
Abstract
Screening against a disease-relevant phenotype to identify compounds that change the outcome of biological pathways, rather than just the activity of specific targets, offers an alternative approach to find modulators of disease characteristics. However, in pain research, use of in vitro phenotypic screens has been impeded by the challenge of sourcing relevant neuronal cell types in sufficient quantity and developing functional end-point measurements with a direct disease link. To overcome these hurdles, we have generated human induced pluripotent stem cell (hiPSC)–derived sensory neurons at a robust production scale using the concept of cryopreserved “near-assay-ready” cells to decouple complex cell production from assay development and screening. hiPSC sensory neurons have then been used for development of a 384-well veratridine-evoked calcium flux assay. This functional assay of neuronal excitability was validated for phenotypic relevance to pain and other hyperexcitability disorders through screening a small targeted validation compound subset. A 2700-compound chemogenomics screen was then conducted to profile the range of target-based mechanisms able to inhibit veratridine-evoked excitability. This report presents the assay development, validation, and screening data. We conclude that high-throughput-compatible pain-relevant phenotypic screening with hiPSC sensory neurons is feasible and ready for application for the identification of new targets, pathways, mechanisms of action, and compounds for modulating neuronal excitability.
Introduction
Cell-based assays using recombinant lines or animal cells are used widely for discovering and profiling new drug candidates. 1 However, poor correlation between the in vitro pharmacology of potential therapeutics measured in reductionist cell-based assays and future clinical effect is likely a factor for efficacy-based failures. 2 To address this lack of predictive clinical potential from early cell-based assays, a key challenge for drug discovery is to adopt earlier use of cell types that more closely resemble those of the patient. Specific cell types made by directed differentiation of human stem cells have great potential, and in recent years, protocols for preparing an increasing range of cell types have been described; examples include neuronal subtypes,3–5 cardiomyocytes, 6 hepatocytes, 7 pancreatic beta cells, 8 erythroid cells, 9 kidney cells, 10 and chondrocytes. 11 Endogenous protein expression levels, intact signaling cascades, and associations between relevant subunits and accessory proteins, together with other factors, are such that these cells will provide a more favorable physiologically relevant cellular context for the study of disease processes and the pharmacology of therapeutics. In addition, such cell types present screening groups with an alternative to conventional target-based assays where compounds are profiled for activity against specific targets of interest, namely, to use native cell types to configure a phenotypic screen where the compound effect on a disease-relevant process or cellular outcome can be measured in place of target protein activity. Phenotypic screens, which have been used widely in the past and which are attracting significant renewed interest, have the potential to identify completely new targets, pathways, or mechanisms of action (MoAs), and ultimately drugs that achieve a desired effect in new and perhaps unexpected ways.12,13
Differentiation protocols and subsequent genomic and functional characterization have identified sensory neurons with a nociceptor phenotype (referred to here as human induced pluripotent stem cell [hiPSC] sensory neurons).5,14 Transcriptomic and electrophysiological characterization of these cells has verified that they express markers of peripheral nociceptors, such as SCN9A, SCN10A, HCN, KCNQ2/3 P2X3, and acid sensing ion channels (ASICs) and are capable of firing multiple action potentials. In addition, it has recently been demonstrated that iPSC cells derived from pain patients and differentiated into sensory neurons can be used to model disease characteristics in vitro. 15 Despite the availability of differentiation protocols and extensive characterization data for select neuronal cell types, including commercially available sources, higher-throughput compound screening using neurons derived from human stem cells remains uncommon.16–22 This is likely due to several significant technical hurdles, including high cost, the challenge of reproducibly scaling up differentiation protocols and cell production, the poor efficiency of replating into microtiter assay plates at the appropriate state of maturation, and identifying a disease-relevant phenotype suitable for assay development.23–26 If these hurdles were overcome and assays with sufficient throughput identified, then chemogenomic and unbiased phenotypic screening approaches could be applied alongside and in combination with conventional target-based approaches to disease modification.
In the field of pain drug discovery, many years of target-based drug discovery have been unable to identify alternative or improved analgesics acting via different mechanisms to the long-established targets of opiates or NSAIDs. Highly validated genetic targets, such as SCN9A (NaV1.7),27,28 have not yet translated into new analgesics; hence, our desire was to open up new and complementary avenues for target identification, validation, and screening, through a phenotypic approach based on sensory neuron excitability. To enable this, we have addressed the challenge of reproducible, scaled-up human stem cell–differentiated sensory neuron production using a combination of “near-assay-ready” cryopreserved cells and in-assay plate neuronal maturation. We have developed and characterized the phenotypic relevance of a 384-well assay of neuronal hyperexcitability, using a veratridine-evoked depolarization of the cells. 29 Finally, we describe the results from the screening of a small-molecule chemogenomics library 30 to identify molecular mechanisms that modulate the evoked excitability phenotype. The results presented in this report demonstrate that the cells, in combination with phenotypic assays, have the potential for use in pain and other neuroscience drug discovery campaigns.
Materials and Methods
Materials
All cell culture reagents were purchased from Life Technologies (Waltham, MA) unless stated otherwise. All chemicals were purchased from Sigma-Aldrich (St. Louis, MO) unless stated otherwise.
iPSC Cell Culture
iPS cells were cultured on human embryonic stem cell (hESC)-qualified Matrigel (Corning Life Sciences, Flintshire, UK) as colonies using mTesr1 (Stemcell Technologies, Cambridge, UK) media with daily changes. To passage, cells were washed with Dulbecco’s modified Eagle’s medium (DMEM)/F12 and incubated with 80 µL/cm2 dispase (StemCell Technologies) at 37 °C for 4 min. The dispase was removed before washing three times with DMEM/F12 and scraping cells into 80 µL/cm2 DMEM/F12. Cells were centrifuged at 200g for 3 min and the pellet triturated in 600 µL of mTesr1 until the clumps of cells held in solution without sinking. The dissociated cells were transferred into a new vessel, with fresh hESC-qualified Matrigel, at the appropriate split ratio.
Sensory Neuron Differentiation
This method was adapted for large-scale cell production from the work previously described by Chambers et al. 5 Two days prior to commencing differentiation, iPSC cultures were dissociated using Accutase and centrifuged for 4 min at 200g. The cells were resuspended in mTesR1 containing the ROCK inhibitor Y-27632 at 10 uM and plated at 25,000 cells/cm2 in new T175 vessels precoated with growth factor–reduced Matrigel (Corning). Cells were fed with the following media to the timings outlined in Supplementary Table S1 :
KSR medium: 500 mL of KnockOut DMEM, 130 mL of KSR-XF, 1× NEAA, 1× Glutamax, and 0.01 mM β-mercaptoethanol
N2B27 medium: 500 mL of Neurobasal medium, 5 mL of N-2 supplement, 10 mL of B-27 supplement without vitamin A, 0.01 mM β-mercaptoethanol, and 1× Glutamax
2i supplement: 100 nM LDN193189 and 10 µM SB-431542
5i supplement: 100 nM LDN193189, 10 µM SB-431542, 10 µM DAPT, 10 µM SU-5402, and 3 µM CHIR99021
3i supplement: 10 µM SB-431542, 10 µM DAPT, 10 µM SU-5402, and 3 µM CHIR99021
Cryopreservation of iPSC Sensory Neurons
On day 11 of the differentiation protocol, cells were washed in phosphate-buffered saline (PBS), harvested with Accutase, centrifuged at 200g for 4 min, and resuspended in N2B27 medium with 10 µM Y-27632. The cells were then recentrifuged and resuspended in cold cryostor CS10 medium (Stemcell Technologies) containing 10 µM Y-27632 at a density between 1 × 107 and 5 × 107 cells/mL. Freezing was performed in cryovials in Coolcells (BioCision, San Rafael, CA) at −80 °C prior to transfer to liquid nitrogen storage.
Recovery and Plating of Cryopreserved iPSC Sensory Neurons
The required number of vials containing cryopreserved iPSC sensory neurons were rapidly thawed and diluted to the required cell density using plating medium (Neurobasal medium with N-2 and B-27 supplements, 1 mM Glutamax, 0.1 mM β-mercaptoethanol, 10 µM Y-27632, and 30 µg/mL gentamicin). Cells were seeded at 40 µL/well into clear-base 384-well tissue culture microwell plates coated with growth factor–reduced Matrigel (Greiner Bio-One Ltd., Stonehouse, UK). After 24 h the medium was replaced with neural growth medium (Neurobasal medium with N-2 and B-27 supplements, 1 mM Glutamax, 0.1 mM β-mercaptoethanol, 30 µg/mL gentamicin, 25 ng/mL nerve growth factor (NGF), 25 ng/mL glial-derived neurotrophic factor (GDNF), 10 ng/mL brain-derived neurotrophic factor (BDNF), and 10 ng/mL NT-3 [all human recombinant growth factors were purchased from Peprotech, Rocky Hill, NJ]). The neural growth medium was replaced every 3–4 days throughout the required culture time. Medium removal was achieved by inversion and gentle tapping onto sterile tissue paper. A 2 h treatment with 1 µg/mL mitomycin C was performed 4 days after plating.
Immunocytochemistry
Day 11 iPSC-derived sensory neurons were thawed as described above and seeded at 100,000 cells/well into 96-well plates coated with growth factor–reduced Matrigel in plating medium. After 2 weeks in culture, media was removed and cells were washed with PBS without calcium or magnesium and fixed with 4% formaldehyde for 10 min. Following two further washes with PBS, wells were blocked using 5% normal donkey serum in 0.3% Triton X-100/PBS (NDS/TX/PBS) for 1 h. Primary antibodies (rabbit anti-BRN3A [Sigma, B9684] at 1:200, goat anti-PRPH [Santa Cruz, Santa Cruz, CA, SC-7604] at 1:200, mouse anti-ISL1 [Abcam, Cambridge, UK, AB86501] at 1:200, and mouse anti-NEUN [Millipore, Burlington, MA, Mab377] at 1:100) or equivalent concentrations of isotype controls (rabbit isotype [Vector, Burlingame, CA, I-1000] at 1:20,000 [0.25 µg/mL], mouse isotype [Dako, Santa Clara, CA, M0744] at 1:100 [2.6 µg/mL], and goat isotype [R&D, Minneapolis, MN, ab-108c] at 1:1000 [1 µg/mL]) in 5% NDS/TX/PBS were applied to triplicate wells and incubated at 4 °C overnight. Wells were washed three times with 0.3% TX/PBS and incubated for 1 h at room temperature with 1:200 donkey Alexa-Fluor 488 (Thermo Fisher, Waltham, MA) against the appropriate species in NDS/TX/PBS. After three further PBS washes, wells were stained with 2 µg/mL Hoechst stain, washed again, and imaged on a Zeiss Axio Observer microscope.
Preparation of Test Compounds
Compound stocks were prepared at 30 mM in 100% DMSO. Prior to the assay, compounds were dispensed using an Echo 550 (Labcyte, Dublin, Ireland) and diluted to a 10× final assay concentration in DMEM/F12 containing 1% DMSO.
Veratridine-Induced Excitability Assay
Neural growth medium was replaced with 1× calcium 5 indicator dye (Molecular Devices, Berkshire, UK) prepared in DMEM/F12 (1:1) basal medium, and the plate was incubated at 37 °C for 15 min. Using an FDSS6000 (Hamamatsu, Welwyn Garden City, UK), 5 µL of test compound was added to each well and the plate incubated for a further 15 min. The fluorescence signal (Ex480:Em540) was measured preaddition and at 1 s intervals for 3 min postaddition of 5 µL of 50 µM veratridine (5 µM final assay concentration). Percent inhibition of the veratridine-induced calcium response in each well was calculated using control well treatments of 1 µM tetrodotoxin (TTX) to define maximum or 100% effect (HPE) and DMSO vehicle to define minimum or 0% effect (ZPE). The iPSC-derived sensory neurons were found to be sensitive to addition artifacts, and hence the FDSS reader internal temperature was maintained at 37 °C and a slow dispense speed of 2.5 µL/s, from 2.5 mm above the cell monolayer, was used.
Chemogenomics Screening and Data Analysis
The Pfizer Chemogenomics 3 (CG3) subset was recently described by Jones and Bunnage 30 and consists of 2746 unique small-molecule tool compounds, largely handpicked by medicinal chemist experts as probes for all major, currently ligandable target families. The manually curated target list for these compounds comprises more than 800 different molecular targets. Typically, compound activity against a designated target is in the low-nanomolar range, but at higher screening concentrations a compound will interact with many additional targets. For example, when potency annotations (Ki, IC50, and EC50) ≤5 µM from both internal and external databases are considered in addition to the manually curated targets, the overall number of targets modulated by the CG3 set amounts to 1111 and the average number of targets per compound increases from 1.4 to 4.3. As a richly pharmacologically annotated compound set, the CG3 library can therefore be used to elucidate mechanisms that evoke or modulate a phenotype of interest.
The CG3 library was screened in the veratridine-induced excitability assay at a single point concentration of 10 µM. Hits were determined using standard high-throughput screening analysis, and the data were then used as outlined below to generate a list of potential molecular targets capable of modulating the excitability phenotype. Note that targets for the compounds in the CG3 sets were defined using both manual curations and annotations from bioactivity databases (potency ≤5 µM). Also, note that hits were commonly known to modulate multiple targets, and in these cases, all of the targets were noted as active in the described enrichment calculation.
Screening data were deconvoluted and analyzed as follows: (1) Active (a) and inactive (i) compounds were determined for the whole CG3 subset. (2) For each target t, the active (at) and inactive (it) compounds were compiled. (3) Where at least one ligand for target t was active, a statistical test (one-sided Fisher’s exact test) was applied to compare the number of active and inactive compounds for target t to the number of active and inactive compounds for all other targets (defined as a – at and i – it, respectively). Fisher’s exact test was used to detect whether ligands for target t were significantly overrepresented among the hits from the phenotypic screen and highlight a potential association between target t and the phenotype. Fisher’s exact test returned a p value that could be interpreted as the probability that the observed screening result was obtained “by chance” without any underlying association between the target and phenotype. A low p value indicated that the modulation of target t could be responsible for the observed phenotype. The chance of finding at least one false-positive target at a given p-value threshold increased with the number of target hypotheses that were evaluated. To account for this, a Bonferroni correction was applied such that all p values were multiplied by the number of hypotheses that were tested. Here, all targets with a corrected p value ≤0.05 were considered significantly enriched.
Results
Validation of Near-Assay-Ready Cryopreserved iPSC Sensory Neurons and In-Plate Culture
The goal of this work was to develop a 384-well plate-based phenotypic assay with iPSC-derived sensory neurons. Critical to the success of this work was ensuring that differentiated and appropriately matured sensory neurons could be produced at a scale and quality that would enable efficient, high-throughput-compatible screening. Because of the complexity and time frame involved in generating differentiated sensory neurons, we believed that decoupling large-scale cell production from the assays was an essential prerequisite to screening. Hence, differentiated but immature neurons were cryopreserved at day 11 of the existing sensory neuron differentiation protocol. Following cryopreservation, cells were resuscitated and seeded directly into assay plates, where the maturation process could be performed without the need for further dissociation and plating.
Three separate batches of cryopreserved cells were produced for the assay development and screening work described, with the largest comprising 100 cryovials of 6 million viable cells, enough for approximately 20,000 wells of 384-well compound screening. Trypan blue viability estimates were performed routinely upon thawing and showed consistently high viability (mean of 91% ± 1.6 from n = 8; data not shown). An in-plate culture to further mature the plated neurons for up to 28 days within the 384-well assay plate proved practical using the simple described method of changing the medium by inversion onto absorbent paper. Critically, the cryopreservation step did not noticeably alter the neuronal phenotype or maturation process compared with equivalent “freshly differentiated” noncryopreserved cells used in other studies.5,14,15 An abundance of neuronal processes was routinely evident at 24 h postplating, with
Figure 1A
showing the typical microscopic appearance of the cells from 2 to 28 days’ maturation postplating. The cells were consistent in appearance between wells at a given time point, and we experienced no issues with microbial contamination. Neuronal morphology changed predictably with time in the 384-well plate, and by 28 days, a significant increase in the number of cells with larger-diameter soma was evident (
Fig. 1A
). Maturation times greater than 28 days resulted in the monolayer starting to contract away from the edges of the wells, and hence they were considered not suitable for screening applications. A brief mitomycin C treatment was required to prevent significant proliferation of nonneuronal cells and was essential for culture beyond 1 week. Peripherin (a type III intermediate filament located in sensory neurons of the peripheral nervous system), Brn3a, Islet 1 (homeodomain transcription factors that are coexpressed in sensory neurons), and NeuN (antigen localized in nuclei and perinuclear cytoplasm of mammalian neurons) have previously been identified as markers of an iPS-derived nociceptor phenotype and used to validate the technical success of the differentiation procedure.5,14 Immunocytochemistry demonstrated that neurons matured from cryopreserved cells were positively stained for the above markers in a manner consistent with previous publications (
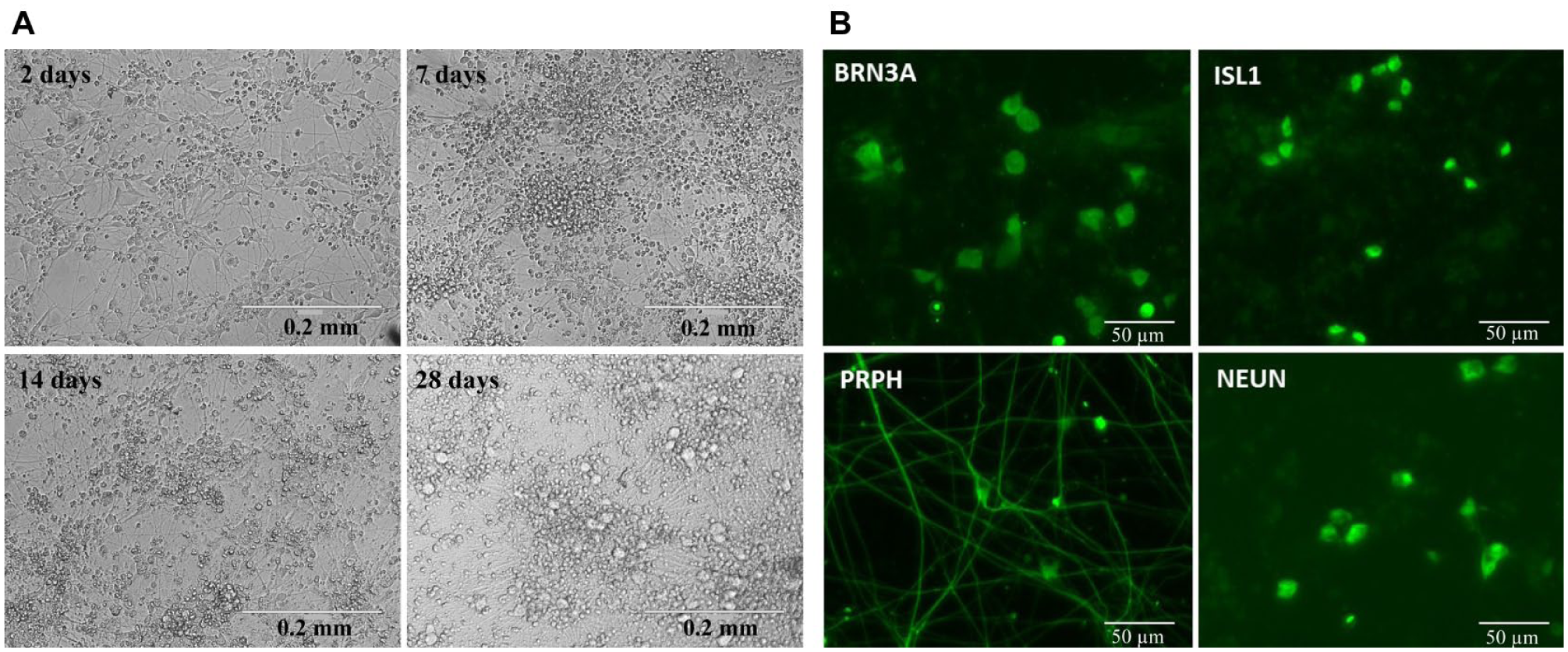
Microscopic characterization of cryopreserved iPSC sensory neuron recovery and maturation in assay plates. (
Development of a 384-Well Calcium Flux Assay Using Veratridine-Induced Depolarization of iPSC Sensory Neurons
We established a robust 384-well, high-throughput-compatible screening assay that measured veratridine-induced depolarization via the resultant intracellular calcium increase. The assay itself therefore resembled a conventional fluorescent kinetic calcium read commonly used throughout the high-throughput screening field.
Two days postplating, veratridine induced a large, reproducible Ca2+ increase in the iPSC sensory neurons with an approximate EC50 of 600 nM (
Fig. 2A
).
Figure 2B
demonstrates that this response was fully antagonized by blockers of voltage-gated sodium channels (NaV), such as TTX.
Figure 2
also shows the impact of cell number on assay quality and compound activity. The efficacy of the veratridine-induced Ca2+ response increased as expected with cell number (
Fig. 2A
), and we obtained good concentration–response inhibition curves to TTX, with all cell numbers in the range of 5000–40,000 cells/well (
Fig. 2B
). Neither the EC50 of veratridine nor the IC50 of TTX changed significantly with cell number (
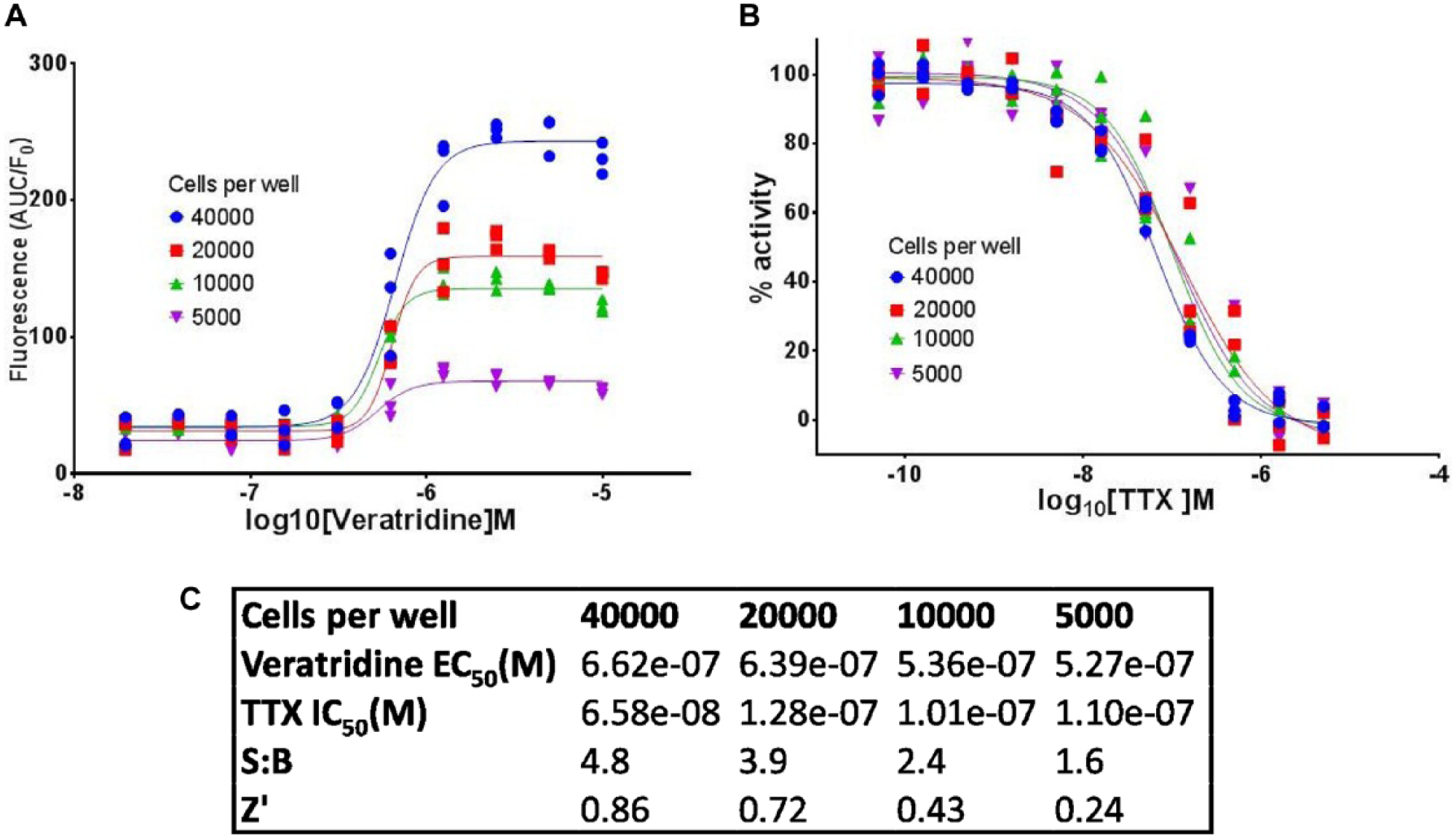
iPSC sensory neuron seeding density optimization in a 384-well-format Ca2+ flux assay. (
Figure 3 shows the kinetic traces for increase in intracellular Ca2+, in response to stimulation by veratridine and KCl (which is commonly used as a positive control for functional neuronal excitability). Both stimuli evoked a similar level of increase, albeit with different kinetics that resulted in a larger area under the curve (AUC) for veratridine. Importantly, veratridine, but not KCl-evoked Ca2+ changes were sensitive to TTX. This confirmed that the change in intracellular calcium evoked by veratridine was a consequence of NaV channel activity and not a result of direct opening of voltage-gated calcium channels, giving us confidence in the phenotypic relevance of the assay to pain as a crude mimicking of action potential firing or general cellular excitability. In addition, despite the complex cellular production process, Figure 3 highlights the low level of intraplate variability in cellular responses to the excitable stimulus.
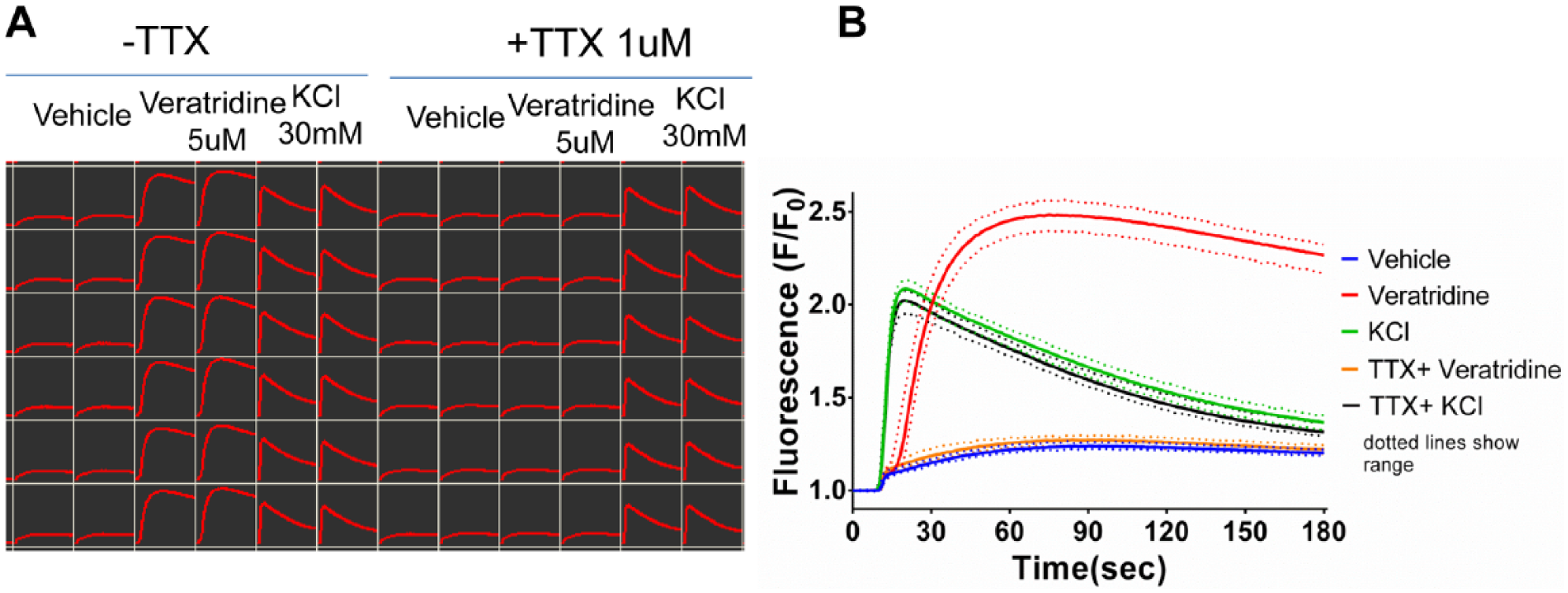
Demonstration of within-plate reproducibility in 384-well-format Ca2+ flux veratridine assay using replicate wells of key controls. (
Figure 4 shows the change in veratridine-induced excitability with in-plate maturation time. The efficacy of response to veratridine increased markedly upon neuronal maturation; however, we found that the veratridine EC50 and consequent TTX IC50 values did not change significantly when measured throughout the 28-day maturation time course.
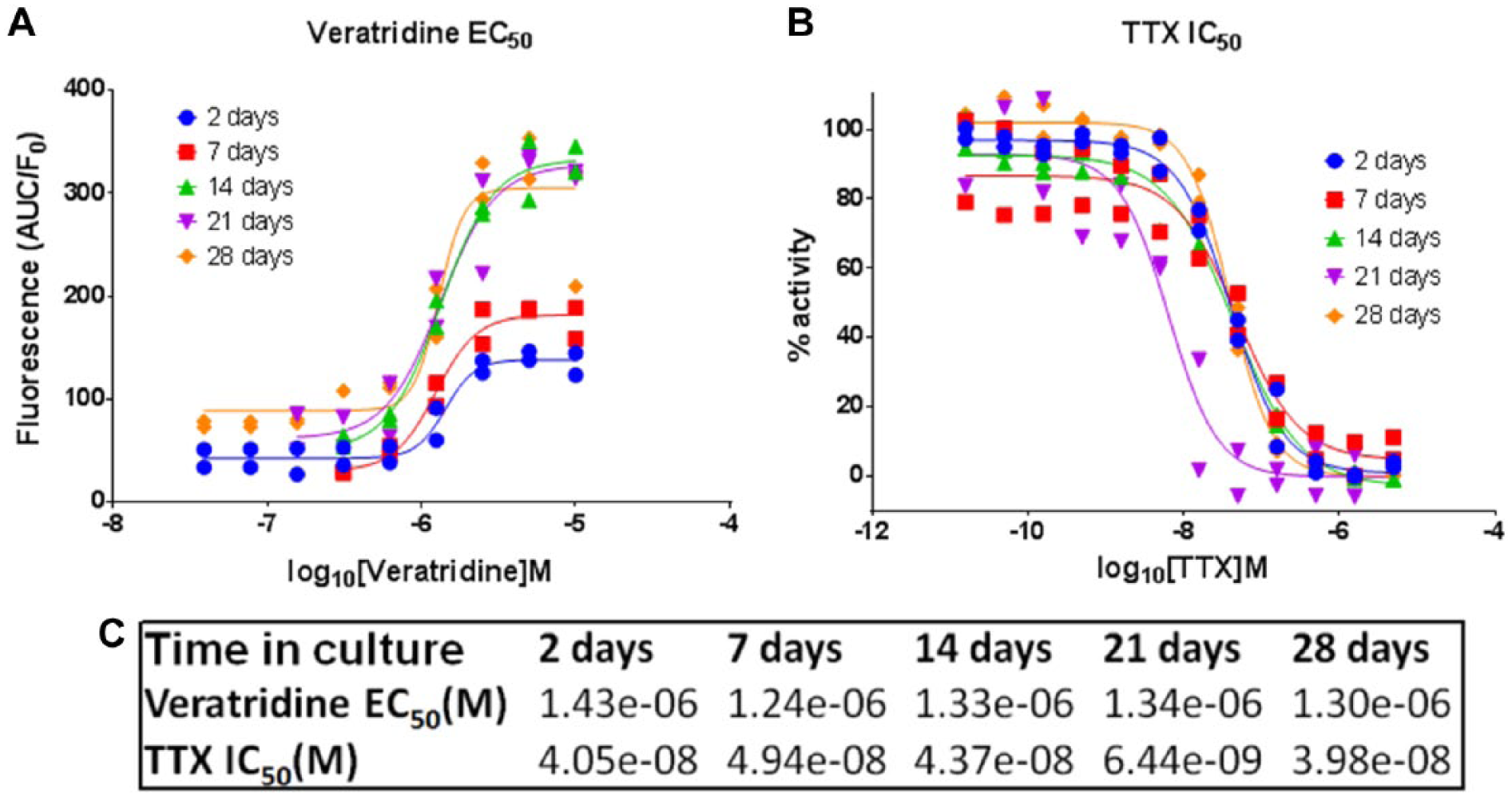
Effect of iPSC sensory neuron maturation time on veratridine-induced stimulation and TTX inhibition of Ca2+ flux. (
In summary, the optimized assay, using 35,000 cells/well and between 2 and 28 days postthaw and postplating of the cells, was suitable to support both single and full concentration–response curve screenings.
Screening of a Neuronal Hyperexcitability Phenotypic Validation Subset
To further characterize the assay’s phenotypic relevance to pain and other hyperexcitability disorders, a bespoke validation set of 350 compounds was collated (the “pain subset”). Specifically, this subset contained clinically relevant analgesics, a range of approved neurological drugs, available tool compounds targeting validated or proposed analgesic targets or pathways, and available tool compounds for the ion channel family. An initial 384-well screen was performed with this validation set using hiPSC sensory neurons at 2 days postplating and compound test concentrations of 30 nM, 300 nM, 3 µM, and 30 µM. The resulting distribution of activity in the assay across these test concentrations is shown in Figure 5A . These data indicated that a 3 µM screening concentration provided the optimum balance between sensitivity to inhibition and minimizing the level of off-target activity that induced the high hit rate observed at 30 µM. The interplate reproducibility of 3 µM data is shown in Figure 5B , with replicates showing a high degree of correlation (R2 = 0.88). Hence, we opted to continue using this validation subset at 3 µM to characterize the influence of increased neuronal maturation time on compound activity. The resulting distribution of activity in the assay across the 4-week maturation time course is shown in Figure 5C and revealed a general reduction in compound effect with maturation time, particularly noticeable at 4 weeks postplating. Figure 5D shows a comparison of compound activity for the entire validation subset at the 2- and 28-day time points, with very few compounds showing equivalent levels of activity at the later time point. The Z′ values of the assay plates steadily reduced as the time course proceeded but remained acceptable throughout (ranging from 0.70 at 48 h to 0.61 at 4 weeks).
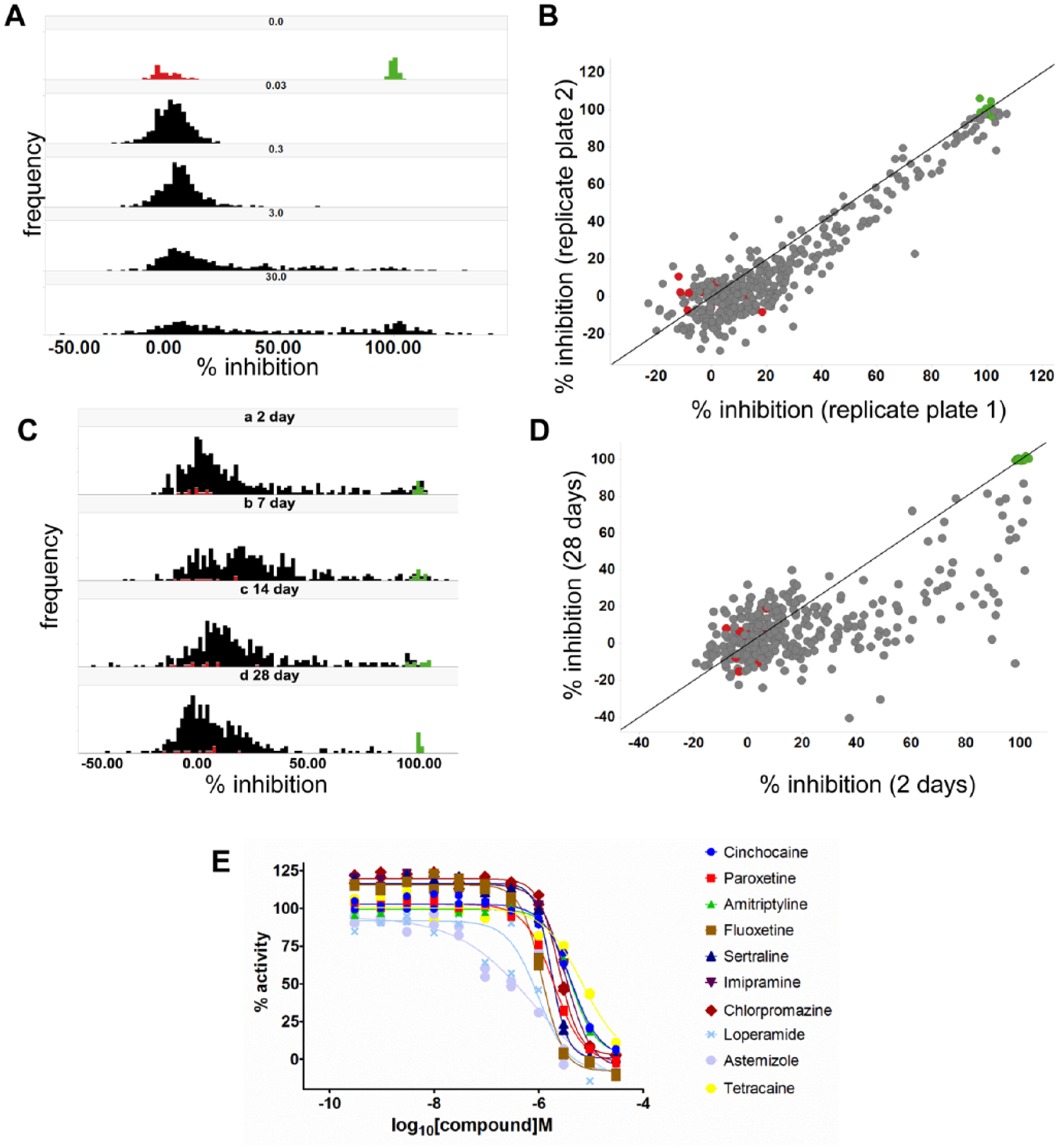
Validation screen of the pain subset. (
To investigate the mechanisms identified as inhibitors of the veratridine-evoked excitability phenotype, active compounds were characterized in concentration–response format. Figure 5E shows IC50 curves for selected active compounds that are known drugs with an established capacity to modulate neuronal excitability. Across all compounds, the IC50 determined in this phenotypic assay was right-shifted relative to the known target-based potency of the molecules (exemplified in Supplementary Table S2 ).
The purpose of the validation set was to confirm that a significant number of known mechanisms or targets of hyperexcitability were detected. To further test and apply the assay for new target or compound identification, we screened Pfizer’s CG3 subset.
Screening of CG3 Subset and Data Deconvolution to Obtain a List of Potential Targets and Mechanisms Relevant to Sensory Neuron Hyperexcitability
After successfully validating the assay using the pain subset, we proceeded to screen the larger, more diverse CG3 compound set with the aim of validating the assay’s capacity to identify new potential targets, mechanisms, or MoA. We used a compound concentration of 10 µM initially and then retested compounds with >80% inhibition using 10 and 1 µM concentrations in duplicate. The activity distribution of all compounds is shown in Figure 6 . Seven percent of compounds (185 compounds) caused >80% inhibition at 10 µM, and most were active when retested, with >95% of actives showing >50% inhibition upon retest. The screen scaled well to this library size, continuing to be reproducible with Z′ values of 0.6–0.9 (data not shown).
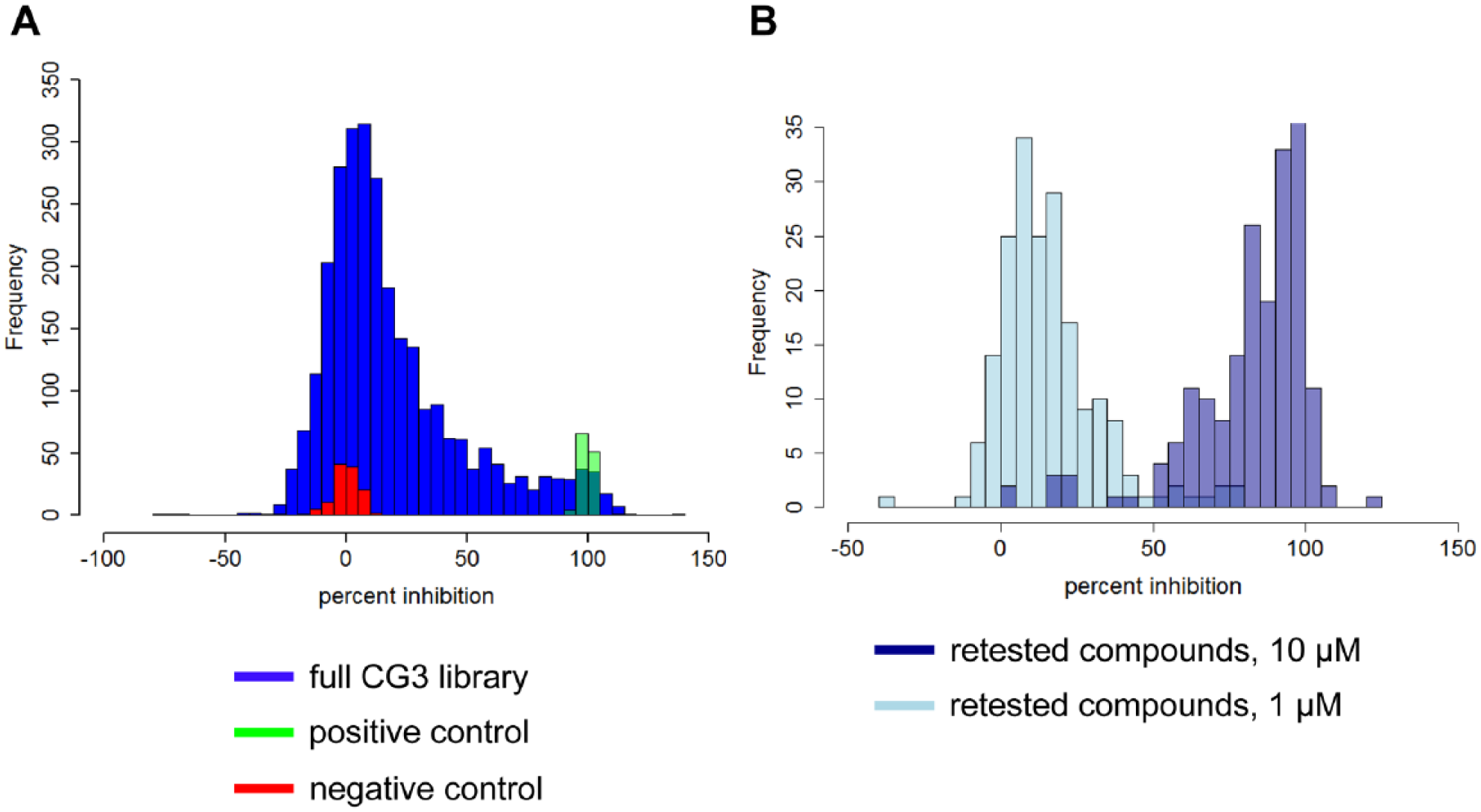
Distribution of percent inhibition activity for compounds in the 2700-compound CG3 set. (
To obtain a list of possible active targets from these results, a statistical analysis was performed, which correlated the veratridine screen results of all compounds with their known target activities as far as these were available. As hits showed good reproducibility when retested, we used the 185 active molecules from the initial screen to determine potential targets of interest. A Fisher’s exact test identified 40 targets whose ligands were significantly enriched among the hits (Bonferroni-corrected p value ≤0.05). The full list of target names from this analysis is shown in Supplementary Table S3 . Many of these targets have known links to pain, for example, µ opioid receptor, 31 tachykinin receptors, 32 melanocortin receptors, 33 serotonin receptors, 34 and voltage-gated sodium channels, including NaV1.7. 35
For some known pain targets, few ligands were available in the chemogenomics set, and hence it was challenging to achieve statistical significance in the Fisher’s exact test as the calculated significance is dependent on the sample size. To ensure inclusion of interesting targets that were underpowered in our study because of their limited number of ligands, we created a second list of 54 further potential targets. For this list, shown in Supplementary Table S4 , we required that at least 50% of the ligands of a target were hits in the phenotypic assay. This “rescued” some known pain targets, such as the neuropeptide Y receptor and the neuropeptide FF receptor 2.36,37
An in-house literature search engine was used to qualify links between the targets on our two hit lists and pain. For 50 of the overall 94 targets, at least three articles could be found that mentioned the target and the term pain together in their title or in several sentences in the abstract.
For the screen conducted at 1 µM compound concentration, we used 40% inhibition as a more stringent cutoff to determine hits. This resulted in the identification of 13 active compounds with the following manually curated targets: 5-serotonin receptor 6, µ opioid receptor, voltage-gated potassium channels KCNC2 and KCNA4, voltage-dependent N-type calcium channel CACNA1B, integrins α4 and β1, farnesyltransferases CAAX box α and β, neuropeptide FF receptor 2, tachykinin receptor 1, interleukin 2, and chemokine (C-C motif) receptor 1 (CCR1). That means that, similarly to the observations made for the 10 µM screen, known pain-related targets populate the list, verifying that the screen is picking up relevant mechanisms.
Thus, the pain-relevant hiPSC sensory neurons in conjunction with a phenotypic end point (block of veratridine-evoked excitability) have been used in a high-throughput-compatible context to identify, among other hits, known targets of pain drug discovery. This assay can now, therefore, form the basis of a high-throughput phenotypic screening campaign and be further developed, for example, by the use of patient-derived cells and/or different stimuli, to evoke excitability.
Discussion
In recent years, there has been a renewed interest in phenotypic screening for the purpose of identifying therapeutics with unknown or unpredicted MoA. 2 However, the significant challenge of scaling and validating assays with disease-relevant cells is a bottleneck to success, and in the field of pain research, this barrier has limited work on in vitro–based phenotypic screens for pain.
Hence, our first challenge was to build on recent publications that described the differentiation and characterization of hiPSC-derived sensory neurons,5,14 to enable scaled-up and screening-compatible cell supply. Decoupling cell supply from cell-based assays through cryopreserved assay-ready cells is a standard approach for screening in the pharmaceutical industry, 38 and we have shown that this can be extended to hiPSC sensory neurons. Our early experiments showed that cryopreserved cells were indistinguishable from equivalent freshly differentiated cells, and since we considered coordinating repeated directed differentiation runs with compound screening to be impractical, we chose to exclude their further use in this study. Our data indicate that batch-to-batch variability of large-scale cryopreserved batches was not a significant problem, and we predict that this would translate into larger screening campaigns through combining batches of QCed cells.
Terminally differentiated neurons from hiPSCs undergo further maturation or functional change in culture over time.14,15 Therefore, we ensured that the cell culture and assay was flexible to support the screening of neurons at different maturation time points. To avoid the negative impact on cell health of replating differentiated neurons, we cultured and matured the cells postcryopreservation directly in the 384-well assay plates. This was successful up to 28 days with simple media changes that minimized potential variability and problems through intrusive pipetting or aspiration near the monolayer, although further investigation of automated media changes would be desirable. For larger-scale screening campaigns, maturation of the neurons at scale in flasks would be advantageous, but represents a high technical hurdle with no guarantee of success due to the aggregation of neuronal cell bodies and the abundance of tangled neuronal processes. Development of such protocols could also enable screening at maturation time points beyond 28 days, which would be desirable since previously published work demonstrates that the phenotype continues to change in culture.14,15
For the development of a phenotypic screen using iPSC-derived sensory neurons, we employed veratridine, as a nonspecific opener of NaV channels, to evoke membrane depolarization and mimic the depolarization induced by nociceptive stimuli via sensory receptors. The downstream events of NaV-mediated depolarization are the consequent increase in intracellular Ca2+ via CaV channel activation and triggering of neurotransmitter release. We opted to measure neuronal function via Ca2+ changes since such transient fluctuations of intracellular Ca2+ levels are often used as a measure of neuronal excitability and in a plate-based context are amenable to higher-throughput, fluorescent plate-based assays. The 384-well calcium flux assay was robust and reproducible, demonstrating that signal size was not an inherent problem with this native cell type and resulted in clear identification of known blockers of NaV activity through antagonism of the veratridine-induced depolarization. The relatively high concentration of veratridine used in this assay was an important factor in determining the observed assay robustness but was also a likely contributor to the observed right shift in potency of NaV blockers tested ( Suppl. Table S2 ). It will be interesting to observe whether future experiments conducted with a lower veratridine concentration or alternative, more physiological stimulus will result in increased potency of both NaV channel blockers and other mechanisms. Of further relevance to assay robustness, we paid careful attention to technical precautions to minimize temperature or mechanical artifacts with these potentially sensitive cells; specifically, 37 °C was maintained throughout incubation with Ca2+ indicator dye, and inside the FDSS plate reader, and a slow dispense from 2.5 mm above the cells was used.
For any phenotypic screening campaign, it is crucial to understand the physiological relevance of the assay (in terms of both the cell type and end point). Since veratridine causes persistent activation of sodium channels that contrasts with physiological state-dependent voltage activation, we were keen to validate that compounds acting via a range of relevant mechanisms were capable of blocking the Ca2+ increase induced by a high veratridine concentration. For the validation work, we collated a (384-well) plate of test compounds (the pain subset) that was designed to enable characterization of the assay phenotype under a variety of conditions (e.g., different maturation times). The screening data from the pain validation subset showed that maturation times of 2–28 days did not yield greater differences in the nature of the hits identified, despite a trend toward loss of both potency and number of active compounds with maturation. This likely reflects the observed increase in efficacy of veratridine-evoked excitability with time, as well as suggesting no fundamental shifts in the molecular mechanisms that are responsible for the responses measured. From screening of the pain subset, a range of neuronal modulators were identified, in addition to specific NaV channel blockers. As expected, some known analgesic mechanisms were inactive, for example, NSAIDs likely due to the lack of nonneuronal cells that are a significant site of action and TrkA blockers due to the acute compound incubation time not allowing changes in sensitization to result in changes to excitability (data not shown). In addition, the potency of active compounds was right-shifted relative to known target-based potency, which could be a result of the different end point measured or the “harsh” nature of the veratridine stimulus. However, the data gave us sufficient confidence to screen the larger and more diverse chemogenomics compound subset.
The Pfizer CG3 library is 2700 compounds covering more than 1000 distinct biological targets. 30 Many of these ligands are well characterized, with detailed pharmacological data available, thus making it possible to deconvolute phenotypic screening results to obtain a list of potential targets. Through an analysis of the correlation between the activity of compounds in the veratridine screen and their activity in a broad range of other assays in which the compounds had previously been tested, we were able to generate a list of potential targets for further investigation. Many of these targets contained a preexisting link to pain, and hence the results have given us confidence in the phenotypic relevance of the cells and 384-well assay for continued use in pain and other neuroscience research.
If the assay is to be employed as a screen for the identification of active compounds with unknown modes of action, then the development of further secondary assays using the same cell preparation will be an important next step in order to characterize hits mechanistically. Secondary assays could range from relatively simple assessment of direct effects on calcium channels by evaluating the blocking of KCl-induced calcium responses through to more complex and technically challenging assays, such as those discussed below. Despite the encouraging results presented in this report, veratridine-induced stimulation is not a truly disease-relevant stimulus. Future studies could improve the relevance of the assay stimulus; for example, electric field stimulation (EFS) using a multielectrode array platform would be one interesting possibility. This work was conducted with cells derived from “normal” human donors; however, similar studies using iPSC lines derived from genetically characterized individuals, such as patients with erythromelalgia, 15 will further our understanding of how excitability is changed in the disease state. Although beyond the scope of the work described here, it is also likely that in addition to the source of the iPSC lines, further development of physiological relevance will be possible through techniques involving coculture with other cell types 39 and the use of more in vivo–like culture conditions (media, extracellular matrix, and other environmental and mechanical stimuli). However, we consider this report to mark the beginning of a path that will lead to the development of highly relevant disease-modeled in vitro phenotypic screens, solving some of the technical hurdles and, in the meantime, providing researchers in the pain field with some early ways to capitalize on the application of the recently described hiPSC sensory neurons for pain research.
Supplemental Material
DS_DISC764678 – Supplemental material for Plate-Based Phenotypic Screening for Pain Using Human iPSC-Derived Sensory Neurons
Supplemental material, DS_DISC764678 for Plate-Based Phenotypic Screening for Pain Using Human iPSC-Derived Sensory Neurons by Peter Stacey, Anne Mai Wassermann, Laura Kammonen, Emma Impey, Anna Wilbrey and Darren Cawkill in SLAS Discovery
Footnotes
Supplementary material is available online with this article.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: All authors were employees of Pfizer who completed this work in the course of their employment. No external funding was used.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.