
The examination of new protein targets to identify potent lead compounds has traditionally been conducted by screening compound libraries of drug-like molecules. Although the overall success rate in identifying compounds that bind to a given target with high affinity is generally quite low, the increasing use of robotics to rapidly screen large, million-compound libraries continues to yield useful initial hits that can be further developed. An alternative approach, the screening of small compound libraries containing low molecular weight compounds (herein called “fragments”) with relatively few functional groups and limited drug-like properties, has some advantages over the traditional screening approach. The size of these fragment libraries is several orders of magnitude smaller than the large drug libraries, requiring less time and resources to screen. Because these fragments are smaller than typical drug-like molecules, the binding affinities are expected to be much weaker than drug library hits, and the specificity for the target protein is likely to be much lower. However, the success rates can be much higher for fragment libraries since different criteria are used to identify promising initial hits. Unlike screening with drug libraries, the criterion used to identify successful hits in fragment library screening is not high binding affinity but high ligand efficiency. Ligand efficiency (LE) is a measure of binding affinity that is adjusted for the smaller size of these fragments and is defined 3 as
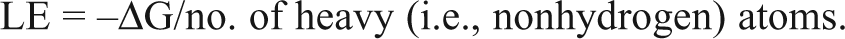
Most successful drug candidates have nanomolar affinities for their targets but also possess ligand efficiencies of 0.3 kcal/mol/heavy atom or greater. The goal for fragment library screening is to identify compounds that bind to the protein target with at least modest affinities but with initial LE values of 0.3 or greater. As the size and complexity of these initial hits are increased through structural elaborations and fragment coupling, maintaining high LE is used as the selection criterion to guide the development of lead compounds. 4
The aspartate biosynthetic pathway is essential for microorganism survival, but this entire pathway is absent in mammals, thus requiring the acquisition of the products of this pathway from dietary sources. Aspartate β-semialdehyde dehydrogenase (ASADH) catalyzes the second step in the aspartate pathway, a reductive dephosphorylation that converts β-aspartyl phosphate to aspartate β-semialdehyde (
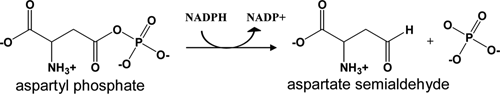
Physiological reaction catalyzed by aspartate β-semialdehyde dehydrogenase (ASADH).
The aspartate biosynthetic pathway presents a number of attractive and as yet untested targets for novel drug intervention. The identification of selective inhibitors of these critical microbial enzymes can lead to the development of new classes of antimicrobials that can be highly effective against the growing threat from multidrug-resistant infectious organisms. Structures have been determined of ASADHs isolated from a variety of microbial species, and these structures are organized into related groups based on the source of the enzyme. All of the ASADHs studied to date possess the same set of substrate binding and active site catalytic groups 15,16 despite sequence homologies ranging from more than 90% to less than 10%. The enzymes from gram-negative bacteria are closely related and share many common structural features. 17,18 Domain and secondary structural comparisons of the archael 19 and fungal 20 forms of ASADH show that they are most closely related to each other and have the greatest differences with the gram-negative enzyme forms. The gram-positive bacterial ASADHs possess structural elements that are situated between those of the fungal and the gram-negative enzyme families. 21 The combined application of kinetic and mutagenic studies, along with structural characterization of key catalytic intermediates, 21,22 has contributed to a detailed understanding of the mechanism of action of ASADH. Because of its crucial position at an early branch point in this essential microbial pathway, identifying new inhibitors against ASADH can be a valuable strategy for the development of novel antibacterial and antifungal drugs. In this study, ASADHs from the gram-positive Streptococcus pneumoniae (spASADH) and the gram-negative Vibrio cholerae (vcASADH) bacterium, as well as from a yeast species, Candida albicans (caASADH), were each screened against small fragment libraries to find initial hits that inhibit ASADH activity with high ligand efficiencies and, in some cases, with unexpected selectivities.
Materials And Methods
Materials
ASADHs from S. pneumonia, V. cholerae, and C. albicans were cloned, expressed, and purified as previously described 21,23,24 and then concentrated and stored in 50 mM HEPES (pH 7) with 1 mM EDTA and dithiothreitol (DTT) at −20°C. Aspartate β-semialdehyde (ASA) was synthesized as previously described 25 and stored in 4 M HCl at −20°C due to the instability of this compound under basic conditions. Working solutions of ASA were neutralized prior to addition to the assay mixtures.
Fragment libraries
Two fragment libraries were assembled by including a range of different low molecular weight compounds to probe binding to the target enzymes. The water-soluble fragment library (SFL) is composed of 384 compounds equally divided among 4 different compound classes:
Amino acids and derivatives—natural and unnatural amino acids; N-derivatized and amino acid esters; halo- and phospho-amino acids; dipeptides
Metabolites and analogues—mono-, di-, and tricarboxylic acids; halo acids; phosphorylated metabolites
Carbohydrates and bases—sugars, sugar acids, and amino sugars; nucleobases
Water-soluble organics and aromatics—5- and 6-membered heterocyclic derivatives; amino alcohols; halo acids and amides; nitro- and hydroxybenzyl derivatives
The average molecular weight of these compounds is ~155 Da, with 90% of the compounds between 90 and 250 Da. An organic fragment library (OFL) of 384 compounds was also assembled from 4 different classes of compounds:
Benzene derivatives—halo, cyano, and hydroxybenzenes; benzaldehydes and benzoic acids
Five-membered heterocycles—oxygen-, nitrogen-, and sulfur-containing heterocycles; halo, amino, carboxy, and methyl derivatives
Six-membered heterocycles—nitrogen- and oxygen- containing heterocycles; halo, amino, nitro, hydroxy, and carboxy derivatives
Fused/multiple ring systems—naphthalenes and indoles; quinines and benzyls; diphenyl and dibenzyl derivatives; cycloalkanes
The average molecular weight of the OFL members is 160 Da, with 90% of the compounds having molecular weights between 90 and 240 Da. Stock solutions of each compound were prepared in water (200 mM) or DMSO (400 mM), with some pH adjustment as needed to solubilize acids or bases in the SFL. For screening purposes, fragment cocktails were assembled by mixing 1 compound from each class, resulting in 96 fragment cocktails containing 4 compounds for each library.
Kinetic assay
ASADH catalyzes the reductive dephosphorylation of an acyl phosphate to generate an aldehyde product (
Due to the instability of aspartyl phosphate, this reaction is most easily followed in the reverse direction by monitoring the production of NADPH at 340 nm. Initial velocity kinetics for inhibition of ASADH were carried out at 25°C with the reaction mixture composed of 120 mM CHES (pH 8.6), 200 mM KCl, and the substrates ASA and NADP added at twice their respective Km values. To identify ASADH inhibitors from the fragment libraries, this reaction was examined in the presence of different 4-compound fragment library cocktails.
Because the organic fragment library members are dissolved in 100% DMSO, it was necessary to identify the optimal enzyme assay conditions in the presence of this organic solvent. Fortunately, the ASADHs are quite stable and active in aqueous DMSO mixtures, retaining nearly 70% of their native activity in DMSO concentrations as high as 50%. Typical assays were run in 20% DMSO in the same reaction mixtures as was used to screen the SFL.
Library screening
Earlier studies had shown that the active sites are highly conserved throughout the whole ASADH family, 21 and the optimum reaction pH for the Escherichia coli enzyme (ecASADH) is between 8 and 9. Therefore, the pH of each fragment cocktail was adjusted to 8~9 before screening by titrating with NaOH or HCl. To improve the screening efficiency, the reactions were monitored in a 96-well plate by using a SPECTRAmax® 340PC Microplate Spectrophotometer (Molecular Devices, Sunnyvale, CA). For each row of the plate, 160 µL of the stock buffer and substrate solution (final substrate concentrations: 1 mM ASA, 1.5 mM NADP, 20 mM phosphate) was added to each well with an 8-channel multitip pipettor, followed by 20 µL of each fragment cocktail solution. The reaction was initiated by addition of 20 µL of enzyme solution to each well in the row. To minimize the variation between the reactions in different rows, the initial velocity of the reaction was compared to an internal standard by assuming the reaction with the highest initial velocity rate in each row has no inhibitor present and then comparing those values to controls run in the absence of inhibitor. Good inhibitors of ASADH were defined as those compounds that resulted in less than 10% of the initial rate compared to the control reactions. Fragments from the cocktails containing these inhibitors were then screened individually by using the same protocol but now varying the concentration of each inhibitor. Inhibition constants (Ki values) were determined by a Dixon analysis 26 in which the inhibitor is assumed to compete with one of the substrates for binding to the enzyme. To eliminate false-positive readings, stock solutions of each individual inhibitor were scanned from 300 to 500 nm, and the concentration of any strongly absorbing inhibitors was decreased to allow the enzyme assay to remain in a linear absorbance range.
Results and Discussion
Fragment library screening
Earlier studies had identified several substrate analogue inhibitors against the target enzyme, ASADH obtained from E. coli, including S-methyl-L-cysteine sulfoxide, an active site-directed inactivator, and aspartyl β-difluorophosphonate, a reversible competitive inhibitor.
27-29
To introduce diversity and novelty into the list of ASADH inhibitors, a custom fragment library of water-soluble compounds (SFL) was assembled (as described in the Materials and Methods section) to test this experimental approach to inhibitor development against different members of the ASADH enzyme family. This SFL was screened kinetically against representative gram-negative (V. cholerae), gram-positive (S. pneumoniae), and fungal (C. albicans) forms of ASADH in 96 four-compound cocktails, containing 1 member from each compound class at 20-mM final concentrations. These ASADHs have very similar overall structures, reasonably high-sequence homologies, and an identical constellation of active site functional groups.
15,16
However, surprisingly, quite different inhibition patterns were observed against these enzymes that use an identical mechanism to catalyze the same chemical reaction with the same substrates. Only 4 of the 96 cocktails in this initial screen contained a compound that showed good inhibition against all 3 enzyme forms (

Fragment library screening against Vibrio cholerae (vcASADH), Streptococcus pneumoniae (spASADH), and Candida albicans (caASADH). (
A second 384-compound, DMSO-soluble OFL was used to probe the aromatic binding selectivity between these different ASADHs. ASADH retains more than half of its catalytic activity in 20% DMSO, which is sufficient to maintain the solubility of the compounds in this OFL and expands the capability to measure inhibition by these water-insoluble compounds. Again, significant differences were observed in the inhibition patterns between the representative ASADHs. In contrast to the results obtained from the SFL screening, in this case 24 different cocktails from this OFL showed good inhibition against all 3 forms of ASADHs (
Inhibitor identification
The individual compounds present in these inhibitory cocktails from each fragment library were examined separately to identify the inhibitory component and also in various combinations if the individual compounds from each of these cocktails did not cause appreciable inhibition. As expected, in a number of cases, the individual compounds strongly absorbed at 340 nm, especially those compounds from the OFL, thus causing interference with the assay and leading to false-positive readings during cocktail screening. These compounds were each rescreened at the highest concentrations that still allowed accurate rate measurements to determine if any of the highly absorbent compounds were able to function as enzyme inhibitors. Inhibition constants (Ki values) were measured for each of the inhibitory compounds, first against the enzyme form identified from the initial screening studies and then against the other enzyme forms to measure their relative selectivities. From these kinetic studies, a total of 8 compounds were identified that inhibited spASADH, 13 compounds that inhibited vcASADH, and only 5 that inhibited caASADH with measureable affinities (
Aspartate β-Semialdehyde Dehydrogenase Inhibitors Identified from Fragment Library Screening
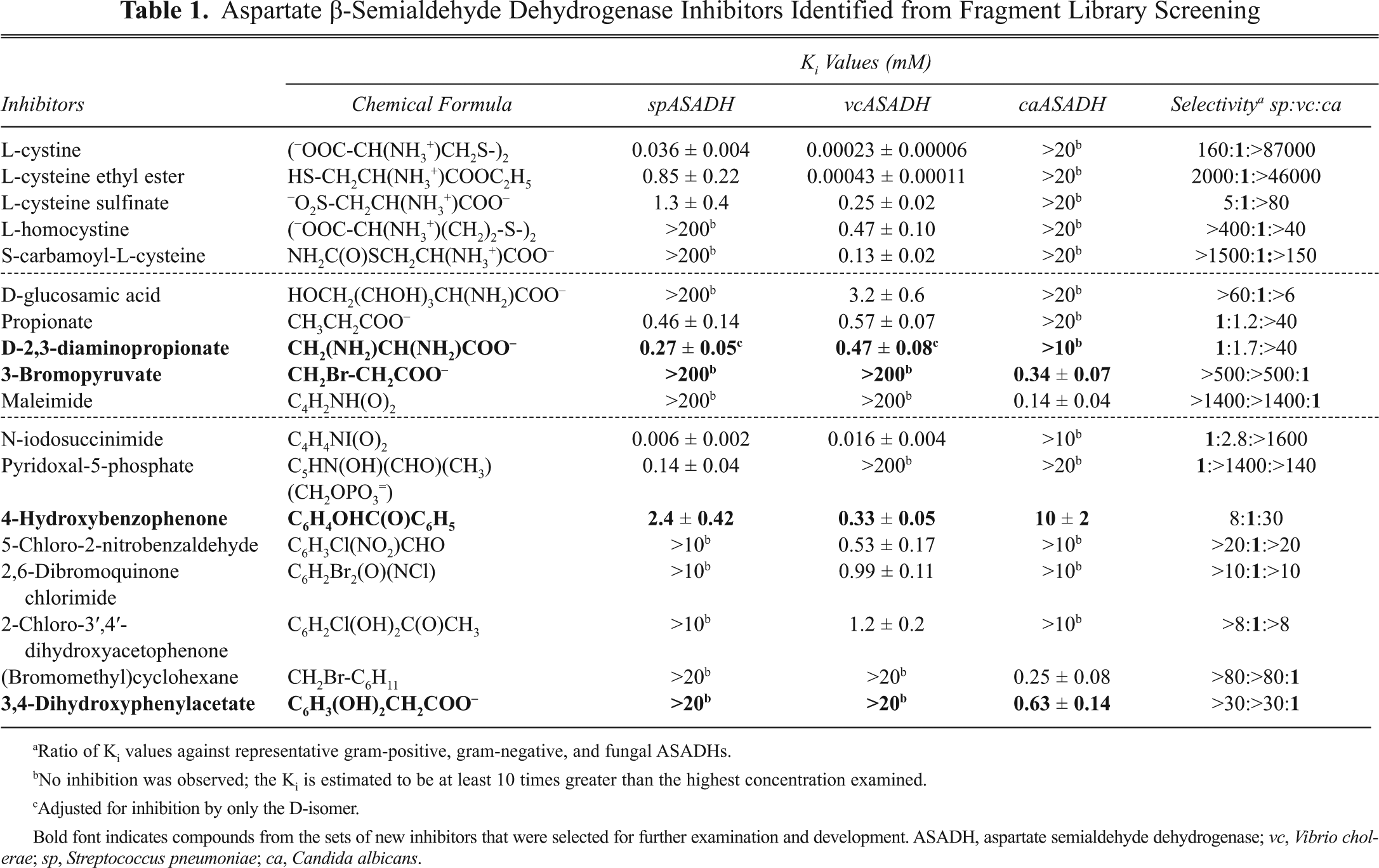
Ratio of Ki values against representative gram-positive, gram-negative, and fungal ASADHs.
No inhibition was observed; the Ki is estimated to be at least 10 times greater than the highest concentration examined.
Adjusted for inhibition by only the D-isomer.
Bold font indicates compounds from the sets of new inhibitors that were selected for further examination and development. ASADH, aspartate semialdehyde dehydrogenase; vc, Vibrio cholerae; sp, Streptococcus pneumoniae; ca, Candida albicans.
Overall, 10 inhibitory compounds were identified from the aqueous SFL (a 2.6% hit rate) and 8 from the DMSO-soluble OFL (a 2.1% hit rate). In most cases, the observed inhibition constants were generally quite modest, typically in the low millimolar range, as would be expected for these lower molecular weight fragments. However, several compounds were found to inhibit one of the ASADHs with Ki values in the low micromolar range (
Selectivity of ASADH inhibitors
On the basis of our initial screening studies, significant differences were observed in the affinity of fragment library hits for each of these representative enzymes despite the presence of a highly conserved active site throughout the ASADH family. The affinity differences between the gram-negative and gram-positive bacterial enzymes suggest that some of the spASADH inhibitors are accessing a binding pocket or a combination of binding groups that are either not present or are less accessible in vcASADH. The complete lack of overlap between the initial hits for the yeast form of ASADH indicates that there must be unique interactions in caASADH that are not shared by the bacterial forms of this enzyme and can potentially be exploited for the development of antifungal agents against this enzyme target.
Some of these new inhibitors, such as propionate and 2,3-diaminopropionate, do not show appreciable selectivity between the bacterial forms of ASADH; however, a number of other compounds show considerable selectivity between the gram-negative and gram-positive ASADHs. S-carbamoyl-L-cysteine has more than a 1500-fold preference for vcASADH, and L-homocystine has more than a 400-fold higher affinity, whereas L-cysteine and L-cysteine ethyl ester are each more than 40,000-fold more potent against the gram-positive spASADH enzyme form (
Inhibitor development
Several interesting compounds from these sets of new inhibitors (
Amino Acid Inhibitors of Bacterial ASADHs
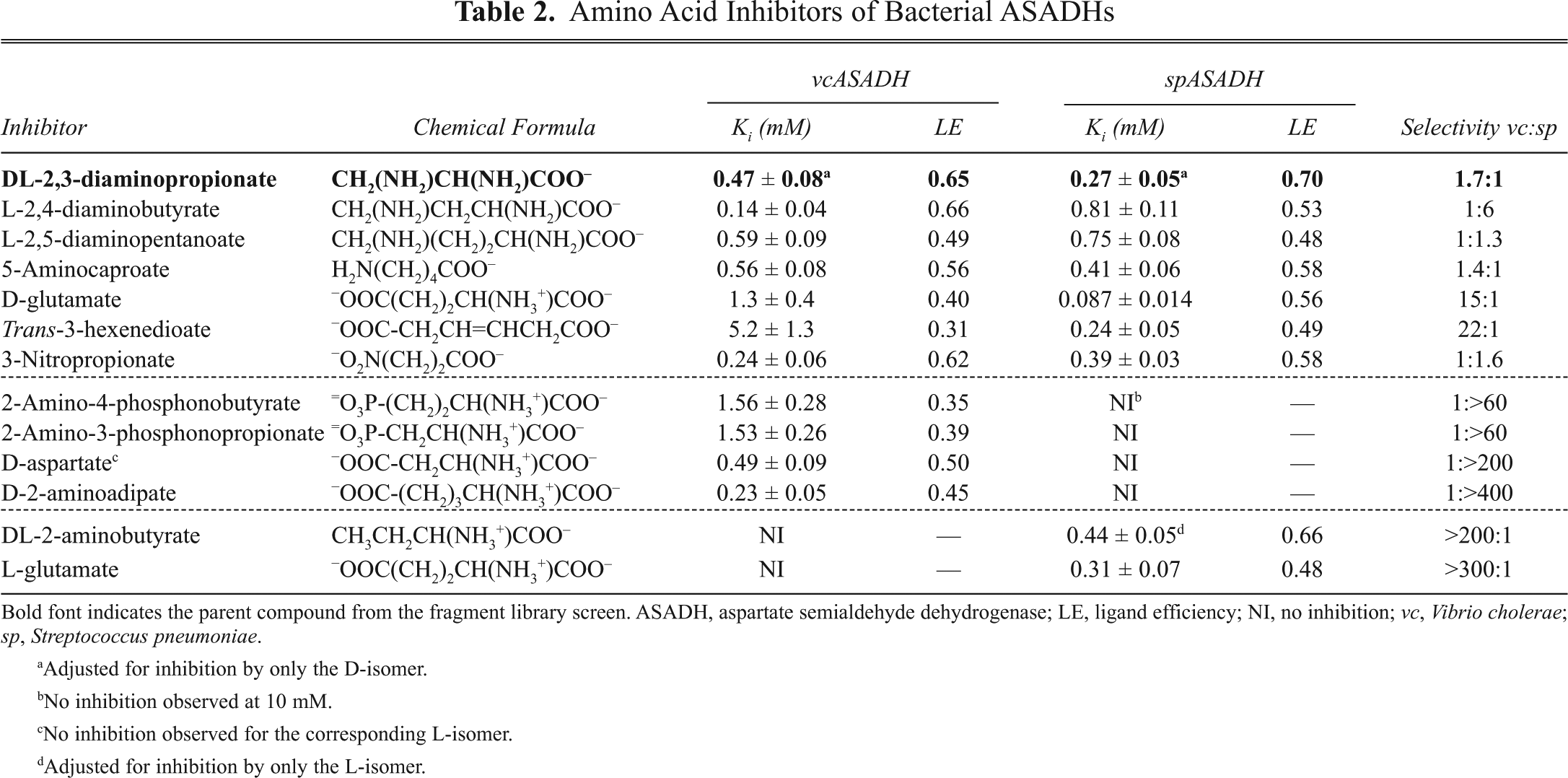
Bold font indicates the parent compound from the fragment library screen. ASADH, aspartate semialdehyde dehydrogenase; LE, ligand efficiency; NI, no inhibition; vc, Vibrio cholerae; sp, Streptococcus pneumoniae.
Adjusted for inhibition by only the D-isomer.
No inhibition observed at 10 mM.
No inhibition observed for the corresponding L-isomer.
Adjusted for inhibition by only the L-isomer.
The 4-carbon dicarboxylic acid succinate does not inhibit either enzyme form, but a nitro analogue of succinate (3-nitropropionate) is a strong inhibitor of both enzymes with high LE values. A conformationally constrained, 6-carbon dicarboxylic acid (trans-3-hexenedioic acid) was also examined and shows >20-fold preference for binding to the gram-positive enzyme form and diminished LE to the gram-negative enzyme. Although the Ki values for these compounds vary by a factor of about 60 from the most to least potent, each of these inhibitors still shows good ligand efficiency values (
Although these particular aminocarboxylate inhibitors show only modest selectivities between these bacterial enzymes, several highly selective inhibitors were identified for both the gram-negative and the gram-positive forms of ASADH. Aminophosphonates have low millimolar inhibition for only vcASADH, and the D-isomers of aspartate and 2-aminoadipate are also selective inhibitors of only vcASADH (
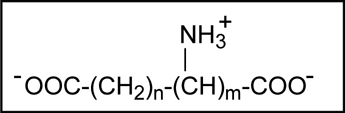
Minimal inhibitory amino acid pharmacophore (n = 0-3, m = 1-2).
The L-stereoisomers of these amino acids show preferential inhibition of the gram-positive ASADH, whereas the D-isomers are selective inhibitors of the gram-negative enzyme form. This chiral selection was unexpected because the substrate for this enzyme family (ASA) requires L-stereochemistry at the α-carbon. The basis for this stereoselectivity between these structurally and functionally related enzymes must be established by structural studies of the respective enzyme-inhibitor complexes.
4-Hydroxybenzophenone shows some selectivity (8:1) for gram-negative versus gram-positive bacterial ASADHs and only very weak inhibition of the fungal enzyme (
Benzophenone Inhibitors of vcASADH
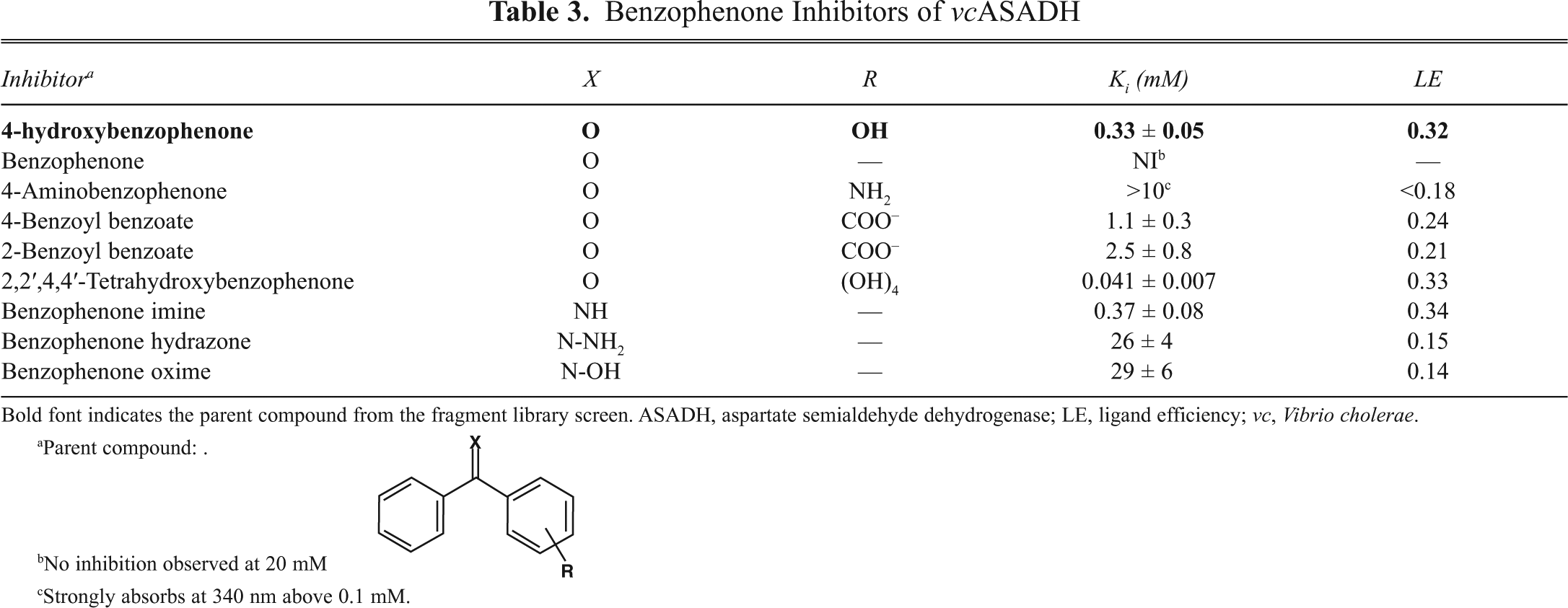
Bold font indicates the parent compound from the fragment library screen. ASADH, aspartate semialdehyde dehydrogenase; LE, ligand efficiency; vc, Vibrio cholerae.
Parent compound:  .
.
No inhibition observed at 20 mM
Strongly absorbs at 340 nm above 0.1 mM.
The fungal form of ASADH has a similar overall structure to that of the bacterial enzymes
20
but shows a very different fragment library screening profile in which each of the inhibitors identified against caASADH was found to be noninhibitory toward the bacterial enzymes (
Organic Acid Inhibitors of caASADH
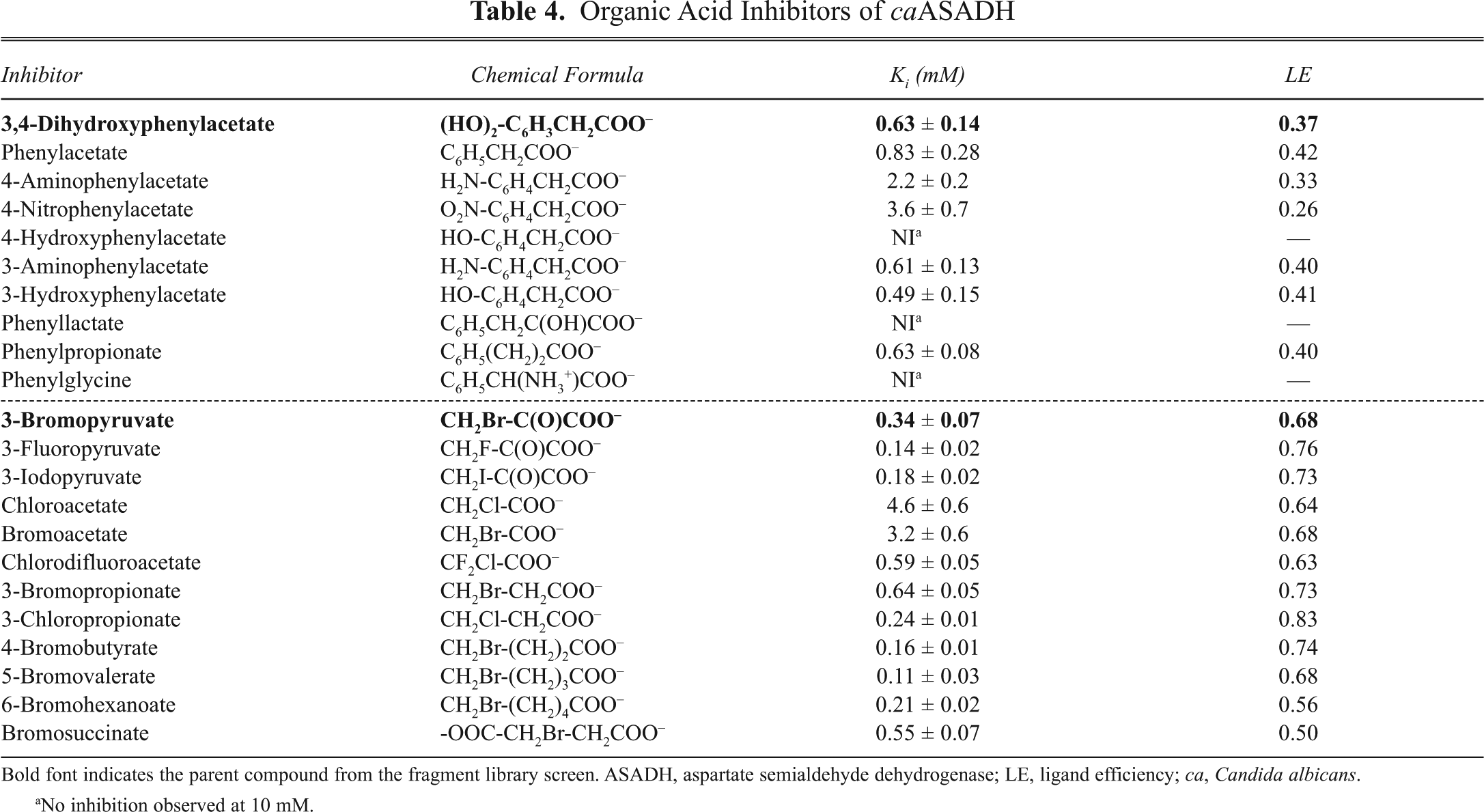
Bold font indicates the parent compound from the fragment library screen. ASADH, aspartate semialdehyde dehydrogenase; LE, ligand efficiency; ca, Candida albicans.
No inhibition observed at 10 mM.
3-Bromopyruvate from the SFL is a sub-millimolar inhibitor of caASADH, and its small size leads to a very high ligand efficiency value (
As the nature of the interactions between these low molecular weight inhibitors and the different forms of ASADH is elucidated, elaboration and functionalization of these different core structures will produce compounds with more drug-like properties. In addition, coupling of fragments that bind at adjacent sites on the target enzyme will increase the complexity of these inhibitors and is expected to lead to the development of lead compounds with increased potency and enhanced selectivity.
Footnotes
Acknowledgements
The authors thank DeMarco Camper and Buenafe Arachea for providing purified samples of the V. cholerae and C. albicans forms of ASADH, as well as Drs. Paul Erhardt and Rahul Khupse (University of Toledo, Center for Drug Design and Development) for helpful discussions on the synthesis of new enzyme inhibitors.
This work was supported by a grant from the National Institutes of Health (AI077720).